

Abstract
There is a remarkable heterogeneity in the functional profile (quality) of T cell responses. Importantly, the magnitude and/or quality of a response required for protection may be different depending on the infection. Here, we assessed the capacity of different Toll like receptor (TLR)-binding compounds to influence T helper cell (Th)1 and CD8+ T cell responses when used as adjuvants in nonhuman primates (NHP) with HIV Gag as a model antigen. NHP were immunized with HIV Gag protein emulsified in Montanide ISA 51, an oil-based adjuvant, with or without a TLR7/8 agonist, a TLR8 agonist, or the TLR9 ligand cytosine phosphate guanosine oligodeoxynucleotides (CpG ODN), and boosted 12 wk later with a replication-defective adenovirus-expressing HIV-Gag (rAD-Gag). Animals vaccinated with HIV Gag protein/Montanide and CpG ODN or the TLR7/8 agonist had higher frequencies of Th1 responses after primary immunization compared to all other vaccine groups. Although the rAD-Gag boost did not elevate the frequency of Th1 memory cytokine responses, there was a striking increase in HIV Gag-specific CD8+ T cell responses after the boost in all animals that had received a primary immunization with any of the TLR adjuvants. Importantly, the presence and type of TLR adjuvant used during primary immunization conferred stability and dramatically influenced the magnitude and quality of the Th1 and CD8+ T cell responses after the rAD-Gag boost. These data provide insights for designing prime-boost immunization regimens to optimize Th1 and CD8+ T cell responses.
There are no successful vaccines to prevent tuberculosis, malaria, or HIV infection. In terms of immune correlates of protection for such infections, it is likely that induction of antibodies, T cell responses, or both will be required (1). Live vaccines have been the gold standard for eliciting durable humoral and cellular responses in humans; however, despite the widespread use of Bacillus Calmette-Guerin as a vaccine against tuberculosis, it is not universally effective for preventing pulmonary tuberculosis. Furthermore, although live-attenuated HIV vaccines have been shown to confer protection in nonhuman primates (NHP), they are precluded for use as the result of potential safety constraints (2). Hence, there is an urgent need to develop safe and effective vaccine formulations that can mimic or improve upon the immunogenicity elicited by live vaccines.
Currently, prime-boost immunization using a variety of heterologous vaccine formulations is an effective approach for eliciting strong cellular immune responses. In NHP, for example, vaccination with plasmid DNA–encoding HIV envelope or structural proteins followed by replication-defective adenovirus (rAD) encoding the same proteins elicits protective responses against SHIV challenge (3, 4). Although this approach is promising, further improvements in immunogenicity are desirable. In particular, primary immunization with plasmid DNA vaccines elicits less robust T cell and antibody responses in humans than observed in rodents or NHP (5). Moreover, the capacity of rAD to induce T cell responses is limited by preexisiting immunity from prior adenoviral infection of the same serotype (6, 7). Finally, the rAD itself will induce neutralizing antibodies, thereby restricting its use for further boosting. Hence, a nonlive vaccine that elicits potent humoral and cellular immune responses would be useful for optimizing the primary immunization and could also be useful to sustain or enhance immunity after the rAD boost.
A recent strategy for developing such a vaccine formulation is to combine protein antigen with adjuvants that have the capacity to activate DCs and B cells. Indeed, we have recently shown that NHP immunized with HIV Gag protein and a Toll-like receptor (TLR)9 ligand (cytosine phosphate guanosine oligodeoxynucleotides [CpG ODN]), or a TLR7/8 agonist elicit increased Gag-specific antibody and Th1 responses compared with immunization with HIV Gag protein alone (8). Importantly, animals immunized with HIV Gag protein conjugated to the TLR7/8 agonist had substantially increased Th1 responses, as well as induction of CD8+ T cell responses, compared with animals immunized with HIV Gag protein and soluble TLR7/8 agonist (8). Moreover, the quality of such responses as defined on the basis of IL-2, IFN-γ, or TNF-α production was distinct in NHP immunized with the conjugate vaccine. Such animals had a high frequency of polyfunctional Th1 and CD8+ T cells secreting all three cytokines after immunization. This study suggested that both the vaccine formulation and the use of specific TLR agonists that have the capacity to activate distinct subsets of DCs will have a critical influence on the type of T cell responses generated in NHP.
In this report, we assessed the role of altering vaccine formulation with HIV Gag protein and specific TLR agonists in the context of prime-boost immunization with rAD-Gag in NHP. A specific focus was how primary immunization influenced the magnitude and quality of effector and memory T cell responses after rAD-Gag boost. HIV Gag protein mixed with a TLR7/8, TLR8 agonist, or a TLR9 ligand and emulsified in Montanide ISA 51 (9), was administered once to animals as a primary immunization. This formulation provides a depot of antigen and adjuvant at the site of immunization. Animals were boosted with a rAD-Gag and the immune responses were assessed for up to1 yr. Multiparameter flow cytometry was used to analyze the magnitude (quantity) of Gag-specific IL-2, TNF-α, or IFN-γ–producing T cells as well as the quality of such responses as defined by the composition of these cytokines in any combination at the single cell level. Overall, the data reveal that the specific TLR adjuvant used during the primary immunization strongly influences the T cell response profiles in terms of magnitude, quality, and Th1/CD8 balance during prime-boost immunization. This study provides a tool for fine-tuning the immune functions to elicit the optimal protective response needed for a given pathogen.
RESULTS
Sustained IFN-γ and IL-2 responses are elicited after primary immunization
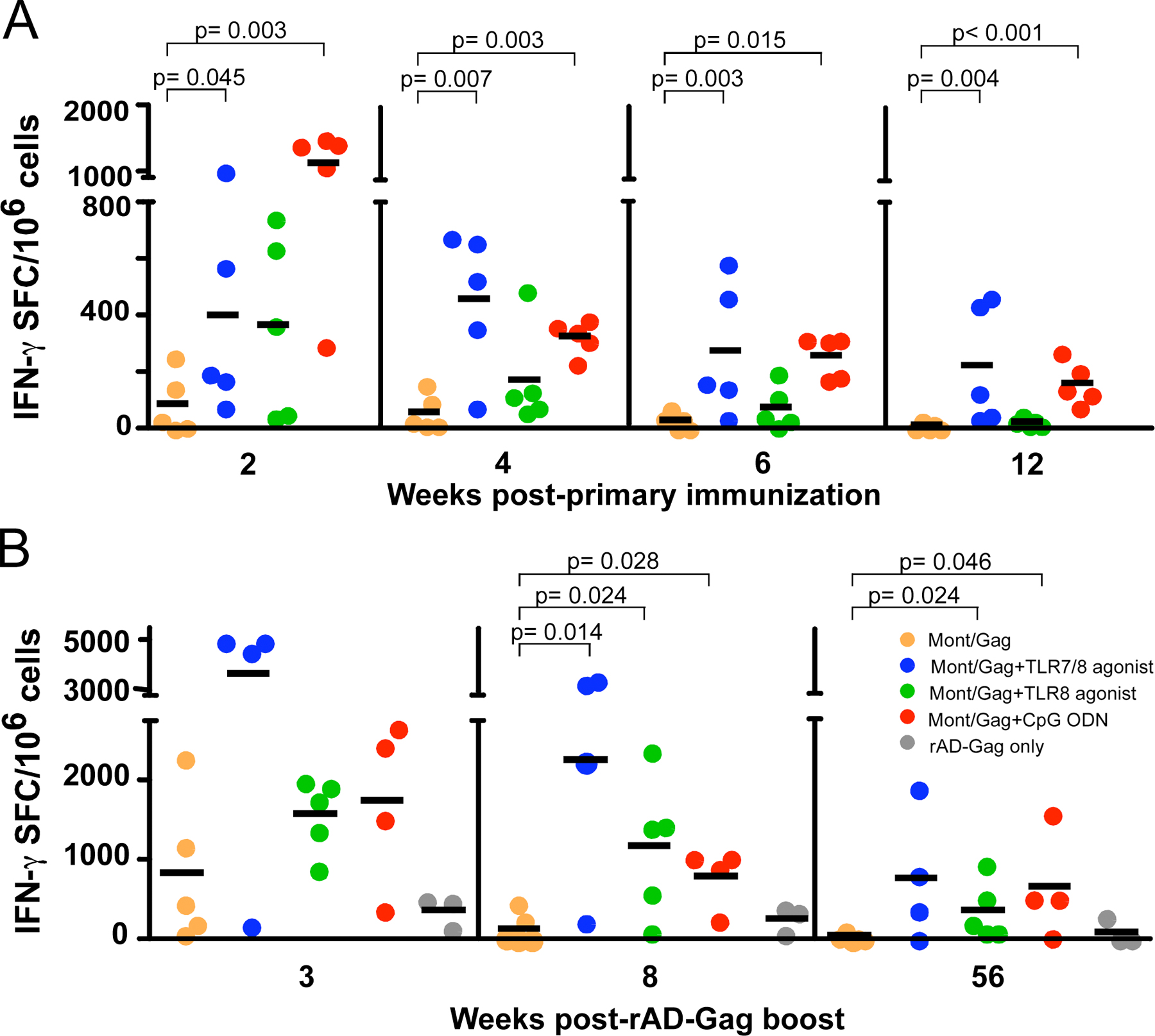
NHP were immunized once with HIV Gag protein emulsified in Montanide, an oil-based adjuvant (Montanide/Gag), with or without a TLR7/8 or TLR8 agonist, or CpG ODN. We used TLR7/8, TLR8, and TLR9 agonists or ligands based on their ability to selectively activate plasmacytoid (TLR7/8 agonist or CpG ODN), conventional DCs (TLR7/8 or TLR8 agonist) or both (TLR7/8 agonist) in vitro (8). This approach allowed an assessment of how the activation of specific DC subsets could influence T cell responses in vivo. 2 wk after immunization, there was a striking increase in the frequency of IFN-γ–producing cells (>1,000 SFC/106 cells) in NHP that received Montanide/Gag plus CpG ODN compared with animals immunized with Montanide/Gag alone (Fig. 1 A). Within 2 wk, such responses rapidly decreased and then remained stable through week 12. Immunization with Montanide/Gag and the TLR7/8, but not the TLR8 agonist also significantly enhanced the frequency of IFN-γ–producing cells throughout 12 wk compared with responses in animals immunized with Montanide/Gag alone (Fig. 1 A). Of note, although the peak IFN-γ responses in animals immunized with the TLR7/8 agonist were lower than in animals immunized with CpG ODN, there was little decrease in such responses throughout the 12 wk. Together, these data are consistent with our previous study showing that the TLR7/8 agonist or CpG ODN, but not the TLR8 agonist, are effective adjuvants for enhancing IFN-γ responses in NHP in vivo (8).
Figure 1.

The frequency of IFN-γ and IL-2–producing cells after immunization with HIV Gag protein and TLR agonists/ligands administered in Montanide. NHP were immunized once with HIV Gag protein with or without CpG ODN, a TLR7/8 or TLR8 agonist emulsified in Montanide, and boosted with rAD-Gag at 12 wk after primary immunization. PBMCs were analyzed for production of IFN-γ and IL-2 by ELISPOT assay at various times after primary immunization (A and B), or 3, 8, and 56 wk after rAD Gag boost (C and D). As controls, animals immunized with PBS, the TLR8 agonist, or CpG ODN without HIV Gag protein were boosted with rAD-Gag. Shown are the means ± SD of five monkeys (A and B), four to five monkeys (C and D), or three monkeys (rAD-Gag only) per group. *, P < 0.05 compared with Montanide/Gag group. After primary immunization, background responses for IFN-γ and IL-2 in animals that received Montanide/PBS, or a TLR agonist alone without HIV Gag protein were <10 SFC/106 cells (not depicted).
In contrast with what was observed for IFN-γ, the frequency of IL-2–producing cells was similar in all groups at 4 wk after immunization (Fig. 1 B). However, although the number of IL-2–producing cells remained increased in animals immunized with the TLR7/8, TLR8 agonist, or CpG ODN, they were markedly decreased over the following 2 wk in animals immunized with Montanide/Gag only. Thus, all TLR adjuvants used in this study conferred durability to the IL-2 response. Finally, we were unable to detect Gag-specific IL-4–producing cells by ELISPOT analysis, or IL-4 and IL-10 protein secretion from PBMCs in any of the vaccine groups after in vitro stimulation (unpublished data). Collectively, these data show that emulsifying HIV Gag protein in Montanide with CpG ODN or the TLR7/8 agonist induces potent and sustained IFN-γ or IL-2 responses after a single immunization. Frequencies of IFN-γ or IL-2–producing cells for the individual animals are shown in Figs. S1 and S2 (available at http://www.jem.org/cgi/content/full/jem.20052433/DC1).
IFN-γ and IL-2 responses are increased after rAD-Gag boost
All NHP were boosted 12 wk after primary immunization with rAD-Gag. As a control, three animals that had not received a primary immunization with HIV Gag protein were immunized with rAD-Gag, allowing a comparison between prime-boost immunization and boost alone. The frequency of IFN-γ–producing cells was dramatically increased in NHP immunized with Montanide/Gag plus any of the TLR adjuvants compared with animals immunized with Montanide/Gag or rAD-Gag alone at 8 and 56 wk after boost (Fig. 1 C). Furthermore, animals immunized with Montanide/Gag plus the TLR7/8 agonist had increased numbers of IL-2–producing cells compared with all other groups for all time points after the boost (Fig. 1 D). It should be noted that one out of four animals immunized with CpG ODN or TLR7/8 agonist did not have an increased IFN-γ responses after the rAD-Gag boost. Thus, this limited a statistical difference compared with animals immunized with Montanide/Gag alone at some of the time points. Nevertheless, there were dramatic differences in the other animals as shown in Figs. S1 and S2.
Assessment of Gag-specific T cell cytokine responses by multiparameter flow cytometry
To characterize how the magnitude and quality of the CD4+ or CD8+ T cell cytokine responses were altered during prime-boost immunization, 10-color flow cytometric analysis was used at 2, 6, and 10 wk after primary immunization, and 3 and 56 wk after the rAD-Gag boost. A gating tree from a representative animal immunized with Montanide/Gag plus the TLR7/8 agonist is shown in Fig. 2. CD4+ and CD8+ T cells were separated into CD45RA+CD95− and CD45RA−CD95+. Expression of CD95 has been used as a specific marker in NHP to distinguish between naive and memory cells (10). Essentially all cytokine-producing T cells were CD45RA−CD95+. This population was used to calculate the total magnitude and quality of the cytokine response, and will be referred to as memory cells. Within the gated CD4+ or CD8+ CD45RA−CD95+ memory T cell population, cells were segregated into IFN-γ− or IFN-γ+ cells and further assessed for production of IL-2, TNF-α, or both. This analysis reveals seven functionally distinct populations producing IL-2, IFN-γ, and TNF-α individually or in any combination. Together, such populations comprise 100% of the total CD4 or CD8 memory cytokine response and are represented pictorially by the pie charts to compare the quality of the responses between groups.
Figure 2.

Phenotypic and functional characterization of memory T cells by multiparameter flow cytometry. PBMCs were analyzed after primary immunization and rAD-Gag boost by multiparameter flow cytometry. CD4+ and CD8+ T cells are first segregated into CD45RA+CD95− and CD45RA−CD95+ T cells. Within the CD45RA−CD95+ CD4+ or CD8+ T cell population, cells were separated into seven distinct populations based on production of IFN-γ, IL-2, or TNF-α in any combination, represented pictorially by the pie charts.
The magnitude and quality of Th1 responses after prime-boost immunization
To first define the magnitude of the responses, the frequency of the total Gag-specific CD45RA−CD95+ CD4+ T cell cytokine responses comprising IL-2, IFN-γ, or TNF-α–producing cells are presented from individual animals from each vaccine group 6 wk after the primary immunization (Fig. 3 A), and 3 and 56 wk after the rAD-Gag boost (Fig. 3 B). Consistent with our ELISPOT data, immunization with Montanide/Gag and the TLR7/8 agonist induced the highest frequency of cytokine-producing memory T cells prior and after rAD-Gag boost. Importantly, administration of rAD-Gag did not increase the total frequency of memory CD4+ T cell cytokine responses in animals that had received a primary immunization (Fig. 3, B and C).
Figure 3.

The magnitude of Gag-specific CD4+ T cell cytokine responses after prime-boost immunization. PBMCs were analyzed by multiparameter flow cytometry as described in Fig. 2. The frequency of total cytokine-producing CD45RA−CD95+ CD4+ T cells (IFN-γ, IL-2, or TNF-α) was analyzed at 6 wk after primary immunization (A) and 3 and 56 wk after rAD Gag boost (B). Data are shown for the individual animals in each group (A and B), or as the means of the three to five animals per group (C) shown in A and B. N/A: nonapplicable.
As described for Fig. 2, the quality of the CD45RA−CD95+ CD4+ T cell cytokine response was determined on the composition of IL-2, IFN-γ, or TNF-α production at the single cell level. 2 wk after primary immunization, ∼35% of Th1 cells from NHP vaccinated with Gag/Montanide and CpG ODN or the TLR8 agonist produced IFN-γ only or IFN-γ and IL-2, whereas animals immunized with the TLR7/8 agonist had very few of such cells (Fig. 4 A). By 6 and 10 wk after immunization, there were progressively fewer cells expressing IFN-γ with or without IL-2 in all vaccine groups; cytokine responses from animals immunized with a TLR adjuvant were mostly comprised of cells secreting IL-2 and TNF-α. The percentage of cells positive for all three cytokines (IFN-γ, TNF-α, IL-2) remained stable in all vaccine groups from weeks 2 to 10 after primary immunization (Fig. 4 A). Of note, NHP immunized with HIV Gag/Montanide without a TLR adjuvant had a response mainly comprised of cells positive for IL-2 only, with very few IFN-γ–producing CD4+ T cells (Fig. 4 A). Moreover, the low frequencies of total cytokine-producing cells in these animals 1 yr after the rAD-Gag boost (Fig. 3 B) shows that the addition of a TLR adjuvant during primary immunization provides durability to the Th1 response even after the rAD-Gag boost.
Figure 4.

The quality of the Th1 response after prime-boost immunization. The total CD45RA−CD95+ CD4+ T cell cytokine response was divided into seven distinct subpopulations producing any combination of IFN-γ, IL-2, or TNF-α (Fig. 2); these data are shown as the means of their respective percentages from those animals with sufficient responses to allow for a consistent assessment of the quality (n = 4 per group, except Montanide/Gag + TLR7/8 agonist at week 56 (n = 3 animals), Montanide/Gag + TLR8 agonist at week 2 (n = 2 animals) and week 56 (n = 5 animals), rAD-Gag only group at weeks 2 and 56 (n = 2 animals), and Gag only at week 6 (n = 3 animals)). *, low number of positive events did not allow for a consistent qualitative assessment; N/A: nonapplicable. Additional analysis of the quality of the T cell responses from individual animals at 3 wk after rAD-Gag boost is shown in Fig. S3 (available at http://www.jem.org/cgi/content/full/jem.20052433/DC1).
Although the magnitude of the memory Th1 cytokine response did not increase after the rAD-Gag boost (Fig. 3, B and C), there was a dramatic change in the quality of such responses, and this was strongly influenced by the TLR agonist used in the primary immunization. This was most evident 1 yr after the rAD-Gag boost in NHP immunized with Montanide/Gag and the TLR7/8 agonist, with a response comprised of ∼50% of polyfunctional cells producing IFN-γ, IL-2, and TNF-α (Fig. 4 B). Collectively, these data show that TLR adjuvants administered during primary immunization markedly affected the quality and stability of the Th1 response before as well as after the rAD-Gag boost.
The magnitude and quality of Gag-specific CD8+ T cell cytokine responses after immunization
In addition to generating Th1 responses, it would be desirable if a nonlive vaccine could also elicit CD8+ T cell responses. Indeed, we have recently shown that antigen-specific CD8+ T cell responses are induced in NHP after several immunizations with HIV-Gag protein conjugated to the TLR7/8 agonist, but not when the protein was administered with the free TLR7/8, TLR8 agonist, or CpG ODN (8). Consistent with this, animals immunized with Montanide/Gag and the TLR7/8 agonist but not the TLR8 agonist or CpG ODN had detectable frequencies of cytokine-producing CD45RA−CD95+ CD8+ T cells (IFN-γ, IL-2, or TNF-α) after a single immunization (Fig. 5 A). Thus, the TLR7/8 agonist is sufficient to mediate cross-presentation in NHP, provided that the vaccine formulation is optimized. After the rAD-Gag boost, there was a striking increase in the total Gag-specific CD45RA−CD95+ CD8+ T cell cytokine response in animals immunized with Montanide/Gag and any of the TLR adjuvants (Fig. 5, B and C) compared with animals immunized with Montanide/Gag alone. Moreover, animals immunized with the TLR7/8 agonist had the highest frequencies of cytokine-producing memory CD8+ T cells for at least 1 yr after the adenoviral boost.
Figure 5.

The magnitude of Gag-specific CD8+ T cells after prime-boost immunization. The frequency of cytokine-producing CD45RA−CD95+ CD8+ T cells (IFN-γ, IL-2, or TNF-α) was analyzed at 6 wk after primary immunization (A), and 3 and 56 wk after rAD Gag boost (B) by multiparameter flow cytometry as described in Fig. 2. Data are shown for the individual animals in each group (A and B), or as the means from the three to five animals per group (C) shown in A and B. N/A: nonapplicable.
With regard to the quality of such responses, animals immunized with Montanide/Gag and the TLR7/8 agonist had 40–60% of cells secreting all three cytokines (IFN-γ, IL-2, or TNF-α) from the peak at 3 wk, to the memory responses at 56 wk after rAD-Gag boost (Fig. 6). The majority of CD45RA−CD95+ CD8+ T cells generated in NHP immunized with Montanide/Gag and CpG ODN or the TLR8 agonist produced IFN-γ and IL-2, or IFN-γ only after the boost. Of note, without a TLR adjuvant during primary immunization, such responses were not sustained after the rAD-Gag boost. These data show that the presence of a TLR agonist was also required to impart stability to memory CD8+ T cells after the rAD-Gag boost.
Figure 6.

The quality of Gag-specific CD8+ T cells after prime-boost immunization. The total CD45RA−CD95+ CD8+ T cell cytokine response was divided into seven distinct subpopulations producing any combination of IFN-γ, IL-2, or TNF-α (Fig. 2); these data are shown as the means of their respective percentages from the four animals per group, except rAD-Gag only (n = 2 animals), Montanide/Gag + TLR7/8 agonist at week 56 (n = 3 animals), and Montanide/Gag + TLR8 agonist at week 56 (n = 5 animals). *, low number of positive events did not allow for a consistent qualitative assessment.
Prime-boost immunization generates a high frequency of central memory T cells
After exposure to antigen, two subsets termed effector and central memory cells can be identified. Although both subsets have effector function, there is evidence that CD8+ central memory T cells have a greater capacity to proliferate after secondary exposure to antigen. Such cells have been described to be more efficient in mediating protection against pathogens in the mice (11) and NHP (12). For NHP, differential expression of CD28 within a memory population has been used to distinguish central and effector memory cells (10). Thus, to assess whether the vaccines used in our study induced central or effector memory cells, the total memory CD45RA−CD95+ CD4+ or CD8+ T cell cytokine response was further segregated into CD28+ (central memory) and CD28− (effector memory) cells. There was a relatively small fraction of CD4+ effector memory cells at 2 wk after primary immunization and 3 wk after rAD-Gag boost in animals vaccinated with Montanide/Gag and CpG ODN. 10 wk after primary immunization and 56 wk after rAD-Gag boost, essentially all cytokine-producing Th1 cells were CD28+ central memory cells (Fig. 7). Furthermore, there was a large number of cytokine-producing effector memory CD8+ T cells at 3 wk after rAD-Gag boost; however, 1 yr after the boost, the majority of CD8+ memory T cells were central memory cells (Fig. 7). Overall, the prime-boost immunization elicited a high frequency of central memory Th1 and CD8+ T cell responses throughout the course of the study.
Figure 7.

Characterization of central and effector memory Gag-specific T cells after prime-boost immunization. Cells were gated on CD28 and CD95, and cytokine-producing cells (IFN-γ, IL-2, or TNF-α) were overlayed as colored dots on the total CD4 or CD8 distributions shown in gray. For each vaccine group, a representative animal is shown.
Gag-specific antibody responses after prime-boost immunization
Montanide has been shown to be an effective adjuvant for enhancing antibody responses when administered with different protein vaccines (13, 14). Thus, we measured endpoint Gag-specific antibody titers throughout the course of prime-boost immunization. Animals immunized with Montanide/Gag alone had antibody titers of >10,000 after primary immunization that increased ∼1 log after the rAD-Gag boost (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20052433/DC1). There was a significant increase in antibody responses in NHP immunized with the TLR7/8 agonist compared with animals immunized with Montanide/Gag only. Animals immunized with rAD-Gag alone without prior priming injection had endpoint titers of 1:500 (unpublished data). These data show that potent humoral and cellular immune responses can be generated with this vaccine regimen.
DISCUSSION
TLRs expressed on antigen-presenting cells play a central role in controlling innate and adaptive immune responses after exposure to infectious pathogens. Hence, TLRs represent potent targets for vaccine development. Here, we used a variety of TLR agonists in the context of a prime-boost vaccine regimen with HIV Gag as a model antigen and found that these immunizations can generate a dramatically different magnitude, quality, and CD4/CD8 balance. The ability to more definitely assess immune responses with multiparameter flow cytometry and to understand how vaccines or adjuvants activate innate immunity will be important for vaccine development and will provide the tools necessary for generating the particular type of T cell response best suited to protect against a given pathogen.
As described previously, heterologous prime-boost immunization with plasmid DNA followed by rAD generates HIV Gag-specific cellular immune responses in NHP (6). Although DNA vaccines are promising, further improvements in immunogenicity are desirable. In the present study, a single immunization of HIV Gag/protein with a TLR7/8 agonist generated T cell IFN-γ responses comparable to those induced after three immunizations of plasmid DNA encoding HIV Gag protein (6).
Importantly, the TLR agonist used in the primary immunization had a strong influence on both the magnitude and quality of the response after the rAD boost. To optimize vaccine delivery, we used Montanide ISA51 as it is easily combined with proteins and TLR adjuvants and has an established safety profile in humans (9). Notably, 12 wk after a single immunization with Montanide/Gag and either the TLR7/8 agonist or CpG ODN, the magnitude of the Th1 responses was similar to responses in NHP 2 wk after four immunizations given without Montanide (8). Mechanisms that could account for the increased efficiency and immunogenicity by the Montanide/TLR agonist include increased duration of antigen/adjuvant at the site of immunization or more synchronous delivery to the APC (8). Thus, this type of vaccine formulation allows for more potent and durable cellular immune responses from a single immunization, making it a potentially cost effective and practical modality.
A potential caveat to this approach is that animals immunized with Montanide/Gag and the TLR7/8 agonist had local reactivity at the site of injection as early as 2 wk after immunization that resolved after 12 wk. This reactivity is not a general feature associated with the TLR7/8 agonist because repeated administration of protein and TLR7/8 agonist without Montanide elicited no local reactivity; the reactivity might be ameliorated by lowering the agonist or protein dose, or by intramuscular administration. Notably, CpG ODN enhanced T cell responses to protein and Montanide with little local reactivity. Indeed, such an approach has been used in humans to enhance CD8+ T cell responses to melanoma peptides (15).
The use of TLR agonists selective for activating plasmacytoid DCs (pDCs), conventional DCs (cDCs), or both in vitro provides insight into how such subsets may be influencing T cell immunity in vivo. The findings that CpG ODN elicited potent CD4+ T cell responses suggests that activating pDCs may be sufficient for generating Th1 responses, possibly through induction of IFN-α. Whether IFN-α produced by pDCs would also affect antigen presentation through maturation of other APCs (i.e., cDCs), or whether pDCs could directly present antigen to CD4+ T cells remains an open question. With regard to the role of cDCs, the TLR8 agonist was a poor adjuvant for generating Th1 responses in vivo despite its potency for eliciting IL-12p70 in vitro (8). It should be noted, however, that NHP immunized with Montanide/Gag and the TLR8 agonist had increased IL-2 responses after the primary immunization and higher CD8+ T cell responses after the rAD-Gag boost than animals immunized with Montanide/Gag alone. Thus, the TLR8 agonist did have functional effects on the T cell response in vivo. Although it remains possible that TLR agonists that activate cDCs only would not be sufficient for eliciting strong Th1 responses in primates in vivo, it is more likely that differences in the stability or optimal dose of the TLR8 agonist when compared with the TLR7/8 agonist could explain its limited adjuvant effect in vivo. Further studies with improved methods to activate and specifically target cDCs will clarify their role for eliciting Th1 responses in vivo. However, as the highest memory T cell responses were detected in NHP immunized with the TLR7/8 agonist, our data suggest that an adjuvant able to activate both pDCs and cDCs will be optimal for enhancing T cell immunity in vivo.
In addition to generating strong Th1 responses, most of the animals immunized with Gag and TLR7/8 agonist in Montanide also had detectable CD8+ T cell responses, providing evidence for cross-presentation. Similarly, animals immunized four times with HIV Gag protein conjugated to the TLR7/8 agonist without Montanide had detectable CD8+ T cell responses (8). Together, these data suggest that induction of CD8+ T cell responses in NHP with a protein vaccine can be achieved with a TLR agonist capable of activating both pDCs and cDCs.
Another major emphasis of this study was to determine how different TLR agonists would alter the quality of memory T cell responses in a prime-boost regimen. 2 wk after primary immunization, animals that received Montanide/Gag with CpG ODN had a striking increase in the frequency of IFN-γ–producing cells compared with animals immunized with Montanide/Gag and the TLR7/8 agonist. However, these responses quickly diminished and, by 12 wk, were similar in both groups. The rapid decrease in the number of IFN-γ–producing cells in NHP immunized with CpG ODN may be explained by the quality of the CD4+ T cell responses 2 wk after primary immunization. These animals had a high frequency of effector memory cells producing only IFN-γ, which would likely undergo cell death (16). Alternatively, the dramatic decrease in CD4+/IFN-γ–producing cells in this vaccine group may be the result of redistribution of such cells to nonlymphoid organs. By 10 wk after primary immunization, the quality of the memory CD4+ T cell responses was nearly identical in animals immunized with CpG ODN or the TLR7/8 agonist. However, the total number of IL-2–producing cells was higher when the TLR7/8 agonist was administered (Fig. 1 B), and this vaccine group had the highest memory T cell responses after the rAD-Gag boost. Collectively, these data suggest that although CpG ODN elicit better short-term effector cells in the primary phase, they are less efficient for inducing “boostable” memory cells than the TLR7/8 agonist. Despite the extensive analysis used in the present study, we did not identify a phenotypic or functional marker of T cells that can predict this boosting potential. However, it is possible that the increased frequency of IL-2–producing cells accounted for the enhanced magnitude and quality of the responses seen with the TLR7/8 agonist.
It is important to note that NHP immunized without a TLR adjuvant had an IL-2–dominated primary CD4+ T cell response (with or without TNF-α). After rAD-Gag boosting, these animals did not show a sustainable increase in IFN-γ–producing cells. This suggests that the primary immunization “imprints” the CD4+ T cell responses and limits their capacity to further differentiate into CD4+/IFN-γ–producing effector cells. This has direct relevance for designing vaccines against diseases requiring both humoral and cellular responses, such as malaria or HIV. In this regard, if a primary immunization with a protein antigen were given with an oil-based or alum adjuvant without a TLR agonist, there would be efficient induction of antibody responses, but the potential of limiting the cellular response after a viral boost.
After the rAD-Gag boost, there was no increase in the frequency of the total CD4 memory responses in all groups of animals immunized with any of the TLR adjuvants. It is possible that the rAD-Gag boost induced further differentiation of the primary Th1 response, resulting in an increased frequency of IFN-γ–producing cells that readily undergo cell death (16), thereby offsetting the expansion of existing or the generation of new antigen-specific cells. Because high doses of rAD-Gag efficiently induce IFN-γ–producing cells, it may be possible to enhance the magnitude of the total CD4 memory responses by using lower doses or by administering rAD by a different route.
In contrast with the CD4 response, the magnitude of the CD8 response dramatically increased in all NHP after the rAD-Gag boost. Primary CD8+ T cell responses were only detected from animals immunized with Montanide/Gag and the TLR7/8 agonist; in this group, the increased magnitude after the rAD-Gag boost may reflect expansion of these cells. However, it is likely that the presence of a Gag-specific CD4+ T cell response significantly enhances the CD8 generation, even in animals without detectable primary CD8+ T cells (17–19). This would extend to animals immunized with Montanide/Gag and CpG ODN or the TLR8 agonists, which had dramatically higher levels of Gag-specific CD8+ T cells after rAD boosting than animals that were not primed or those that were primed with Montanide/Gag alone.
Finally, the Montanide/Gag plus TLR7/8 agonist priming and rAD-Gag boosting induced a high frequency of polyfunctional cells producing IL-2, IFN-γ, and TNF-α; the high responses were sustained over 1 yr. Hence, these data suggest that activating both pDCs and cDCs with the TLR7/8 agonist elicits polyfunctional Th1 and CD8+ T cells. Generating a high frequency of IL-2–producing Th1 and CD8+ T cells may indeed be desirable for an HIV vaccine. In this regard, individuals infected with HIV have better control of infection if their CD4+ and CD8+ T cells sustain their proliferative and/or IL-2–producing capacity (20–24). A recent study extended this finding by demonstrating that long-term nonprogressors had a significantly biased T cell response with more “polyfunctional” CD8+ T cells simultaneously capable of multiple effector functions when compared with progressors (27). Similarly, in the experimental mouse model of Leishmania major infection, vaccines that induce a high frequency of CD4+ T cells simultaneously producing IL-2, TNF-α, and IFN-γ confer the best protection (unpublished data). Together, these results emphasize that multi-functional T cell responses will be beneficial for infections in which Th1 or CD8+ T cells are mediating effector functions. In the current study, we could not determine whether the immune responses generated by the TLR agonist–HIV protein vaccines would be sufficient to mediate protection because HIV Gag responses do not protect against SIV challenge. Future studies using SIV rather than HIV Gag protein together with TLR agonists are planned to assess whether this type of vaccine regimen confers protection.
Overall, these data highlight the remarkable heterogeneity of T cell responses elicited by prime-boost immunization regimens. An important question for vaccine development is whether the quality of the immune response is a better predictor of efficacy than the magnitude of the response. To this end, we show that remarkably different types of T cell responses (cytokine profile, CD4/CD8 balance, as well as magnitude) can be generated through the use of different TLR agonists. Our data provide a framework for using protein-based vaccines with TLR agonists in combination with replication-defective viral-based boosting to induce long-lived polyfunctional Th1 and CD8+ T cell responses. The ability to generate a variety of durable T cell responses will be a focus of future vaccine studies for infections such as HIV, Mycobacterium tuberculosis, Plasmodium falciparum, or L. major.
MATERIALS AND METHODS
Animals.
Indian rhesus macaques were stratified into comparable groups based on age, weight, sex, and frequency of naive T cells. Animals were maintained at the animal facility of the Walter Reed Army Institute of Research/Naval Medical Research Center. All experiments were conducted according to the guidelines of the National Research Council, under protocols approved by the Institutional Animal Care and Use Committee at the Walter Reed Army Institute of Research/Naval Medical Research Center and the National Institutes of Health.
Immunizations.
NHP (n = 5/group) were immunized a single time in two separate sites of the back (s.c.) with 200 μg HIV Gag protein with or without 2 mg of CpG ODN, TLR7/8, or TLR8 agonist emulsified in Montanide ISA 51 (Montanide). As controls during the primary immunization, a single monkey was immunized with Montanide/PBS without HIV Gag protein. In addition, there were three animals that received PBS, the TLR8 agonist or CpG ODN without Montanide, and these were also boosted later with rAD-Gag to control for the immunogenicity of boosting alone. To formulate these vaccines, 5 ml aqueous (200 μg HIV Gag protein plus 2 mg TLR ligand/agonist in PBS) plus 5 ml Montanide were aseptically homogenized to form emulsions as described previously (25). Montanide emulsions formed water-in-oil droplets and dispersed in an oil phase consisting of Marcol 5 paraffin oil (Exxon Co.) and 2% (vol/vol) Montanide 80 surfactant (SEPPIC, Inc.). All emulsions contained droplets with an average distribution of ∼1 micron, as per the manufacturer's (SEPPIC, Inc.) recommendation. 12 wk after primary immunization, animals were boosted with 1010 particles of rAD-Gag intramuscularly (i.m.) in a volume of 1 ml into the right thigh, rectus femoris muscle. Clinical grade rAD-Gag, an adenovirus serotype 5 (Ad5), was obtained from GenVec.
Reagents.
Cytosine phosphate guanosine oligodeoxynucleotides “C” class (CpG ODN, 2395) were purchased from Coley Pharmaceutical Group, the TLR7/8 agonist (3M-012), a structural analogue of 3M-003, and the TLR8 agonist (3M-002) were provided by 3M Pharmaceuticals (26). Purified recombinant HIV Gag p41 protein (HXB2 isolate) was made by the Protein Purification Group at the National Cancer Institute. An extensive functional analysis excluded the presence of contaminating peptides in the HIV Gag protein preparation.
Preparation of PBMCs.
PBMCs were isolated from fresh blood by Ficoll density centrifugation using Accuspin tubes according to the manufacturer's instructions (Sigma-Aldrich). Cells were used immediately or following cryopreservation for ELISPOT analysis or intracellular FACS staining. Similar results were seen using fresh or cryopreserved cells.
Detection of Gag-specific IFN-γ and IL-2–producing cells by ELISPOT assay.
The frequency of IFN-γ and IL-2–producing cells from PBMCs was determined by ELISPOT assay as described previously (8). In general, ELISPOT analysis was performed on all five animals in groups immunized with Montanide/Gag and any of the TLR adjuvants. As a result of the high titers of preexisting neutralizing antibody titers against the Ad5 vector, one animal in each group immunized with the TLR7/8 agonist or CpG ODN was excluded from analysis for all time points after AD-Gag boost. In addition, there were only three animals in the group that received rAD-Gag only without primary immunization.
Polychromatic flow cytometry.
5 × 106 PBMCs were stimulated in complete RPMI 1640 for 6 h with 5 μg/ml αCD28-Ax680, αCD49d and Brefeldin A (10 μg/ml each) with or without 2 μg/ml HIV Gag peptides. After stimulation, cells were stained as described previously (8). In brief, cells were surface stained for CD4, CD8, CD95, and CD45RA; in addition, ethidium monoazide bromide (EMA; 1 μg/ml) was added to identify dead cells. Cells were incubated for 15 min in the dark at room temperature and were exposed for 10 min to fluorescent light to photolink the EMA to the DNA. After washing, fixing, and permeabilizing, cells were stained for IFN-γ, IL-2, TNF-α, and CD3. 6 × 105–106 cells were acquired on a BD LSR II (BD Biosciences) and FACS data were analyzed using FlowJo software (Tree Star).
Statistics.
All group comparisons on ELISPOT analysis were done using Student's t tests with unequal variances on log-transformed data. p-values of <0.05 were considered significant.
Online supplemental material.
Fig. S1 shows the frequencies of IFN-γ–producing cells for the individual animals depicted as means in Fig. 1. Fig. S2 shows the frequencies of IL-2–producing cells for the individual animals depicted as means in Fig. 1. Fig. S3 shows the quality of the Th1 and CD8 T cell response 3 wk after rAD-Gag boost for the individual animals depicted as means in Figs. 4 B and 6. Fig. S4 shows the antibody responses after immunization with HIV Gag and TLR agonists/ligands. HIV Gag-specific antibodies were detected in serum obtained from animals 12 wk after primary immunization and 3 wk after rAD-Gag boost. 96-well plates were coated with HIV Gag protein, serum samples were added in serial dilutions, and after incubation with horseradish peroxidase–conjugated anti-IgG (BD Biosciences), plates were developed using TMB substrate-chromogen (DakoCytomation). *, P < 0.05 when compared with gag group. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20052433/DC1.
Supplemental Material
Acknowledgments
We are grateful to S. Felt for expert husbandry and technical assistance with the vaccinations, to S.P. Perfetto and R. Nguyen for their technical assistance at the LSR, J. Yu and P. Chattopadhyay for reagent manufacture, B. Hartman for helping with the graphics, and to M. Nason for statistical analysis.
This work was supported by the Naval Medical Research Center Work Unit STOF (grant no. 6.2.622787A.0101.870.EFX).
The authors have no conflicting financial interests.
Abbreviations used: cDC, conventional DC; CpG ODN, cytosine phosphate guanosine oligodeoxynucleotides; NHP, nonhuman primates; pDC, plasmacytoid DC; rAD, replication-defective adenovirus; TLR, Toll-like receptor.
R.M. Kedl's present address is Integrated Department of Immunology, University of Colorado Health Center, Denver, CO 80206.
References
- 1.Seder, R.A., and A.V. Hill. 2000. Vaccines against intracellular infections requiring cellular immunity. Nature. 406:793–798. [DOI] [PubMed] [Google Scholar]
- 2.Baba, T.W., Y.S. Jeong, D. Pennick, R. Bronson, M.F. Greene, and R.M. Ruprecht. 1995. Pathogenicity of live, attenuated SIV after mucosal infection of neonatal macaques. Science. 267:1820–1825. [DOI] [PubMed] [Google Scholar]
- 3.Shiver, J.W., T.M. Fu, L. Chen, D.R. Casimiro, M.E. Davies, R.K. Evans, Z.Q. Zhang, A.J. Simon, W.L. Trigona, S.A. Dubey, et al. 2002. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature. 415:331–335. [DOI] [PubMed] [Google Scholar]
- 4.Seaman, M.S., L. Xu, K. Beaudry, K.L. Martin, M.H. Beddall, A. Miura, A. Sambor, B.K. Chakrabarti, Y. Huang, R. Bailer, et al. 2005. Multiclade human immunodeficiency virus type 1 envelope immunogens elicit broad cellular and humoral immunity in rhesus monkeys. J. Virol. 79:2956–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donnelly, J.J., B. Wahren, and M.A. Liu. 2005. DNA vaccines: progress and challenges. J. Immunol. 175:633–639. [DOI] [PubMed] [Google Scholar]
- 6.Casimiro, D.R., L. Chen, T.M. Fu, R.K. Evans, M.J. Caulfield, M.E. Davies, A. Tang, M. Chen, L. Huang, V. Harris, et al. 2003. Comparative immunogenicity in rhesus monkeys of DNA plasmid, recombinant vaccinia virus, and replication-defective adenovirus vectors expressing a human immunodeficiency virus type 1 gag gene. J. Virol. 77:6305–6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santra, S., M.S. Seaman, L. Xu, D.H. Barouch, C.I. Lord, M.A. Lifton, D.A. Gorgone, K.R. Beaudry, K. Svehla, B. Welcher, et al. 2005. Replication-defective adenovirus serotype 5 vectors elicit durable cellular and humoral immune responses in nonhuman primates. J. Virol. 79:6516–6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wille-Reece, U., B.J. Flynn, K. Lore, R.A. Koup, R.M. Kedl, J.J. Mattapallil, W.R. Weiss, M. Roederer, and R.A. Seder. 2005. HIV Gag protein conjugated to a Toll-like receptor 7/8 agonist improves the magnitude and quality of Th1 and CD8+ T cell responses in nonhuman primates. Proc. Natl. Acad. Sci. USA. 102:15190–15194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aucouturier, J., L. Dupuis, S. Deville, S. Ascarateil, and V. Ganne. 2002. Montanide ISA 720 and 51: a new generation of water in oil emulsions as adjuvants for human vaccines. Expert Rev. Vaccines. 1:111–118. [DOI] [PubMed] [Google Scholar]
- 10.Pitcher, C.J., S.I. Hagen, J.M. Walker, R. Lum, B.L. Mitchell, V.C. Maino, M.K. Axthelm, and L.J. Picker. 2002. Development and homeostasis of T cell memory in rhesus macaque. J. Immunol. 168:29–43. [DOI] [PubMed] [Google Scholar]
- 11.Wherry, E.J., V. Teichgraber, T.C. Becker, D. Masopust, S.M. Kaech, R. Antia, U.H. von Andrian, and R. Ahmed. 2003. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 4:225–234. [DOI] [PubMed] [Google Scholar]
- 12.Vaccari, M., C.J. Trindade, D. Venzon, M. Zanetti, and G. Franchini. 2005. Vaccine-induced CD8+ central memory T cells in protection from simian AIDS. J. Immunol. 175:3502–3507. [DOI] [PubMed] [Google Scholar]
- 13.Ahlers, J.D., N. Dunlop, C.D. Pendleton, M. Newman, P.L. Nara, and J.A. Berzofsky. 1996. Candidate HIV type 1 multideterminant cluster peptide-P18MN vaccine constructs elicit type 1 helper T cells, cytotoxic T cells, and neutralizing antibody, all using the same adjuvant immunization. AIDS Res. Hum. Retroviruses. 12:259–272. [DOI] [PubMed] [Google Scholar]
- 14.Kumar, S., T.R. Jones, M.S. Oakley, H. Zheng, S.P. Kuppusamy, A. Taye, A.M. Krieg, A.W. Stowers, D.C. Kaslow, and S.L. Hoffman. 2004. CpG oligodeoxynucleotide and Montanide ISA 51 adjuvant combination enhanced the protective efficacy of a subunit malaria vaccine. Infect. Immun. 72:949–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Speiser, D.E., D. Lienard, N. Rufer, V. Rubio-Godoy, D. Rimoldi, F. Lejeune, A.M. Krieg, J.C. Cerottini, and P. Romero. 2005. Rapid and strong human CD8+ T cell responses to vaccination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J. Clin. Invest. 115:739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu, C.Y., J.R. Kirman, M.J. Rotte, D.F. Davey, S.P. Perfetto, E.G. Rhee, B.L. Freidag, B.J. Hill, D.C. Douek, and R.A. Seder. 2002. Distinct lineages of T(H)1 cells have differential capacities for memory cell generation in vivo. Nat. Immunol. 3:852–858. [DOI] [PubMed] [Google Scholar]
- 17.Sun, J.C., M.A. Williams, and M.J. Bevan. 2004. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat. Immunol. 5:927–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun, J.C., and M.J. Bevan. 2003. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 300:339–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shedlock, D.J., and H. Shen. 2003. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 300:337–339. [DOI] [PubMed] [Google Scholar]
- 20.Zimmerli, S.C., A. Harari, C. Cellerai, F. Vallelian, P.A. Bart, and G. Pantaleo. 2005. HIV-1-specific IFN-γ/IL-2-secreting CD8 T cells support CD4-independent proliferation of HIV-1-specific CD8 T cells. Proc. Natl. Acad. Sci. USA. 102:7239–7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lichterfeld, M., D.E. Kaufmann, X.G. Yu, S.K. Mui, M.M. Addo, M.N. Johnston, D. Cohen, G.K. Robbins, E. Pae, G. Alter, et al. 2004. Loss of HIV-1–specific CD8+ T cell proliferation after acute HIV-1 infection and restoration by vaccine-induced HIV-1–specific CD4+ T cells. J. Exp. Med. 200:701–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Migueles, S.A., A.C. Laborico, W.L. Shupert, M.S. Sabbaghian, R. Rabin, C.W. Hallahan, D. Van Baarle, S. Kostense, F. Miedema, M. McLaughlin, et al. 2002. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat. Immunol. 3:1061–1068. [DOI] [PubMed] [Google Scholar]
- 23.Younes, S.A., B. Yassine-Diab, A.R. Dumont, M.R. Boulassel, Z. Grossman, J.P. Routy, and R.P. Sekaly. 2003. HIV-1 viremia prevents the establishment of interleukin 2–producing HIV-specific memory CD4+ T cells endowed with proliferative capacity. J. Exp. Med. 198:1909–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harari, A., S. Petitpierre, F. Vallelian, and G. Pantaleo. 2004. Skewed representation of functionally distinct populations of virus-specific CD4 T cells in HIV-1-infected subjects with progressive disease: changes after antiretroviral therapy. Blood. 103:966–972. [DOI] [PubMed] [Google Scholar]
- 25.Miles, A.P., H.A. McClellan, K.M. Rausch, D. Zhu, M.D. Whitmore, S. Singh, L.B. Martin, Y. Wu, B.K. Giersing, A.W. Stowers, et al. 2005. Montanide ISA 720 vaccines: quality control of emulsions, stability of formulated antigens, and comparative immunogenicity of vaccine formulations. Vaccine. 23:2530–2539. [DOI] [PubMed] [Google Scholar]
- 26.Gorden, K.B., K.S. Gorski, S.J. Gibson, R.M. Kedl, W.C. Kieper, X. Qiu, M.A. Tomai, S.S. Alkan, and J.P. Vasilakos. 2005. Synthetic TLR agonists reveal functional differences between human TLR7 and TLR 8. J. Immunol. 174:1259–1268. [DOI] [PubMed] [Google Scholar]
- 27.Betts, M.R., M.C. Nason, S.M. West, S.C. DeRosa, S.A. Migueles, J. Abraham, M.M. Lederman, J.M. Benito, P.A. Goepfert, M. Connors, M. Roederer, and R.A. Koup. 2006. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood. In press. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}