

Abstract
Although the immune system evolved to fight infections, it may also attack and destroy solid tumors. In most cases, tumor rejection is initiated by CD8+ cytotoxic T lymphocytes (CTLs), which infiltrate solid tumors, recognize tumor antigens, and kill tumor cells. We use a combination of two-photon intravital microscopy and immunofluorescence on ordered sequential sections to analyze the infiltration and destruction of solid tumors by CTLs. We show that in the periphery of a thymoma growing subcutaneously, activated CTLs migrate with high instantaneous velocities. The CTLs arrest in close contact to tumor cells expressing their cognate antigen. In regions where most tumor cells are dead, CTLs resume migration, sometimes following collagen fibers or blood vessels. CTLs migrating along blood vessels preferentially adopt an elongated morphology. CTLs also infiltrate tumors in depth, but only when the tumor cells express the cognate CTL antigen. In tumors that do not express the cognate antigen, CTL infiltration is restricted to peripheral regions, and lymphocytes neither stop moving nor kill tumor cells. Antigen expression by tumor cells therefore determines both CTL motility within the tumor and profound tumor infiltration.
T cell–mediated immune responses require the active migration of T cells in tissues and the establishment of specific cell–cell contacts with other cell types. T cell migration was analyzed intravitally in the last few years using noninvasive two-photon microscopy (1–3). In lymph nodes, T lymphocytes move using an “ameboid”- like migration mode and display extremely high instantaneous velocities (up to 25 μm/min) (4, 5) compared with DCs or tumor cells (6, 7). The resulting high mean velocities allow naive T cells to “scan” high numbers of DCs seeking for their MHC-peptide ligands (8, 9). Once a specific ligand was encountered, T cells stop their migration and establish antigen-specific contacts with DCs. The duration of these contacts is determined by the maturation state of the interacting DCs and the eventual presence of regulatory T cells. In most experimental systems, long lasting (hours) contacts occurred during the induction of effective priming (4, 5, 9), whereas short interactions (minutes) were observed in tolerogenic conditions (10, 11) or in the presence of regulatory T cells (12, 13). Shakar et al. (14), however, only found modest differences in the mobility of CD4+ T cells during the induction of priming and tolerance.
During antiviral, antitumor, or autoimmune responses, for example, differentiated effector T cells migrate in nonlymphoid tissues to interact with target cells. Similar to lymph nodes, effector T cell migration in rat spinal cord sections during experimental autoimmune encephalomyelitis displays “ameboid” characteristics and high mean velocities, showing antigen-specific arrests upon autoantigen recognition within the spinal cord (15). Few studies also analyzed the effector phases of immune responses by NK T cells or NK cells (16, 17). The killing of target cells by cytotoxic T lymphocytes was recently imaged in lymph nodes. Regulatory T cells were thus shown to reduce the killing efficiency of CTLs by interfering with the polarized secretion of cytotoxic granules (18). The trafficking of CD8+ cytotoxic T cells in tumor- bearing mice was analyzed recently using magnetic resonance or positron-emission tomography imaging (19–21), but the actual resolution of these methods is too low for single cell imaging. CTL-mediated cytotoxicity in solid tumors, in contrast, has not been imaged thus far.
Although adoptive transfer of T cells induces the rejection of established solid tumors, both in murine models and in cancer patients (22–25), very little is known about the actual mechanisms of tumor invasion by CTLs and rejection. For example, the relative contribution of direct tumor cell killing by cytotoxic T cells compared with killing by other effectors of innate immunity is still uncertain (26, 27). Various hematopoietic cells, including macrophages, natural killers, and neutrophiles, were shown to infiltrate the tumors after CTL responses (28, 29).
To analyze the infiltration and tumor cell killing in solid tumors, we used a combination of tumor cells and CTLs expressing recombinant fluorescent proteins. Both conventional and intravital, one- and two-photon microscopy allowed us to image the dynamics of CTL motility and tumor infiltration by CTLs. We thus describe a complex, antigen-dependent topography of invasion and motility of CTLs in solid tumors.
RESULTS
OT1-GFP cells infiltrate and reject OVA-expressing tumors
To analyze the role of antigen recognition in tumor infiltration and in CTL motility within tumors, we inoculated individual C57BL/6 mice s.c. with two tumors: the EL4 thymoma in one flank and the EG7 (OVA-expressing EL4 cells) in the other flank. Naive GFP-expressing, OVA-specific, TCR-transgenic OT1 cells (OT1-GFP) were adoptively transferred to the mice when the EL4 and EG7 tumors were 500–1,000 mm3 (around day 10). OT1-GFP cells induced the complete rejection of EG7, but not of EL4 tumors, within 6–8 d (Fig. 1 A). After the adoptive transfer of OT1-GFP cells, the tumors continued to grow for 3 d. After 3–4 d, the tumor stops growing before the size actually starts decreasing at days 5–6. At days 7–8, the tumors continued to shrink and disappeared completely. We defined two periods for the analysis of the active rejection process: the “early” rejection phase corresponds to days 3–4 after adoptive transfer when the tumor stops growing; the “late” rejection phase corresponds to days 5–6 when the size of the tumors starts to decrease, but before complete rejection (which occurs at days 7–8).
Figure 1.

Activation of specific T cells in antigen-expressing tumors. C57Bl6 mice were injected s.c. with five 105 EG7 and EL4 tumor cells in either flank. 8–10 d later, the mice were adoptively transferred with 107 purified naive CD8+ cells from OT1-GFP mice. (A) Evolution of EG7 (▪) and EL4 (□) tumor size after adoptive transfer. (mean ± SD, n = 5 independent experiments). Early phase (day 3–4) and late phase (day 5–6) were defined in Material and methods. (B) Density of OT1-GFP cells within EG7 and EL4 at early (day 3) and late (day 6) phase of tumor rejection were calculated from tumor section by counting the number of OT1-GFP cells per mm2. (C) Expression of CD69 (hatch marked) and CD62L (gray) activation markers in GFP+ cells as analyzed by flow cytometry (dLN, draining LN) (mean ± SD, n = 3–6). (D) IFN-γ production of GFP+ cells in EG7 and EL4 tumors after 3 h of treatment with Brefeldin A without antigen restimulation (mean ± SD, n = 5). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
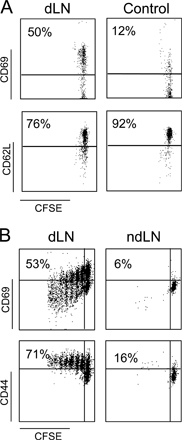
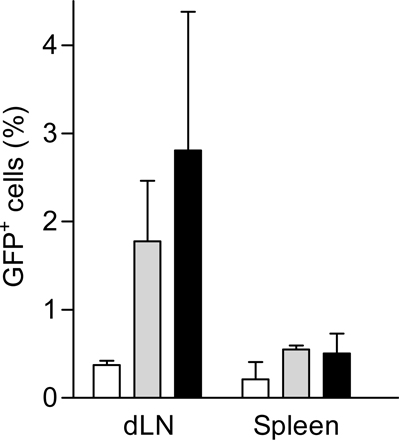
We first sought to analyze the activation of the transferred OT1 cells in the draining lymph node or nondraining lymph node EG7 tumors. As shown in Fig. S1 (available at http://www.jem.org/cgi/content/full/jem.20061890/DC1), OT1-GFP cells are primed in EG7 draining lymph nodes but not in the nondraining ones. The transferred cells transiently up-regulate CD69, maintain high levels of CD44, and display decreased CD62L expression (Fig. S1, A and B and reference 30). OT1 activation resulted in the expansion of the OT1-GFP cells up to fivefold at day 3 (0.37% ± 0.02 versus 1.8% ± 0.3 of total cells; P = 0.005) (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20061890/DC1). The slight increase of OT1-GFP cell numbers in the spleen (0.20% ± 0.11 to 0.55% ± 0.1; P = 0.006) is probably caused by the recirculation of activated cells rather than a local antigen-induced expansion (30). Therefore, transferred OT1 cells are selectively activated in the antigen-expressing tumor draining lymph nodes.
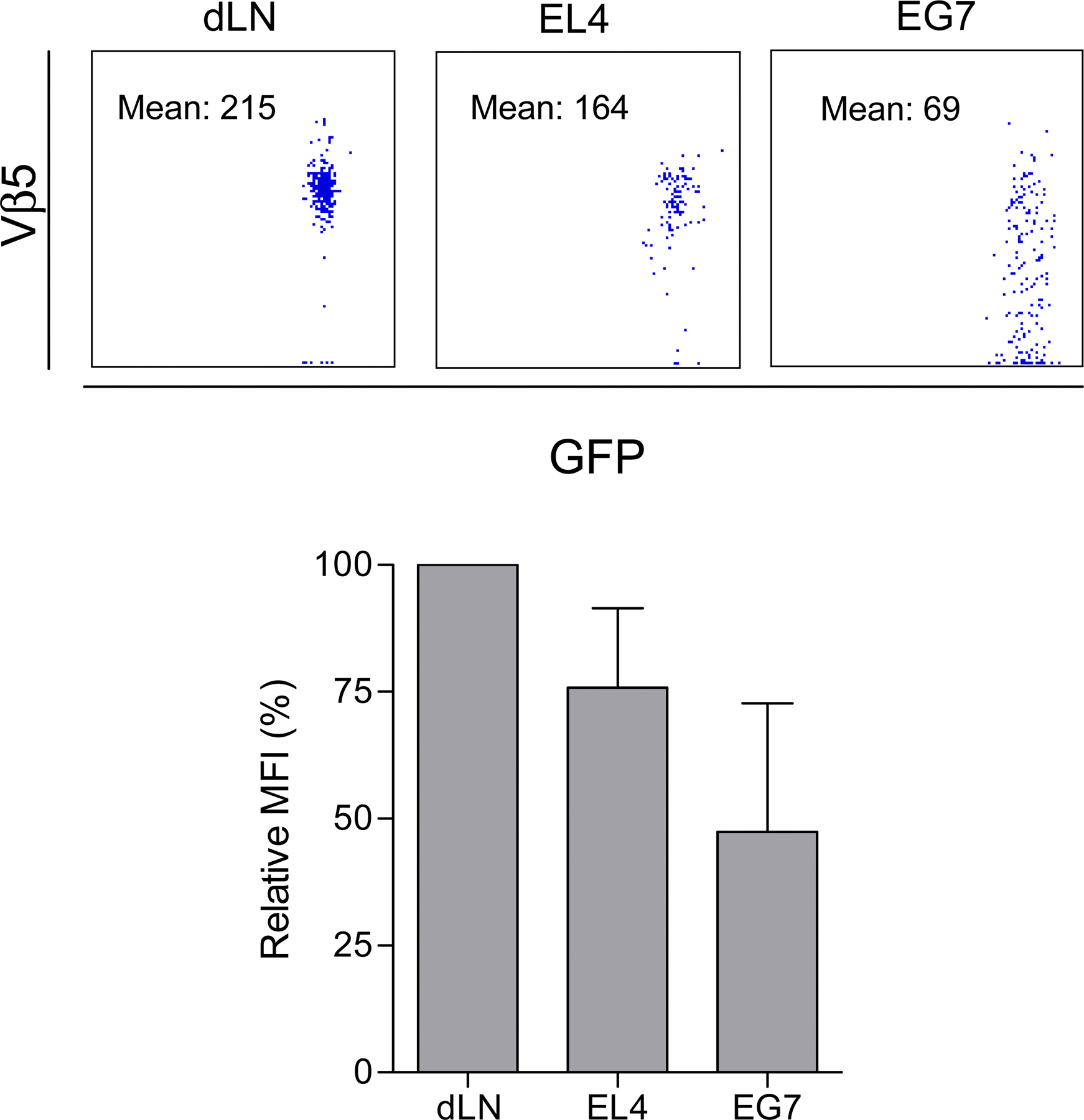
The infiltration of the tumors by the OT1 cells was analyzed next by measuring the density of OT1-GFP cells in frozen sections of either tumor at days 3–4 after adoptive transfer. Both EG7 and EL4 tumors were infiltrated with OT1-GFP cells (Fig. 1 B), consistent with the observation that activated primary CD8+ T cells migrate to nonlymphoid tissue regardless of their site of activation (31). The number of OT1-GFP cells infiltrating EG7 was, as expected, higher than the number infiltrating EL4, especially at day 6 after adoptive transfer (327 ± 50 cells/mm2 versus 27 ± 2 cells/mm2) (Fig. 1 B). The OT1-GFP cells infiltrating both EL4 and EG7 tumors expressed low levels of CD62L, attesting that both cell populations had been previously activated (Fig. 1 C). In contrast, only the OT1-GFP cells infiltrating OVA-expressing EG7 tumors, but not those infiltrating the EL4 tumors, reexpressed high levels of the early activation marker CD69 (Fig. 1 C) and produced IFN-γ (Fig. 1 D). The EG7-infiltrating OT1-GFP cells, but not OT1-GFP cells infiltrating EL4, also down modulated their TCR, as attested by the decreased expression of Vβ5 (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20061890/DC1). We conclude that the parallel inoculation of antigen-expressing and antigen-nonexpressing tumors to the same mouse induced the infiltration of both tumors, although the T cells accumulated preferentially in the antigen-expressing tumors. T cell activation and effector function (CD69 expression, IFN-γ production, and killing), however, were only detectable in CTLs infiltrating the antigen-bearing tumors.
Antigen expression determines the motility of CTLs in tumors
To investigate the role of antigen recognition in the motility of antigen-specific CTLs within tumors, we next performed intravital time-lapse two-photon imaging. 8–10 d after s.c. injection of the tumors, OT1-GFP cells were adoptively transferred into tumor-bearing mice. At days 3–4 (early phase of rejection) or 5–6 (late phase of rejection) after adoptive transfer of OT1-GFP cells, the mice were anesthetized and injected i.v. with fluorescent dextran to visualize blood vessels. As shown in Videos S1 and S2 (available at http://www.jem.org/cgi/content/full/jem.20061890/DC1), during the early phase of rejection OT1-GFP moved actively in EL4 tumors but had reduced motility in EG7 tumors. During the late phase of rejection, OT1-GFP cells moved actively in both tumors (Videos S3 and S4, available at http://www.jem.org/cgi/content/full/jem.20061890/DC1). Individual cell trajectories were tracked (representative examples are shown in Fig. 2, A and B), and the mean velocity, arrest coefficient (the proportion of time every individual remains arrested), and confinement ratio (the ratio of the distance between the initial and the final positions of each cell to the total distance covered by that cell) were determined (Fig. 2 C).
Figure 2.

CTLs motility during tumor infiltration and clearance. (A) TPLSM images (260 × 260 μm) of OT1-GFP cells (green) within tumors (EG7 and EL4) of anesthetized mice at early (day 3) and late (day 5) time points after adoptive transfer. Vascular vessels are labeled by 70 kD rhodamine-dextran (red); typical migratory path (white) examples are shown. Bar, 47 μm. (B) Overlay of 50 individual T cell tracks plotted after aligning their starting positions. (C) Scatter plots of T cell mean velocity, confinement ratio, and arrest coefficients of all cells analyzed. Data from two to four independent experiments were pooled (day 3: EG7, n = 675; EL4, n = 275; day 5: EG7, n = 1249; EL4, n = 126). ns, not significant. ***, P < 0.001.
During the early phase of rejection, OT1-GFP cells moved with a mean velocity of 4 ± 2 μm/min in the EG7 tumors compared with 10 ± 4 μm/min in the EL4 tumors. The migration trajectories were more restrained in the EG7 compared with the EL4 tumors (confinement ratios of 0.4 ± 0.2 versus 0.6 ± 0.2). Consistently, the arrest coefficient was higher in EG7 (33 ± 17%) than in EL4 (13 ± 17%) tumors. During the late phase of rejection, the motility of OT1-GFP cells in EG7 increased to 8 ± 3 μm/min−1 to become similar to the mean velocity of OT1-GFP cells in EL4 tumors (10 ± 5 μm/min−1). The migration trajectories also became less confined (0.6 ± 0.2) and identical to the ones observed in EL4 tumors. Finally, the arrest coefficient in EG7 tumors dropped to 14 ± 16%, which is close to that of OT1-GFP cells in EL4 tumors (16 ± 21%). We conclude that CTLs transiently stop moving in antigen-expressing tumors during the early phase and then resume their migration during the late phase when the size of the tumor starts to decrease.
OT1-GFP stop in contact with OVA-expressing tumor cells
To investigate if this transient arrest of migration in the antigen-expressing tumors is related to the killing of tumor cells by the CTLs, EG7 and EL4 tumor cells were transfected with GFP-encoding plasmid. The GFP-expressing tumors behaved like their GFP-negative counterparts in terms of tumor growth and rejection after transfer of OT1 cells. Tumor cell viability in vivo was determined at different time points by the level and the distribution of intracellular GFP using two-photon microscopy. During the early phase of tumor rejection, tumor cells formed a dense network of bright living cells in both EL4-GFP and EG7-GFP tumors, similar to that observed in the absence of OT1 cells (Fig. 3 A). In the late phases of rejection, in contrast, EG7 tumors, but not EL4 tumors, were mainly composed of residual dead tumor cells— i.e., cells displaying lower GFP levels and a GFP-negative, compact nucleus. In EG7 tumors during the late phase, the enhanced second harmonic generation (SHG) signal reflected an increase in the density of collagen fibers (Fig. 3 A), suggesting increased fibrosis in this region of the tumor.
Figure 3.

Tumor cell clearance during early and late phase. (A) Representative TPLSM images (160 × 160 μm) of GFP-EG7 or GFP-EL4 tumors during early (day 3) and late phase (day 6) of tumor rejection after adoptive transfer of 107 OT1 cells. Vessels (red) are imaged by intravenous injection of 70 kD rhodamine-dextran (2.5 μg/ml) and collagen fibers (blue) using SHG signals. Capture parameters were identical for all images. Bar, 44 μm. (B) TPLSM images (150 × 150 μm) of GFP-EG7 (green) during early (day 4) and late (day 6) phase of rejection after intratumoral injection of propidium iodide (blue). Bar, 41 μm.
To determine if the cells expressing low levels of GFP observed in the late phase were dead tumor cells or macrophages having engulfed dead tumor cells, we injected the tumor-bearing mice with propidium iodide (PI). As shown in Fig. 3 B, in EG7 tumors during the early phase, when most tumor cells are still alive, few cells are stained with PI. During the late phase, in contrast, all cells displaying low levels of GFP with dark GFP-negative nuclei are labeled by PI (Fig. 3 B). We conclude that during tumor rejection, dead tumor cells accumulate in the periphery of the tumors, most likely before being cleared by macrophages.
The change in the dynamics of OT1 cell motility in the EG7 tumors between the early and late phases could therefore correspond to the arrests of OT1 cells during tumor cell killing. To investigate this possibility, we analyzed the dynamics of OT1 cells expressing cyan fluorescent protein (OT1-CFP) in tumors expressing GFP during the early or late rejection phases (Videos S5 and S6, available at http://www.jem.org/cgi/content/full/jem.20061890/DC1). As shown in Fig. 4 A and Video S5, during the early phase the majority of the arrested OT1-CFP cells are in contact with living EG7-GFP tumor cells. During the late phase, OT1-CFP cells seem to resume motility in regions where the tumor cells are dead (Fig. 4 B and Video S6). We therefore compared the dynamic of the OT1 cells in regions where the tumor cells are alive or dead. As shown in Fig. 4 C, the OT1-CFP cells displayed lower mean velocity (1.1 ± 0.4 μm/min) and confinement ratios (0.15 ± 0.05) and higher arrest coefficients (82 ± 11%) in regions where the tumor cells are alive during early phase than in regions where the tumor cells are dead during late phase (mean velocity, 6.4 ± 2.5 μm/min; confinement ratio, 0.7 ± 0.2; and arrest coefficient, 17 ± 18%). During the late phase, we eventually found regions of the tumors where at least parts of the EG7-GFP cells are still alive. OT1-CFP cells in these regions only partially resumed motility compared with regions where most of the tumor cells were dead (Fig. 4 C).
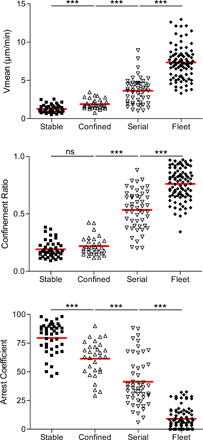
Figure 4.

Dynamics of CTL interactions with tumor cells. TPLSM images of OT1-CFP cells (A, blue; B, magenta) within EG7-GFP tumors (green) during early phase (day 4) and late phase (day 5) of tumor rejection. Collagen fibers (blue) are imaged by SHG. Examples of typical migratory paths (red) are shown. Bars: (A) 27 μm; (B) 39 μm. (C) Scatter plots of T cell mean velocity, confinement ratio, and arrest coefficients of all cells analyzed. Data from three to four independent experiments were pooled. (Alive-Early, n = 20; Dead-Late, n = 121; Alive-Late, n = 77). ***, P < 0.001. (D) TPLSM images (90 × 90 μm) representing typical migratory paths (red) for each class of interaction as described in Material and methods (Stable, black; Confined, dark gray; Serial, light gray; Fleet, white). Bar, 28 μm. (E) Histograms representing the relative fraction of the different classes of interaction in areas displaying alive or dead tumor cells during the early (day 4) and the late phase (day 5) of tumor regression.
The interactions between OT1-CFP and tumor cells were analyzed in more details by defining four types of trajectories. Fig. 4 D shows representative examples of cell trajectories falling into each of these four categories. The first trajectory corresponds to cells arrested in close contact with GFP-positive tumor cells (Fig. 4 D, Stable). The second one corresponds to cells that are not completely arrested, but move around a GFP-positive tumor cell, or interact with several neighboring tumor cells (Fig. 4 D, Confined). The third one includes OT1 cells that interact transiently with several distant tumor cells (Fig. 4 D, Serial), and the fourth one corresponds to OT1 cells that do not establish any measurable interaction with a tumor cell (Fig. 4 D, Fleet). After sorting the cells into these four categories, we analyzed their mean velocity, confinement ratio, and arrest coefficient (Fig. S4). As expected, the two former parameters increased from categories 1 to 4, whereas the arrest coefficient decreased.
The relative proportions of each type of OT1 categories were quantified during the early phase (in regions of the tumors where tumor cells are alive) and during the late phase (in regions where most tumor cells are dead). As shown in Fig. 4 E, the proportion of OT1 cells in the first two categories (Fig. 4 D, Stable and Confined) represents 95% in regions of the tumor where the tumor cells are alive. In contrast, in regions where most tumor cells are dead, categories 3 and 4 (Fig. 4 D, Serial and Fleet) represent ∼90% of the OT1 cells. Importantly, during the late phase, in regions of the tumor where part of the tumor cells were alive, OT1-CFP cells adopted an intermediate behavior, indicating again that resumed motility OT1-CFP cells was dictated by the viability of the tumor cells rather than the day after adoptive transfer.
We conclude that during the early phase of tumor rejection, OT1 cells are in contact with living tumor cells, either completely arrested or following very constrained trajectories, suggesting that they interact with several neighboring tumor cells. During the late phase of tumor rejection, in the regions where most tumor cells are dead, OT1 cells either make very short arrests or migrate rapidly within the tumor with no apparent stops. Therefore, resumed T cell motility in antigen-expressing tumors occurs preferentially in regions where the tumor cells are dead.
Modes of CTL migration in tumors
We next sought to characterize this resumed CTL migration in more detail. Resumed CTL motility in regions of the tumor where the tumor cells are dead followed an amoeboid pattern (Videos S6 and S7, available at http://www.jem.org/cgi/content/full/jem.20061890/DC1). We observed important changes in the cell shape, suggesting a strong adaptation of the CTLs to the extracellular matrix and their ability to squeeze through narrow spaces as observed in artificial three-dimensional collagen matrix (32) or around stromal and tumor cells. CTLs occasionally followed collagen fibers, which were visualized by their SHG signals (Video S6). We also observed OT1 cells dividing inside the EG7 tumors (0.8% of the tracked OT1 cells, n = 1,249; Video S8). Because our videos last 30 min, the proportion of divisions over 24 h could be relatively high (up to 30–40% of the OT1 cells, assuming the rate of divisions remains the same over this period of time). Divisions might have gone unnoticed in the EL4 tumors because of the near 10-fold lower T cell population. Accumulation of tumor cells around blood vessels was observed occasionally (Video S9), suggesting either regions of preferential CTL extravasation or chemotactism of CTLs for the blood vasculature. Three-dimensional reconstitution shows that CTLs are in close contact to blood vessels (Video S10). Strikingly, CTLs moved frequently along blood vessels (Video S7), adopting a more elongated morphology and undergoing less important changes in their shape.
To characterize and quantify the cells migrating along blood vessels in more detail, we took advantage of certain regions of the tumor periphery that displayed a highly organized blood vessel pattern orientated along one major axis (Fig. 5 A). In these areas, numerous CTLs maintained close contact with vessels during migration (Video S11, available at http://www.jem.org/cgi/content/full/jem.20061890/DC1) for relatively long distances (114 ± 63 μm, n = 17). The CTLs adopted an anisotropic migration pattern along the axis of the blood vessels as indicated by a higher correlation coefficient (no-contact, r = 0.31 and contact, r = 0.73; P < 0.001) (Fig. 5 A, bottom). The ability of several cells to migrate from a blood vessel to another confirmed that the CTLs are probably not in the lumen of the vessels, but either in the tissue or within the “sheath” around the vessels. Migration along blood vessels proceeds with a mean velocity similar to that of the CTLs migrating at distance of visible blood vessels (8.2 ± 2.4 μm/min and 8.6 ± 2.8 μm/min, respectively; Fig. 5 B).
Figure 5.

Distinct migratory patterns of T cells within tumors. (A) TPLSM images of OT1-GFP cells (green) within an EG7 tumor during the late phase of tumor rejection (day 5). Vessels (red) are labeled by 70 kD rhodamine-dextran. Several representative migration tracks of OT1-GFP cells are shown for cells either in close contact or not with blood vessels (green); individual tracks are plotted after normalization of their initial positions (bottom). Bar, 120 μm. (B) Scatter plot of the mean velocities of OT1 cells either in contact or not with blood vessels. ns, not significant. (C) Representative cell shapes of cells in close contact (right) or not (left) with the blood vessels (45 × 45 μm). Cells at distance or in close contact with blood vessels were tracked (mean = 10 min in both conditions), and the means of their elongation index during their migratory path were plotted (bottom). ***, P < 0.001. Bar, 28 μm. (D) Top graph shows the elongation index along time of representative cells migrating at distance (black) or in close contact (red) with blood vessels. Straight lines indicate the mean value of the elongation index for cells in close contact (1.9, red) or at distance (1.6, black) from blood vessels. Bottom graph shows the elongation index along time of a representative cell that migrates first within the tumor matrix (black) and thereafter in close contact with a blood vessel (red). Straight line indicates the mean value of its elongation index (1.9, black).
The shape of CTLs during migration along blood vessels seemed to differ from their morphology in other areas of the tumor (examples are shown in Fig. 5 C, top, and Video S12, available at http://www.jem.org/cgi/content/full/jem.20061890/DC1). Fig. 5 D, top, shows the evolution of the elongation index (ratio between the length and the breadth of a cell) of two representative cells migrating either in contact or at distance from a blood vessel. Cells in contact with blood vessels were more elongated, displaying a higher elongation index (1.9 ± 0.2) than cells crawling at distance from visible blood vessels (1.6 ± 0.1) (Fig. 5 C, bottom). To investigate if the shape of individual cells changes when they migrate in contact or at distance of a blood vessel, we followed the elongation index of individual cells migrating in the same videos in contact or at distance of a vessel (Fig. 5 D, bottom). As long as the cell is at distance from any visible vessel, its elongation index is <2 for most of the time, and occasionally rises over this value for short periods of time (<1 min). When the cell starts migrating in close vicinity to the blood vessel, its elongation index rises to values >2 for several min. The elongation index then falls to ∼2, whereas the cell is still migrating along the same blood vessel. For this particular cell, the difference in the elongation index before and after the initiation of the contact with the blood vessel is statistically significant (P = 0.0015). A significant increase in the elongation index before and after the initiation of the contact with the blood vessel, however, was only observed in three out of five cells analyzed.
We conclude that single CTLs can migrate along blood vessels using high or low elongation indexes. Elongations indexes higher than 2 lasting for several min were only observed in cells migrating along blood vessels and not in cells migrating at distance of blood vessels. In the latter, the elongation index was very variable and eventually increased over 2 for short periods of time, reflecting a higher deformability of the cells, most likely a result of constraints imposed by the extracellular matrix encountered during the crawling.
Deep infiltration in antigen-expressing tumors
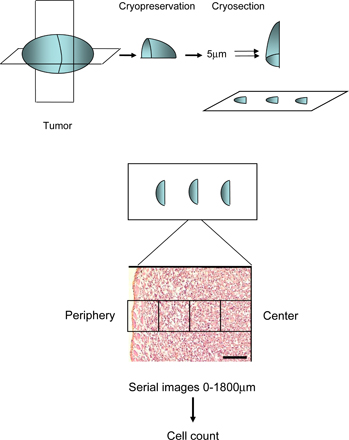
Because two-photon imaging is technically restricted to peripheral area of the tumors, we next sought to analyze the topography of CTL infiltration deeper into the tumor and to analyze the role of antigen recognition in this process. To do so, we had to quantify infiltration by CTLs in regions of the tumor beyond the penetration limit of the two-photon microscope. We therefore made ordered sequential frozen sections of either EG7 or EL4 tumors at different time points after adoptive transfer of GFP-OT1 cells (Fig. S5). The number of GFP-OT1 cells was quantified according to the distance from the surface of the tumor (Fig. S5). Representative examples of OT1-GFP cells in tumor sections are shown in Fig. 6 A. During the early phase of rejection, most GFP-OT1 cells were found within the first 200-μm peripheral sections of both EL4 and EG7 tumors (Fig. 6 B, top left). An accumulation of lymphocytes in the periphery of solid tumors has been reported previously in other tumor models (33, 34), but the actual mechanisms for this preferential infiltration through the periphery are still unclear. To address possible macroscopic heterogeneities in tumor vessel distribution that might account for the peripheral infiltration pattern, we stained endothelial cells in tumor sections using anti-CD31 antibody. We did not detect any significant difference in the density of blood vessels (Fig. S6). This result, of course, does not exclude functional differences in the “permeability” of blood vessels to lymphocytes in different regions of the tumors.
Figure 6.

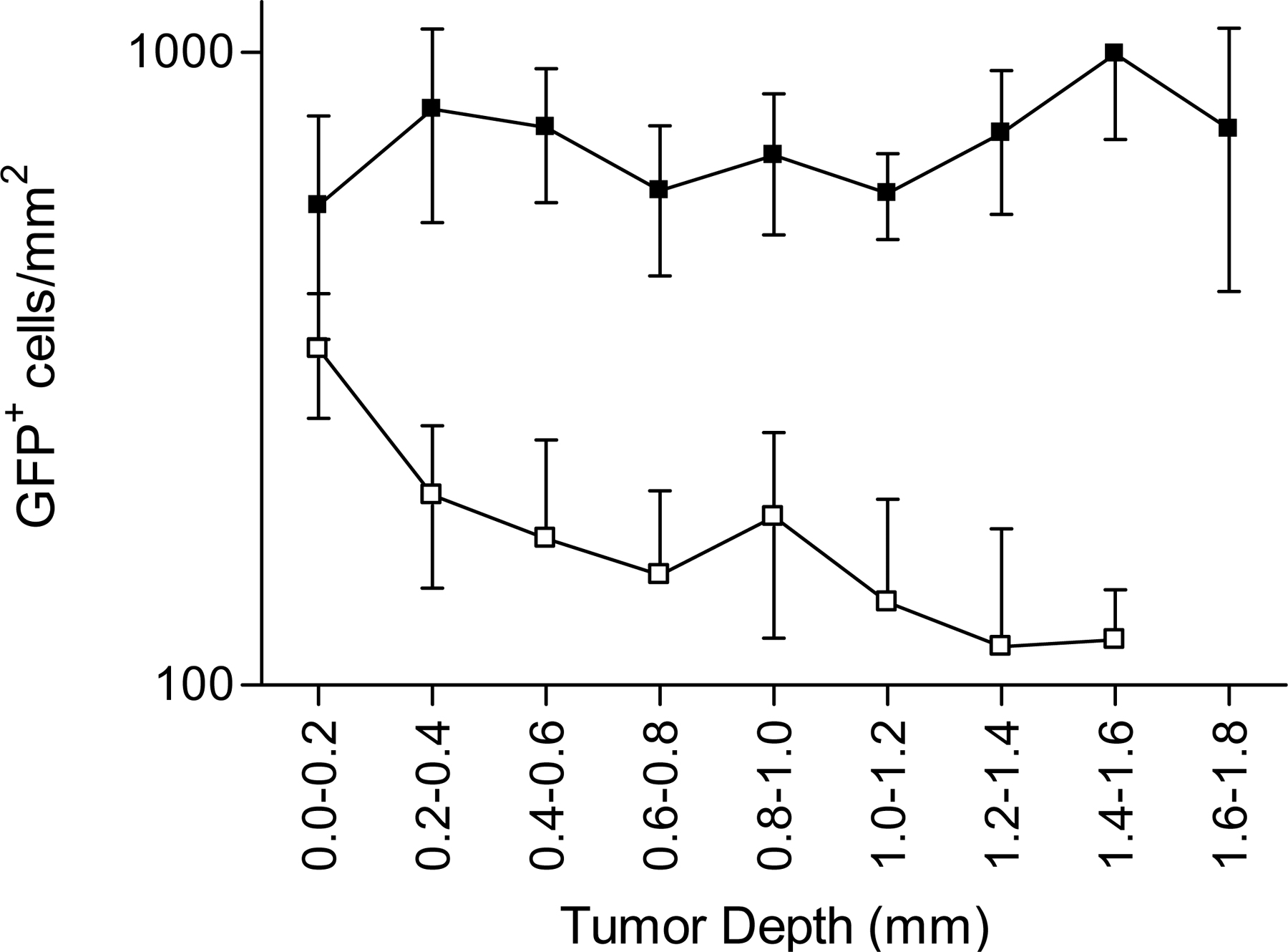
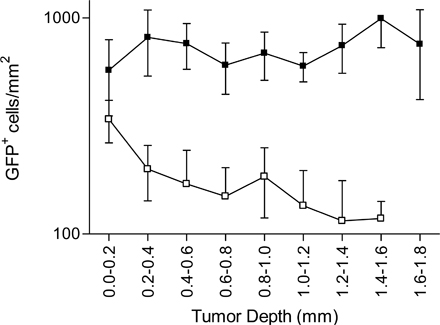
T cell infiltration through the periphery of tumors. OVA-specific OT1-GFP infiltration in EG7 and EL4 tumors (Thymoma) or MCA tumors (Fibrosarcoma), expressing or not the OVA epitope, during the early (day 3) and the late (day 6) phase of tumor rejection. (A) Representative epifluorescence micrographs of EG7 and EL4 tumor sections (900 × 670 μm). The white line delimits tumor periphery. Bar, 210 μm. (B) Distribution of OT1-GFP cells across OVA-expressing (black) or non–OVA-expressing tumors (white), during the early and the late phase of tumor rejection, 3 d (round) and 6 d (square) after adoptive transfer (mean ± SD, n = 3–8). ns, not significant. ***, P < 0.001.
In the late phase of rejection, similar to the early phase, most OT1-GFP cells infiltrating EL4 tumors remain in the periphery (Fig. 6 B, top right). In contrast, in the OVA-expressing EG7 tumors, an important redistribution of the CTLs is observed during the late phase. The density of OT1-GFP cells in more central regions of the tumor increases, resulting in a homogeneous distribution from the periphery to the tumor center. We conclude that CTLs infiltrate the thymomas through the periphery and that deep tumor infiltration during rejection requires antigen expression by the tumor cells (since the OVA-negative EL4 tumors were not infiltrated deeply).
Antigen expression within the tumor causes the local activation (increased CD69 expression), IFN-γ production, and cytotoxicity (tumor rejection) of OT1 cells (Fig. 1). To investigate if deep tumor infiltration requires antigen recognition by individual T cells or is the consequence of overall modifications in the tumor environment caused by antigen recognition by the CTLs, we analyzed the infiltration of EG7 tumors by polyclonal, GFP-expressing T cells. The polyclonal T cells were activated in vitro and then adoptively transferred to EG7-bearing mice in the presence or absence of OT1 cells. At days 5–6 after transfer, tumor rejection started only in mice injected with polyclonal T cells and OT1 cells (unpublished data). Strikingly, these polyclonal T cells accumulated and infiltrated the tumors deeply only in the presence of OT1 cells (Fig. S7). We conclude that OT1 cells induce a change in the antigen-expressing tumors, which makes them permissive to infiltration by other activated T cells.
If tumor infiltration occurs through the periphery of the tumors, tumor cell killing by CTLs should also start in the periphery. To address this possibility, we performed ordered frozen sections as in Fig. 6 and labeled apoptotic cells using tunnel. Apoptotic cells were most abundant in the periphery of EG7 tumors at the end of the early phase (Fig. 7 A). The number of tunnel-positive cells was counted in sequential sections and plotted as a function of the distance from the surface of the tumor. As shown in Fig. 7 B, tunnel-positive cells accumulated in the periphery of the EG7 tumors. Reduced accumulation of apoptotic cells was detected in the EL4 tumors. The low numbers of tunnel-positive cells detected in the periphery of EL4 are most likely caused by the endogenous immune response. We conclude that apoptotic tumor cells accumulate in the periphery of the tumor, consistent with the peripheral infiltration of the CTLs evidenced in Fig. 6.
Figure 7.

Apoptotic cells after tumor T cell infiltration. Tunnel assay after adoptive transfer of OVA-specific OT1 T cells in EG7 or MCA-OVA tumor-bearing mice. (A) Typical epifluorescence images (900 × 670 μm, right and left; 225 × 167 μm, middle) of MCA-OVA tumor sections 4 d after transfer or not of OT1 cells: cell nuclei were labeled with Dapi (blue) and apoptotic cells (green) were labeled by using the FITC-Tunnel detection kit. Bar, 204 μm. (B) Representative quantification of apoptosis in thymoma or fibrosarcoma (MCA-101) tumors expressing (closed symbols) or not (open symbols) the OVA epitope 4 d after OT1 cell transfer or not (control, crosses) (mean ± SD, three to four different tumors). The statistical differences between peripheral and deep regions for EG7 and MCA-OVA are indicated. ***, P < 0.001.
To extend our conclusions about the role of antigen in tumor infiltration to another tumor model, we used a fibrosarcoma tumor cell line, MCA-101, expressing (MCA-OVA) or not expressing (MCA) recombinant OVA. OT1-GFP cells adoptively transferred to tumor-bearing mice induce the rejection of MCA-OVA, but not MCA-tumors, with a kinetic similar to that of EG7 rejection (unpublished data). OT1 cells were recruited to both tumors, although the recruitment to the OVA-expressing tumors was higher compared with the control MCA tumors (Fig. S8). Similar to the thymoma model, OT1-GFP cells infiltrated the periphery of MCA tumors independently of antigen expression (Fig. 6 B, bottom left). OVA expression, however, was required for deep tumor infiltration at days 5–6 (Fig. 6 B, bottom right). Similar to the thymoma model, at days 3–4 tunnel-positive apoptotic tumor cells accumulated in the tumor periphery of antigen-expressing tumors (Fig. 7 B). We conclude that deep tumor infiltration, but not peripheral infiltration, requires antigen expression by fibrosarcoma tumor cells.
DISCUSSION
We show here that antigen expression by thymoma cells plays a critical role in the motility of CTLs, in the extent of infiltration, and in the distribution of the infiltrating CTLs. As suggested previously (33, 34), our results indicate that infiltration by lymphocytes proceeds from the periphery of the tumors. Our results also support a novel working model for the dynamics of tumor infiltration in depth: after diapedesis from blood vessels in periphery of the tumor, CTLs encounter antigen-expressing tumor cells and arrest their migration for killing. Once tumors cells have been cleared in that region of the tumor, CTLs resume motility sometimes guided along collagen fibers and blood vessels until they reach other regions with living tumor cells. Because the killing starts in the tumor periphery, adjacent regions where tumor cells are alive are always toward the center of the tumor. We speculate that when arriving in a new region of the tumor where the tumor cells are alive, CTLs arrest their migration again and kill the antigen-expressing tumor cells. The overall result would be a displacement of the CTL population toward deeper regions of the tumor. The repetition of this sequence of “motility” and “static/cytotoxic” phases would result in the progressive, oriented, antigen-dependent infiltration of the solid tumors that we have observed.
This model is mainly based on observations made on one single tumor—a mouse thymoma growing s.c. The topography of infiltration observed in this thymoma was confirmed in the more relevant chemically induced fibrosarcoma, MCA101. To date, the available imaging techniques impose the use of artificial experimental models, including the transfer of antigen-expressing tumors and the use of high numbers of transgenic T cells specific for transfected antigens. The analysis of mouse solid tumors more relevant to human cancers, spontaneous and growing in other locations, and the use of CTLs recognizing “true” tumor antigens is required to confirm or invalidate our model of CTL invasion.
In any case, this working model is still preliminary. It is unclear from our results whether the critical factor for the observed arrest in CTL motility and the deep tumor infiltration is tumor cell killing or antigen recognition (and the resulting T cell activation). Experiments using control transgenic CTLs with unrelated antigen specificity or CTLs defective for different functions (activation, cytokine secretion, cytotoxicity) should allow nailing down the precise requirements for CTL arrest on tumor cells and for tumor infiltration in depth. Our experiment using polyclonal GFP-expressing cells suggests that T cells that do not induce rejection by themselves and do not infiltrate tumors in depth alone may still infiltrate tumors deeply in the course of rejection by other CTLs (Fig. S7).
It is therefore possible that T cell activation or cytokine secretion within the tumor, rather than tumor cell killing, is determinant for deep infiltration. Indeed, it has been proposed that tumor infiltration by antigen-specific CTLs triggers the invasion by other immune effectors, including neutrophils, macrophages, and NK cells (26, 28, 29). It is therefore possible that this “shell” of immune cells that modifies the tumor microenvironment results in resumed CTL motility and deep infiltration. Our working model could be developed to include the progressive formation of this shell of immune cells, which accompanies CTL infiltration and contributes to regained CTL motility and, thereby, to progression of the CTLs deeper into the tumor.
It is therefore critical to understand the mechanisms underlying the observed peripheral infiltration during the early rejection phase. Although the density of blood vessels is not higher in the tumor periphery (Fig. S6), we cannot exclude that differences in the permeability of the vessels in different regions of the tumor account for the observed CTL infiltration. It is likely, however, that the high permeability of the tumor vasculature results in the facilitated extravasations of the activated lymphocytes as soon as they enter the tumor, favoring the peripheral infiltration. This peripheral infiltration could also be a “feedback” favored by the local inflammation, which is probably stronger in the periphery of the tumor, caused by tumor cell killing by CTLs. These peripheral CTLs would then proceed to infiltrate the tumor in depth through the mechanism depicted above. It is also possible, however, that as the CTLs, and most likely the rest of the antitumor immune response, proceeds deeply into the tumor, the permeability of the vessels is also progressively increased.
Although it has been very difficult thus far, it should be technically possible to analyze tumor cell killing in situ in real-time. Indeed, our study has not addressed the frequency of the killing events, the kinetics of tumor cell killing, or the number of tumor cells killed by each CTL. Studies combining fluorescent tumor and stroma cells should also contribute to better understanding the interactions between the tumor and its close microenvironment. Future studies should also determine if the spatial and temporal organization of the invasion of other tissues by effector T cells, for example, in the course of autoimmune disease or antiinfectious immune responses, follow the same rules that govern the infiltration of solid tumors.
MATERIALS AND METHODS
Mice.
C57BL/6 mice were obtained from Charles River Laboratories. C57BL/6 Rag2−/− TCR (Vα2, Vβ5) transgenic mice (OT1) were crossed in the Curie Institute animal facility with UBI-GFP/BL6 or with the Actβ-ECFP/BL6 (35) to obtain the F1 progeny (OT1-GFP and OT1-CFP mice, respectively). Live animal experiments were done in accordance to the guidelines of the French Veterinary Department.
Cells.
CD8+ OT1 cells (specific for the OVA257-264 peptide in a H2-Kb MHC class I context) were obtained from LNs of OT1 mice (purity ∼96%), and CD8+ OT1-GFP/CFP cells were purified (96% purity) using Myltenyi Biotec CD8+ magnetic cell sorting kit and injected retroorbitally in 6–8-wk- old female C57BL/6 recipient tumor-bearing mice. EG-7 and EL-4 cell lines were stably transfected with a GFP coding plasmid and selected by cell sorting (97% GFP+ cells). The MCA101 (MCA) cell line was stably transfected with an empty plasmid or a plasmid encoding for OVA coupled to the C1C2 domain of MFG-E8 to generate the control MCA and MCA-OVA, respectively. Injected s.c. MCA-OVA is actively rejected after OT1 adoptive transfer (unpublished data). Polyclonal cells were isolated from LNs of UBI-GFP/BL6 mice and were stimulated overnight in flat wells coated with 1 μg/ml purified anti-CD3 and 1 μg/ml anti-CD28. All cell lines were cultured in RPMI 1640+Glutamax I (GIBCO BRL) supplemented with additional antibiotics and 10% FCS. Tumor cells were injected s.c. in the flank of mice (EL4 and EG7 in a contralateral manner), and tumor size was measured three times a week using the following formula:
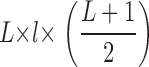 |
Flow cytometry.
Phenotypic characterization of T cells was performed using a FACSCalibur (Becton Dickinson). EG7 and EL4 tumors, lymph node, and spleen were harvested at different days after adoptive transfer of OT1-GFP naive cells. Cell suspensions were prepared in PBS 0.5% BSA, and cells were labeled with anti–CD69-biotin followed by streptavidin–PercP–Cy5.5, anti-CD62L–allophycocyanin, anti-CD44–PE, or anti-Vβ5–PE (Becton Dickinson). For intracellular cytokine staining, cell suspensions were incubated for 3 h at 37°C in the presence of 5 μg/ml Brefeldin A, fixed in 4% PFA, and incubated in PBS 0.5% BSA with saponin at 0.1% in the presence of anti-IFN-γ–APC. CFSE staining was performed by incubating OT1 cells in PBS with 5 μM CFSE (Invitrogen) during 10 min at 37°C. CFSE was quenched and cells washed by adding large volume of culture medium and then centrifuged. Cells were diluted in PBS for injection.
Histological tumor analysis.
The analysis of the distribution of OT1-GFP cells, polyclonal GFP cells, and of CD31- or tunnel-positive cells across the tumors is illustrated in Fig. S5. Tumors were transversally and sagitally sectioned in the middle, fixed, frozen in Tissue-Tek OCT compound, and sectioned (5 μm) with a cryostat (Leica). For GFP quantification, 10 series of 10 contiguous images from the periphery to the center of each tumor, from 6 different sections, spaced from 10 to 20 μm (225 × 170μm) were acquired in the first 1,800 μm for EG7 and 1400 μm for MCA tumors. The periphery of the tumor was delimited by light transmission imaging. The density of OT1 cells across the tumor was calculated by normalizing the relative distribution of OT1 cells in each tumor to the mean of the global density for all tumors. For tunnel experiments, tumor sections were labeled using an in situ cell death detection kit (Roche) following manufacturer's instructions. Endothelial cells were stained using purified anti-CD31 (Becton Dickinson) or isotype control and a secondary antibody coupled with cyanin-3. Tumor sections were additionally stained with DAPI. Quantification was performed by calculating the fluorescent area of tunnel- and CD31-positive cells. A minimum of three different tumors were processed (analyzing one or two different sections per tumor), as for the analysis of GFP-positive cells.
Two-photon laser scanning microscopy.
107 naive OT1-GFP cells were retroorbitally injected to 6–8-wk-old female C57BL/6 recipients. After 3 or 5–6 d after adoptive transfer, mice were anesthetized (initial dose of 80 mg/kg ketamine, 4 mg/kg xylazine, and 0.3 mg/kg flunitrazepan, followed by a half-dose injection every 45 min; mice were kept under anesthesia during 3 h), and the tumor region was surgically exposed for two-photon imaging. Local temperature was monitored and maintained at 37°C. GFP/CFP- labeled T cells could be detected up to 150 μm from the tumor's surface. Vessels were stained by i.v. injection of 200 μl of 2.5 μg/ml rhodamine-Dextran 70 kD (Sigma-Aldrich). In vivo PI staining was performed by a slow intratumoral injection of 50 μl of a PBS solution at 1 mg/ml PI at distance of the analyzed area.
Measurements were performed in at least three independent experiments. The time-lapse two-photon laser-scanning microscopy (TPLSM) set-ups used were either a Fluoview200 (Olympus) in a descanned configuration equipped with a 20 × 0.95 NA dipping objective (Olympus), using a 540 nm dichroic mirror to separate GFP and Rodamine emission spectra, or an LSM510 Meta (Carl Zeiss MicroImaging, Inc.) coupled to a Maitai femtosecond laser (710–920 nm) (Spectra-Physics). With the latter configuration, SHG-GFP–Rhodamine fluorescence (Fig. 3) were separated using the Meta detector and were pixel-wise linear unmixed taken experimental fluorescence spectra of monolabeled samples as an external reference. Otherwise, three fluorescence channels were separated using 495 and 590 nm dichroic mirrors in combination with either 457/50 (SHG) or 472/30 (CFP), 520/35 (GFP), and 624/40 nm bandpass filters. The excitation wavelength was 900 nm. For analysis of cell migration, every 30 s during 30–60 min, four consecutive 560 μm2 images, with 10-μm z-spacing using the 20× objective whose back aperture was artificially underfilled to obtain an axial resolution near 5 μm. Images were average-projected using Image J (National Institutes of Health), and manual tracking of individual cells was performed using Metamorph software. Cells that could not be tracked for more than 5 min were not considered to minimize projection errors inherent to the two-dimensional track analysis. The acquisition and analysis protocols for all experimental conditions to be compared were identical. The arrest coefficient is defined as the proportion of time each cell's instantaneous velocity (calculated for every 30-s interval) is <2 μm/min. The confinement ratio corresponds to the ratio of the distance between the initial and the final positions of each cell to the total distance covered by the same cell. The elongation index is defined by the ratio between length of the long axis of the cell and length of the orthogonal short axis of the cell. Length and breadth of the cells were automatically calculated using Metamorph software after manually delimiting individual cells in each image.
Statistical analysis.
Nonnormal distributions were compared using the Mann-Whitney rank sum test. The distributions of the time points of the trajectories in contact or at distance to the vessels (Fig. 5 A, bottom) were evaluated by calculating the correlation coefficients. For histological analysis, paired Student's t tests were performed to compare the distribution between peripheral region and deep region for each image series (*, P < 0.05; **, P < 0.01; ***, P <0.001; ns, not significant.)
Online supplemental material.
Fig. S1 shows that OT1 cells are activated in the antigen-expressing tumor-draining lymph nodes. Fig. S2 quantifies OT1 T cell expansion in secondary lymphoid organs of tumor-bearing animals. Fig. S3 characterizes the down-regulation of the TCR on OT1-GFP cells infiltrating the antigen-expressing tumors. Fig. S4 depicts the motility characteristics of the four categories of migrating OTI cells found in tumors. Fig. S5 illustrates the procedure of histological analysis used in Figs. 6 and 7 and Figs. S6 and S7. Fig. S6 shows the analyses of the distribution of blood vessels in the tumors. Fig. S7 shows the infiltration of nonspecific T cells in the presence or absence of antigen-specific T cells. Fig. S8 shows the analyses of the density of OT1 cells in MCA-OVA and MCA tumors. Video S1–S4 show migration of OT1-GFP effector cells within EG7 tumor (Videos S1 and S3) and EL4 tumor (Videos S2 and S4) during the early (Videos S1 and S2) and late (Videos S3 and S4) phases after adoptive transfer. Videos S5 and S6 show interaction of OT1-CFP effector cells with GFP-EG7 tumor cells during the early and late phase after adoptive transfer, respectively. Video S7 shows shape modifications of OT1-GFP effector cells migrating within an EG7 tumor during the late phase after adoptive transfer. Video S8 shows OT1-GFP effector cells dividing within an EG7 tumor during the late phase after adoptive transfer. Video S9 shows OT1-GFP effector cells migrating in close contact with blood vessels within an EG7 tumor during the late phase of tumor rejection. Video S10 shows a three-dimensional reconstruction from Video S9. Video S11 shows examples of OT1-GFP effector cells migrating from blood vessels toward the EG7 tumor matrix during the late phase after adoptive transfer. Video S12 shows typical shapes of OT1-GFP effector cells migrating either in contact or not with blood vessels within an EG7 tumor during the late phase after adoptive transfer.
Supplemental Material
Acknowledgments
The authors thank Dr. C. Combadière for providing GFP and CFP transgenic mice.
Alexandre Boissonnas is supported by a fellowship from the Association de la Recherche Contre le Cancer (ARC). This work was supported by funding from Institut National de la Santé et de la Recherche Médicale, (Centre National de la Recherche Scientifique), Ligue de Lutte Contre le Cancer, ARC, and the Institut Curie.
The authors have no conflicting financial interests.
Abbreviations used: CFP, cyan fluorescent protein; PI, propidium iodide; SHG, second harmonic generation; TPLSM, two-photon laser-scanning microscopy.
A. Boissonnas and L. Fetler contributed equally to this work.
References
- 1.Halin, C., J. Rodrigo Mora, C. Sumen, and U.H. von Andrian. 2005. In vivo imaging of lymphocyte trafficking. Annu. Rev. Cell Dev. Biol. 21:581–603. [DOI] [PubMed] [Google Scholar]
- 2.Germain, R.N., M.J. Miller, M.L. Dustin, and M.C. Nussenzweig. 2006. Dynamic imaging of the immune system: progress, pitfalls and promise. Nat. Rev. Immunol. 6:497–507. [DOI] [PubMed] [Google Scholar]
- 3.Cahalan, M.D., and I. Parker. 2006. Imaging the choreography of lymphocyte trafficking and the immune response. Curr. Opin. Immunol. 18:476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller, M.J., S.H. Wei, I. Parker, and M.D. Cahalan. 2002. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science. 296:1869–1873. [DOI] [PubMed] [Google Scholar]
- 5.Stoll, S., J. Delon, T.M. Brotz, and R.N. Germain. 2002. Dynamic imaging of T cell-dendritic cell interactions in lymph nodes. Science. 296:1873–1876. [DOI] [PubMed] [Google Scholar]
- 6.Lindquist, R.L., G. Shakhar, D. Dudziak, H. Wardemann, T. Eisenreich, M.L. Dustin, and M.C. Nussenzweig. 2004. Visualizing dendritic cell networks in vivo. Nat. Immunol. 5:1243–1250. [DOI] [PubMed] [Google Scholar]
- 7.Condeelis, J., and J.E. Segall. 2003. Intravital imaging of cell movement in tumours. Nat. Rev. Cancer. 3:921–930. [DOI] [PubMed] [Google Scholar]
- 8.Bousso, P., and E. Robey. 2003. Dynamics of CD8+ T cell priming by dendritic cells in intact lymph nodes. Nat. Immunol. 4:579–585. [DOI] [PubMed] [Google Scholar]
- 9.Mempel, T.R., S.E. Henrickson, and U.H. Von Andrian. 2004. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 427:154–159. [DOI] [PubMed] [Google Scholar]
- 10.Hugues, S., L. Fetler, L. Bonifaz, J. Helft, F. Amblard, and S. Amigorena. 2004. Distinct T cell dynamics in lymph nodes during the induction of tolerance and immunity. Nat. Immunol. 5:1235–1242. [DOI] [PubMed] [Google Scholar]
- 11.Zinselmeyer, B.H., J. Dempster, A.M. Gurney, D. Wokosin, M. Miller, H. Ho, O.R. Millington, K.M. Smith, C.M. Rush, I. Parker, et al. 2005. In situ characterization of CD4+ T cell behavior in mucosal and systemic lymphoid tissues during the induction of oral priming and tolerance. J. Exp. Med. 201:1815–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang, Q., J.Y. Adams, A.J. Tooley, M. Bi, B.T. Fife, P. Serra, P. Santamaria, R.M. Locksley, M.F. Krummel, and J.A. Bluestone. 2006. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat. Immunol. 7:83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tadokoro, C.E., G. Shakhar, S. Shen, Y. Ding, A.C. Lino, A. Maraver, J.J. Lafaille, and M.L. Dustin. 2006. Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J. Exp. Med. 203:505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shakhar, G., R.L. Lindquist, D. Skokos, D. Dudziak, J.H. Huang, M.C. Nussenzweig, and M.L. Dustin. 2005. Stable T cell-dendritic cell interactions precede the development of both tolerance and immunity in vivo. Nat Immunol. 6:707–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawakami, N., U.V. Nagerl, F. Odoardi, T. Bonhoeffer, H. Wekerle, and A. Flugel. 2005. Live imaging of effector cell trafficking and autoantigen recognition within the unfolding autoimmune encephalomyelitis lesion. J. Exp. Med. 201:1805–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geissmann, F., T.O. Cameron, S. Sidobre, N. Manlongat, M. Kronenberg, M.J. Briskin, M.L. Dustin, and D.R. Littman. 2005. Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol. 3:e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bajenoff, M., B. Breart, A.Y. Huang, H. Qi, J. Cazareth, V.M. Braud, R.N. Germain, and N. Glaichenhaus. 2006. Natural killer cell behavior in lymph nodes revealed by static and real-time imaging. J. Exp. Med. 203:619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mempel, T.R., M.J. Pittet, K. Khazaie, W. Weninger, R. Weissleder, H. von Boehmer, and U.H. von Andrian. 2006. Regulatory T cells reversibly suppress cytotoxic T cell function independent of effector differentiation. Immunity. 25:129–141. [DOI] [PubMed] [Google Scholar]
- 19.Kircher, M.F., J.R. Allport, E.E. Graves, V. Love, L. Josephson, A.H. Lichtman, and R. Weissleder. 2003. In vivo high resolution three-dimensional imaging of antigen-specific cytotoxic T-lymphocyte trafficking to tumors. Cancer Res. 63:6838–6846. [PubMed] [Google Scholar]
- 20.Smirnov, P., E. Lavergne, F. Gazeau, M. Lewin, A. Boissonnas, B.T. Doan, B. Gillet, C. Combadiere, B. Combadiere, and O. Clement. 2006. In vivo cellular imaging of lymphocyte trafficking by MRI: a tumor model approach to cell-based anticancer therapy. Magn. Reson. Med. 56:498–508. [DOI] [PubMed] [Google Scholar]
- 21.Su, H., D.S. Chang, S.S. Gambhir, and J. Braun. 2006. Monitoring the antitumor response of naive and memory CD8 T cells in RAG1−/− mice by positron-emission tomography. J. Immunol. 176:4459–4467. [DOI] [PubMed] [Google Scholar]
- 22.Dobrzanski, M.J., J.B. Reome, and R.W. Dutton. 1999. Therapeutic effects of tumor-reactive type 1 and type 2 CD8+ T cell subpopulations in established pulmonary metastases. J. Immunol. 162:6671–6680. [PubMed] [Google Scholar]
- 23.Hanson, H.L., D.L. Donermeyer, H. Ikeda, J.M. White, V. Shankaran, L.J. Old, H. Shiku, R.D. Schreiber, and P.M. Allen. 2000. Eradication of established tumors by CD8+ T cell adoptive immunotherapy. Immunity. 13:265–276. [DOI] [PubMed] [Google Scholar]
- 24.Rosenberg, S.A. 2001. Progress in human tumour immunology and immunotherapy. Nature. 411:380–384. [DOI] [PubMed] [Google Scholar]
- 25.Dudley, M.E., J.R. Wunderlich, P.F. Robbins, J.C. Yang, P. Hwu, D.J. Schwartzentruber, S.L. Topalian, R. Sherry, N.P. Restifo, A.M. Hubicki, et al. 2002. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 298:850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Helmich, B.K., and R.W. Dutton. 2001. The role of adoptively transferred CD8 T cells and host cells in the control of the growth of the EG7 thymoma: factors that determine the relative effectiveness and homing properties of Tc1 and Tc2 effectors. J. Immunol. 166:6500–6508. [DOI] [PubMed] [Google Scholar]
- 27.Hollenbaugh, J.A., J. Reome, M. Dobrzanski, and R.W. Dutton. 2004. The rate of the CD8-dependent initial reduction in tumor volume is not limited by contact-dependent perforin, Fas ligand, or TNF-mediated cytolysis. J. Immunol. 173:1738–1743. [DOI] [PubMed] [Google Scholar]
- 28.Blohm, U., D. Potthoff, A.J. van der Kogel, and H. Pircher. 2006. Solid tumors “melt” from the inside after successful CD8 T cell attack. Eur. J. Immunol. 36:468–477. [DOI] [PubMed] [Google Scholar]
- 29.Hollenbaugh, J.A., and R.W. Dutton. 2006. IFN-gamma regulates donor CD8 T cell expansion, migration, and leads to apoptosis of cells of a solid tumor. J. Immunol. 177:3004–3011. [DOI] [PubMed] [Google Scholar]
- 30.Boissonnas, A., C. Combadiere, E. Lavergne, M. Maho, C. Blanc, P. Debre, and B. Combadiere. 2004. Antigen distribution drives programmed antitumor CD8 cell migration and determines its efficiency. J. Immunol. 173:222–229. [DOI] [PubMed] [Google Scholar]
- 31.Masopust, D., V. Vezys, E.J. Usherwood, L.S. Cauley, S. Olson, A.L. Marzo, R.L. Ward, D.L. Woodland, and L. Lefrancois. 2004. Activated primary and memory CD8 T cells migrate to nonlymphoid tissues regardless of site of activation or tissue of origin. J. Immunol. 172:4875–4882. [DOI] [PubMed] [Google Scholar]
- 32.Wolf, K., R. Muller, S. Borgmann, E.B. Brocker, and P. Friedl. 2003. Amoeboid shape change and contact guidance: T-lymphocyte crawling through fibrillar collagen is independent of matrix remodeling by MMPs and other proteases. Blood. 102:3262–3269. [DOI] [PubMed] [Google Scholar]
- 33.Mukai, S., J. Kjaergaard, S. Shu, and G.E. Plautz. 1999. Infiltration of tumors by systemically transferred tumor-reactive T lymphocytes is required for antitumor efficacy. Cancer Res. 59:5245–5249. [PubMed] [Google Scholar]
- 34.Lurquin, C., B. Lethe, E. De Plaen, V. Corbiere, I. Theate, N. van Baren, P.G. Coulie, and T. Boon. 2005. Contrasting frequencies of antitumor and anti-vaccine T cells in metastases of a melanoma patient vaccinated with a MAGE tumor antigen. J. Exp. Med. 201:249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hadjantonakis, A.K., and A. Nagy. 2001. The color of mice: in the light of GFP-variant reporters. Histochem. Cell Biol. 115:49–58. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}