

Abstract
c-FLIP proteins (isoforms: c-FLIPL, c-FLIPS, and c-FLIPR) play an essential role in the regulation of death receptor–induced apoptosis. Here, we demonstrate that the cytoplasmic NH2-terminal procaspase-8 cleavage product of c-FLIP (p22-FLIP) found in nonapoptotic malignant cells, primary T and B cells, and mature dendritic cells (DCs) strongly induces nuclear factor κB (NF-κB) activity by interacting with the IκB kinase (IKK) complex via the IKKγ subunit. Thus, in addition to inhibiting apoptosis by binding to the death-inducing signaling complex, our data demonstrate a novel mechanism by which c-FLIP controls NF-κB activation and life/death decisions in lymphocytes and DCs.
c-FLIP is a well-described inhibitor of death receptor–mediated apoptosis (1). At the mRNA level, it can be found in multiple splice variants, whereas at the protein level only three isoforms, c-FLIPL, c-FLIPS, and c-FLIPR, have been detected so far (1–4). All three c-FLIP isoforms contain two death effector domains (DEDs), which are structurally similar to the NH2-terminal part of procaspase-8. c-FLIPL also contains catalytically inactive caspase-like domains (p20 and p12).
c-FLIP proteins are recruited to the death-inducing signaling complex (DISC) by DED interactions (3–5). Both short c-FLIP isoforms, c-FLIPS and c-FLIPR, block death receptor–induced apoptosis by inhibiting procaspase-8 activation at the DISC (2, 3). The role of c-FLIPL at the DISC is still a matter of controversy (6, 7). Some reports describe c-FLIPL as an antiapoptotic molecule, functioning in a way analogous to c-FLIPS, whereas others describe c-FLIPL as a proapoptotic molecule, facilitating the activation of procaspase-8 at the DISC. This proapoptotic role may explain the phenotype of c-FLIP–deficient mice characterized by heart failure and death at embryonic day 10.5. The same phenotype has been reported for caspase-8– and FADD-deficient mice (8–11).
In addition to its antiapoptotic role in death receptor–induced apoptosis, c-FLIP proteins were invoked to play a prominent role in NF-κB signaling (12–14). The transcription factor NF-κB family regulates the expression of genes crucial for innate and adaptive immune responses, cell growth, and apoptosis (15). In mammalian cells, the NF-κB family is composed of five members: RelA, RelB, c-Rel, p50/NF-κB1, and p52/NF-κB2 (16). In most cells, the NF-κB dimer is sequestered in the cytosol by inhibitors of the κB protein (IκB), and its nuclear translocation can be induced by a wide variety of stimuli (16). These stimuli trigger activation of the IκB kinase (IKK) complex, which consists of two catalytic subunits, IKKα and IKKβ, as well as a regulatory subunit, IKKγ/NEMO. When the IKK complex is activated, IκB is phosphorylated, and the IκBs are degraded in a ubiquitin-dependent manner. The NF-κB dimers can then be translocated into the nucleus, where target gene transcription is induced.
Recently, it has been demonstrated that overexpression of c-FLIPL activates NF-κB (13, 17). In another study, upon overexpression, c-FLIPL was shown to interact with established components of the TNFR-mediated NF-κB activation pathway, TRAF1, TRAF2, and RIP (12). In addition, it has been reported that c-FLIPL–mediated NF-κB activation requires cleavage to p43-FLIP, also demonstrated to interact with TRAF2 (18). In TNFR-mediated NF-κB activation, TRAF2 and RIP were described to act upstream of the IKK complex (19, 20).
Here, we show that in nonapoptotic cells, c-FLIP forms heterodimers with procaspase-8 resulting in a novel NH2-terminal fragment of c-FLIP (p22-FLIP). p22-FLIP turned out to be the key mediator of NF-κB activation by direct binding to the IKK complex. These findings provide a new mechanism of c-FLIP–mediated NF-κB activation and shed light on the regulation of life/death decisions made in lymphocytes.
RESULTS
A new form of c-FLIP can be detected in malignant B and T cells
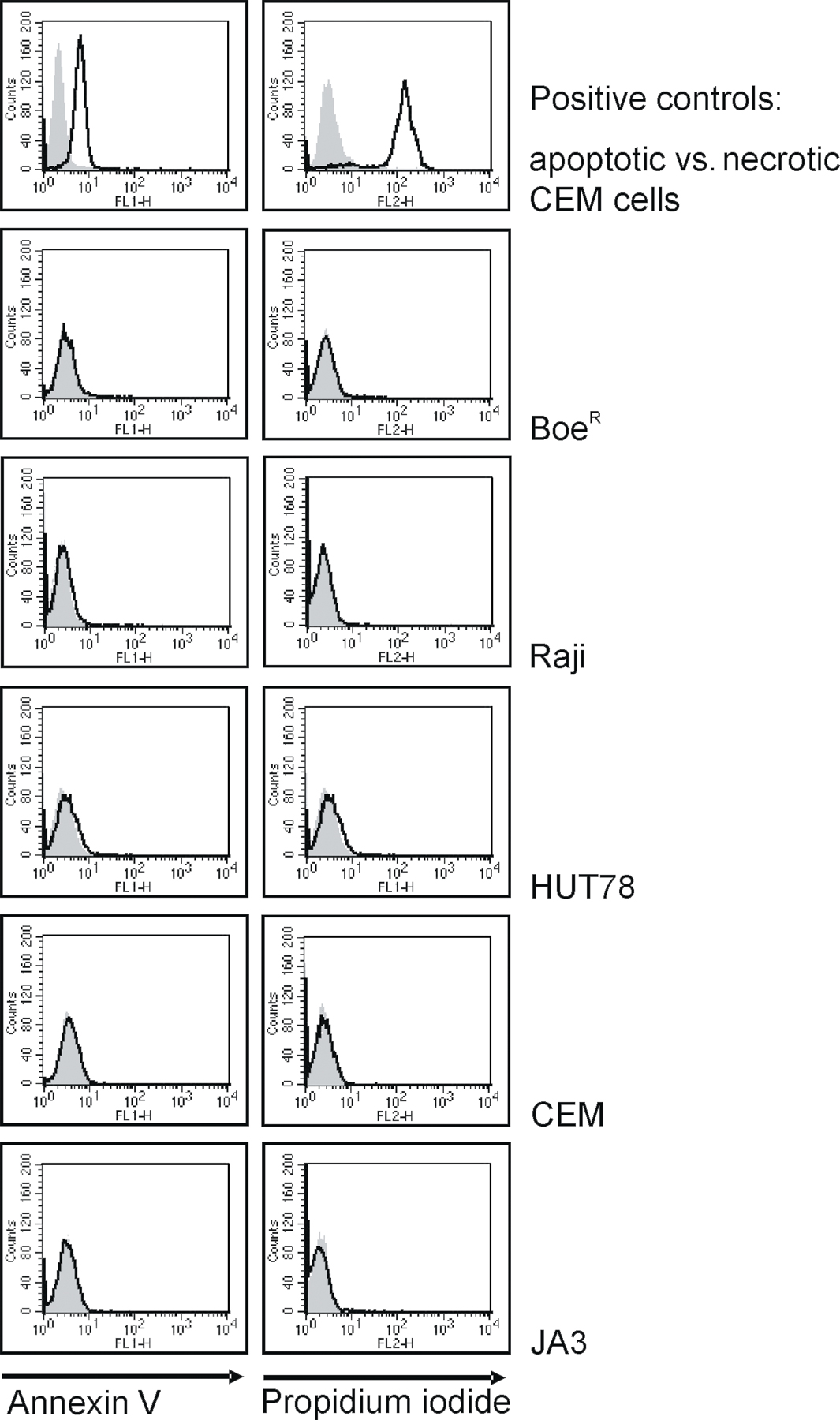
In addition to the three previously described c-FLIP protein isoforms, c-FLIPL, c-FLIPR, and c-FLIPS (2, 3, 21), we have detected a new prominent protein band with the anti-FLIP mAb NF6 directed against the DED region of c-FLIP (Fig. 1 A). The molecular mass of this protein is ∼22 kD. The p22 protein was observed in total cellular lysates (Fig. 1 A) and in immunoprecipitates (Fig. 1 B) from B lymphoblastoid cell lines BoeR and Raji and the T cell lines HUT78 and Jurkat A3, but not in CEM and SKW6.4 cells. The viability of the cells used for analysis was verified by negative propidium iodide and annexin V staining (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20051556/DC1). P22 protein was the most prominent in BoeR cells (Fig. 1, A and B). We call this protein p22-FLIP.
Figure 1.

Caspase-dependent presence of p22-FLIP in tumor cell lines. (A) Total cellular lysates of the indicated T and B cell lines were subjected to 12% SDS-PAGE and Western blot analysis using the anti-FLIP mAb NF6. The positions of c-FLIPL, c-FLIPS/R, and p22-FLIP are indicated. (B) Western blot analysis of c-FLIP proteins after immunoprecipitation from various cell lines (5 × 107 cells each) using anti-FLIP mAb NF6. Positions of c-FLIPS/R and p22-FLIP are indicated. (C) HUT78 and Jurkat A3 cells were preincubated with or without 20 μM zVAD-fmk for 30 min. Analysis of c-FLIP proteins by Western blot was performed as in A. (D) Schematic representation of c-FLIP proteins. DEDs are depicted in black. The cleavage site for generation of p22-FLIP (D198) is shown. The epitope for anti–c-FLIP mAb NF6 is indicated. (E) The NH2 terminus of c-FLIP encoding the amino acids 1–198 was in vitro translated, [35S] labeled, and added in the indicated amounts to the lysates of BoeR cells as well as to immunoprecipitates of c-FLIP from 5 × 107 BoeR cells.
The detection of p22-FLIP with the anti-FLIP mAb NF6 indicated the presence of DEDs in p22-FLIP because the antibody was raised against the NH2 terminus of c-FLIP. Furthermore, p22-FLIP disappeared upon the addition of zVAD-fmk (Fig. 1 C). This suggests that p22-FLIP is likely a caspase-dependent cleavage product of c-FLIP. We then analyzed the primary structure of c-FLIPL/S and found an aspartate residue at position 198 (Fig. 1 D). Cleavage at Asp198 could result in the formation of an NH2-terminal DED-containing cleavage product with a molecular mass of ∼22 kD, corresponding to p22-FLIP.
To test this hypothesis, we generated a cDNA corresponding to the c-FLIP NH2-terminal fragment resulting from cleavage at Asp198. Subsequently, p22-FLIP was translated in vitro and added to cell lysates of BoeR cells, followed by immunoprecipitation with the anti-FLIP mAb NF6. The products of immunoprecipitation as well as the corresponding lysates were analyzed by Western blot (Fig. 1 E). After adding the in vitro–translated c-FLIP NH2-terminal fragment, the band corresponding to p22-FLIP increased considerably. Thus, we conclude that the molecular mass of the in vitro–translated product was indeed identical to endogenous p22-FLIP. These data provide the first evidence that p22-FLIP is an NH2-terminal cleavage product of c-FLIP generated by cleavage at Asp198.
p22-FLIP identification as the NH2-terminal cleavage product of c-FLIP
To study whether p22-FLIP can be generated from both c-FLIPL and c-FLIPS, we performed the following experiment in vitro. c-FLIPL, c-FLIPS, and FLAG-c-FLIPL were translated in vitro, [35S] labeled, and added to total cellular lysates of HUT78 and J16 cells (Fig. 2 A). Upon incubation, all c-FLIPs were cleaved into the NH2-terminal fragment p22-FLIP (for c-FLIPL and c-FLIPS), the NH2-terminal FLAG-p22-FLIP (for FLAG-c-FLIPL), and the COOH-terminal fragment p33-FLIP (for c-FLIPL and FLAG-c-FLIPL). Consistent with the results obtained previously, p22-FLIP was not detected upon the addition of zVAD-fmk. Thus, we observed a caspase-dependent processing of c-FLIPL/S into p22-FLIP in nonapoptotic cells.
Figure 2.

Identification of the p22-FLIP cleavage site. (A) In vitro–translated [35S]-labeled c-FLIPS, c-FLIPL, or FLAG-c-FLIPL was added to the lysates of HUT78 (left) and Jurkat A3 cells (right) and incubated overnight at 4°C in presence or absence of 50 μM zVAD-fmk. Reactions were separated on 12% SDS-PAGE gels, blotted, and subjected to autoradiography. (B) In vitro–translated [35S]-labeled WT-FLAG-c-FLIPL or D198A-FLAG-c-FLIPL was added to the lysates of HUT78 cells and incubated overnight at 4°C in the presence or absence of 50 μM zVAD-fmk and visualized as in A. (C) BoeR cells were transfected with WT-FLAG-c-FLIPL or D198A-FLAG-c-FLIPL and incubated with or without 20 μM zVAD-fmk, and c-FLIP proteins were analyzed by Western blot.
To provide conclusive evidence that p22-FLIP is indeed the cleavage product of c-FLIP resulting from cleavage at Asp198, we generated an uncleavable D198A mutant of FLAG-c-FLIPL. In vitro–translated [35S]-labeled D198A-FLAG-c-FLIPL and WT-FLAG-c-FLIPL were added to the lysates of HUT78 cells (Fig. 2 B). As anticipated, FLAG-p22-FLIP was generated in a caspase-dependent manner only from WT-FLAG-c-FLIP. In addition, BoeR cells were transfected with WT- and D198A-FLAG-c-FLIP constructs, and consistent with the in vitro data, FLAG-p22-FLIP generation was observed only in the cells transfected with the WT construct (Fig. 2 C). Collectively, these data demonstrate that p22-FLIP is the NH2-terminal cleavage product of c-FLIP generated by caspase cleavage at Asp198.
p22-FLIP is generated by procaspase-8 activity and inhibits death receptor–induced apoptosis
To unravel the mechanism of p22-FLIP formation and find the caspase directly involved in c-FLIP processing, we investigated whether procaspase-8 might generate p22-FLIP. Procaspase-8, which, as a proform, was reported to possess catalytic activity (7, 22) and form heterodimers with c-FLIP in the cytosol by DED interactions, represented a likely candidate (6, 23). Therefore, we immunoprecipitated procaspase-8 from HUT78 cells with an anti–caspase-8 mAb and added in vitro–translated [35S]-labeled c-FLIPL (Fig. 3 A, left). Interestingly, we observed cleavage of c-FLIPL into the NH2-terminal fragment p22-FLIP and the COOH-terminal fragment p33-FLIP by procaspase-8. The processing was blocked by zVAD-fmk.
Figure 3.

p22-FLIP is generated by procaspase-8 and inhibits death receptor–induced apoptosis. (A) Procaspase-8 was immunoprecipitated from HUT78 cells using anti–caspase-8 mAb C15 and then incubated for 1 h at 37°C together with in vitro–translated [35S]-labeled c-FLIPL in the presence or absence of zVAD-fmk. c-FLIP processing was analyzed by autoradiography (top left). c-FLIP cleavage products p22 and p33 are indicated. Afterward, the same membrane was subjected to Western blot analysis using anti–caspase-8 mAb C15 (bottom left). [35S]-labeled c-FLIPL was incubated with the indicated concentrations of recombinant caspase-8 for 1 h at 37°C. c-FLIP processing was analyzed byautoradiography (top right). c-FLIP cleavage products p12 and p43 are indicated. Afterward, the same membrane was subjected to Western blot analysis using anti–caspase-8 mAb C15 (bottom right). (B) Analysis of p22-FLIP expression in BJAB cell lines stably overexpressing high or low amounts of p22-FLIP (p22-FLIPhigh or p22-FLIPlow, respectively). Endogenous expression of c-FLIPS is used as a loading control. (C) p22-FLIPhigh, p22-FLIPlow, and vector-transfected BJABs (Ctrl.) were stimulated with 1 μg/ml anti–APO-1 antibodies or 50 μl/ml LZ-CD95L for 16 h. Specific cell death was calculated as described in Materials and methods. (D) p22-FLIPhigh, p22-FLIPlow, and vector-transfected BJABs (Ctrl.) were stimulated with the indicated concentrations of FLAG-TRAIL for 16 h. (E) CD95 DISCs were immunoprecipitated from 5 × 107 cells of p22-FLIPhigh and vector-transfected BJABs (Ctrl.) and analyzed by Western blot with anti–caspase-8 mAb C15, anti-FLIP mAb NF6, anti-CD95 polyclonal antibody C20, and anti-FADD mAb.
To ensure that the mature caspase-8 heterotetramer could not process c-FLIP into p22-FLIP, we added recombinant active caspase-8 to in vitro–translated c-FLIPL (Fig. 3 A, right). As expected, recombinant caspase-8 cleaves c-FLIPL in a “classic apoptotic” fashion and generated p43-FLIP and p12-FLIP. Thus, we show that caspase-8 and procaspase-8 process c-FLIP in two mutually exclusive ways. The active caspase-8 heterotetramer generates the well-characterized p43-FLIP and p12-FLIP cleavage products, whereas procaspase-8 activity induces formation of the novel p22-FLIP cleavage product as well as the COOH-terminal p33-FLIP. This is consistent with a report on different substrate specificities of the caspase-8 proform and active caspase-8 (7). Clearly, the present study is the first to demonstrate two different caspase-8 specificities with respect to c-FLIP cleavage.
c-FLIP proteins are well-established inhibitors of death receptor–mediated apoptosis. To examine the role of p22-FLIP in death receptor–mediated apoptosis, we generated stable BJAB cell lines overexpressing either high (p22-FLIPhigh) or low (p22-FLIPlow) amounts of p22-FLIP. The amount of p22-FLIP in these cell lines was validated by Western blot (Fig. 3 B). Both p22-FLIPhigh and p22-FLIPlow BJAB cells were characterized by reduced sensitivity toward CD95- and TRAIL-induced apoptosis as compared with the vector-transfected control (Fig. 3, C and D, respectively). The reduction in apoptosis was more prominent in the p22-FLIPhigh cells. These data provide evidence that compared with other c-FLIP proteins, p22-FLIP is an even stronger inhibitor of death receptor–induced apoptosis.
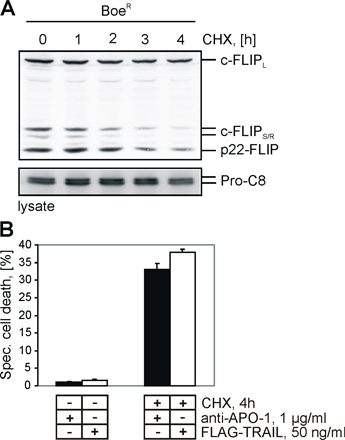
c-FLIP proteins were reported to have a short half-life (24, 25). We and others have demonstrated that cycloheximide (CHX) sensitizes cells toward death receptor–induced apoptosis, which correlates with a decrease of c-FLIP levels (24, 25). To understand whether sensitization also involves a decrease of the p22-FLIP level, we studied Boer cells that contain high levels of p22-FLIP. Treatment of Boer cells with CHX resulted in a substantial decrease of the p22-FLIP level within 4 h (Fig. S2 A, available at http://www.jem.org/cgi/content/full/jem.20051556/DC1). This decrease correlated with an enhanced sensitivity toward CD95- and TRAIL-induced apoptosis (Fig. S2 B). These data provide additional evidence that the p22-FLIP level in Boer cells correlates with the sensitivity toward death receptor–induced apoptosis, pointing toward the inhibitory role of p22-FLIP.
The antiapoptotic action of c-FLIP in death receptor–mediated apoptosis involves inhibition of caspase-8 activation at the DISC (2, 3, 24). To investigate whether p22-FLIP is also recruited to the DISC, we immunoprecipitated the CD95 DISC from p22-FLIPhigh cells. Indeed, p22-FLIP was recruited to the DISC (Fig. 3 E). As expected, the activation of procaspase-8 at the DISC of p22-FLIPhigh cells was lower than in vector-transfected cells. The amount of caspase-8 cleavage products in the DISC, p43/p41, and p18 was markedly reduced in p22-FLIPhigh cells, whereas the amounts of FADD and CD95 were similar in both p22-FLIPhigh and vector-transfected cells. This observation is consistent with the reduced sensitivity of p22-FLIPhigh cells toward CD95-induced apoptosis and shows that p22-FLIP effectively blocks caspase-8 activation at the DISC, thereby inhibiting CD95-induced apoptosis.
p22-FLIP is a strong inducer of NF-κB
We established an inhibitory role of p22-FLIP in death receptor–induced apoptosis; however, the questions of why p22-FLIP is present in nonapoptotic malignant cells and which functional role p22-FLIP might play in the nonapoptotic scenario were not answered. Using NF-κB luciferase activation assays with p22-FLIP and c-FLIPL, we observed that p22-FLIP is a strong inducer of NF-κB (Fig. 4 A). Moreover, p22-FLIP–mediated NF-κB activation was much stronger than that observed with c-FLIPL, even though expression levels of p22-FLIP and c-FLIPL were similar (Fig. 4 A).
Figure 4.

p22-FLIP is a strong inducer of NF-κB. (A) 293T cells were cotransfected with MEKK1, p22-FLIP, c-FLIPL, and luciferase reporter plasmid. GFP transfections were performed to control the transfection efficiency. Western blot analysis using anti-FLIP mAb NF6 was performed to control equal protein expression (right). (B) 293T cells were cotransfected with p22-FLIP and the luciferase reporter plasmid. After the indicated periods of time, cells were lysed and NF-κB luciferase activity was determined (top). Western blot analysis using anti-FLIP mAb NF6 was performed to determine the expression level of p22-FLIP. (C) Nuclear extracts, which were prepared from 293T cells transfected with p22-FLIP or GFP, were subjected to EMSAs using 32P-labeled oligonucleotides containing an NF-κB (left) or an NF-Y (right) binding site. p22-FLIP expression was verified by Western blot.
To clarify whether NF-κB activation correlates with the expression level of p22-FLIP, we performed transient transfections of 293T cells with p22-FLIP (Fig. 4 B). The maximum of NF-κB activity at 10 h paralleled the increase of p22-FLIP expression. Moreover, the observed expression level of p22-FLIP in 293T cells (Fig. 4 B) compared with c-FLIPL was still lower as compared with the ratio p22-FLIP/c-FLIPL in BoeR cells (Fig. 1 A). Thus, we demonstrate that the induction of NF-κB is specific for p22-FLIP and does not depend on a high expression level of p22-FLIP.
We also assayed p22-induced NF-κB activity by electrophoretic mobility shift assay (EMSA; Fig. 4 C), which independently confirmed that p22-FLIP induces NF-κB. Thus, we demonstrated that p22-FLIP is a strong inducer of NF-κB.
p22-FLIP induces NF-κB by direct interaction with the IKK complex
To get more insight into the mechanism of p22-mediated NF-κB induction, we coexpressed p22-FLIP with the inhibitors of NF-κB (IκBα and IκBβ) and with components of the IKK complex (IKKα, IKKβ, DN-IKKα, and DN-IKKβ; Fig. 5 A). Cotransfections of p22-FLIP with increasing amounts of IκBα and IκBβ inhibited NF-κB activation. Looking at more upstream events, we observed that cotransfection with DN-IKKα and DN-IKKβ also led to suppression of NF-κB activation. Collectively, the results indicated that p22-FLIP is a strong inducer of NF-κB acting via the canonical NF-κB pathway.
Figure 5.

p22-FLIP induces NF-κB by direct interaction with the IKK complex. (A) 293T cells were cotransfected with luciferase reporter plasmid and either MEKK1, p22-FLIP, or c-FLIPL (top part of the diagram). 293T cells were cotransfected with p22-FLIP, the luciferase reporter plasmid, and any one of the constructs IκBα, IκBβ, WT-IKKα, WT-IKKβ, mutated IKKα, or IKKβ (bottom part of the diagram). Transfection efficiency was examined using GFP transfections. NF-κB luciferase activity was determined as described in Materials and methods. (B) FLAG or FLIP immunoprecipitations were performed from 293T cells that were transfected with p22-FLIP and any one of the constructs FLAG-IKKα, FLAG-IKKβ, or FLAG-IKKγ. Immunoprecipitated products were subjected to 12% SDS-PAGE gels and analyzed by Western blot using anti-FLIP mAb NF6 and anti-FLAG mAb. (C) 293T cells were cotransfected with MEKK1, p22-FLIP, c-FLIPL, p43-FLIP, and the luciferase reporter plasmid. Transfected cells were incubated for 16 h in the presence of the indicated concentrations of zVAD-fmk and lysed, and NF-κB luciferase activity was determined.
We further examined whether p22-FLIP directly interacts with the IKK complex. FLAG-tagged IKKα, IKKβ, and IKKγ were transiently cotransfected with p22-FLIP into 293T cells, and then immunoprecipitated with anti-FLAG and anti-FLIP antibodies (Fig. 5 B). We did not observe any interaction between p22-FLIP and IKKα or IKKβ. However, p22-FLIP was coimmunoprecipitated with FLAG-IKKγ and vice versa. Thus, we showed that p22-FLIP interacts with the IKK complex via IKKγ.
In addition, we compared NF-κB induction by p22-FLIP, c-FLIPL, and p43-FLIP upon transient transfections in 293T cells (Fig. 5 C). Interestingly, the addition of zVAD-fmk resulted in a decrease in NF-κB activation for p43-FLIP and c-FLIPL, but did not affect p22-FLIP–mediated NF-κB induction. These results strongly indicate that p43-FLIP and c-FLIPL require further cleavage to induce NF-κB activity, whereas p22-FLIP does not. Thus, we show that for the induction of NF-κB activity, both p43-FLIP and c-FLIPL have to be processed into p22-FLIP.
p22-FLIP induces NF-κB during activation of primary lymphocytes and maturation of primary DCs
Next, we examined primary human T and B cells for the presence of p22-FLIP. Interestingly, p22-FLIP was absent in freshly prepared cells, but was generated upon activation of these cells with PHA (Fig. 6 A). The same phenomenon was observed in DCs. p22-FLIP was generated upon LPS stimulation, indicating that p22-FLIP is present during maturation of DCs (Fig. 6 A). Also, the increase of p22-FLIP levels in primary cells correlated with the increase of c-FLIP levels and, correspondingly, with the increase of the ratio of c-FLIP to procaspase-8. This observation provides additional evidence for the proposed mechanism of procaspase-8–mediated c-FLIP cleavage to p22-FLIP (Fig. 3).
Figure 6.

p22-FLIP is a key mediator of NF-κB induction. (A) Primary human T and B cells were stimulated with 1 μg/ml PHA. Primary immature iDCs were stimulated with 500 ng/ml LPS. Western blot analysis was performed using the anti-FLIP mAb NF6, the anti–caspase-8 mAb C15, and the anti-ERK1 mAb. (B) 107 primary human T cells were stimulated with 1 μg/ml PHA for 12 h and lysed, and anti-IKKγ immunoprecipitation was performed. Western blot analysis was performed using anti-IKKα/β antibody and anti-FLIP mAb NF6. (C) Primary human T cells were transiently transfected with double-stranded siRNA oligonucleotides comprising a FLIP-specific sequence (FLIP-1) or a nonspecific sequence (Ctrl.). 48 h after transfection, cells were stimulated with 1 μg/ml PHA for 3 d (right) or 0.5 μg/ml anti-CD3/28 for 24 h (left) or 3 d (right). After incubation, cells were lysed and Western blot analysis was performed using the anti-FLIP mAb NF6 (left), or the proliferation was measured after incorporation of tritiated thymidine ([3H]TdR) during the last 18 h.
To find out whether p22-FLIP also directly interacts with the IKK complex in primary human T cells, we performed immunoprecipitations using an anti-IKKγ antibody (Fig. 6 B). We observed p22-FLIP in the IKK complex. Thus, we conclude that p22-mediated NF-κB induction occurs via the same mechanism in primary cells.
To obtain more insight into the role of p22-FLIP in T cell activation, we studied the proliferation of primary T cells upon silencing of c-FLIP using small interfering RNA (siRNA). The silencing of c-FLIP resulted in down-regulation of c-FLIPL/S/R as well as its cleavage product, p22-FLIP (Fig. 6 C). Primary T cells were stimulated with either PHA or anti-CD3/CD28, and thymidine incorporation was measured after 3 d of additional culture (Fig. 6 C). Interestingly, silencing of c-FLIP led to a complete stop of cell proliferation. Thus, we show that the absence of c-FLIP and, consequently, p22-FLIP led to severe defects in T cell proliferation.
DISCUSSION
c-FLIP proteins were demonstrated to induce NF-κB activation (12–14). However, the exact underlying mechanism of this process has not been established yet. In this study, we identified a new mechanism of c-FLIP–mediated NF-κB activation and showed that NF-κB activation requires c-FLIP processing into the NH2-terminal DED-containing fragment, p22-FLIP (Fig. 7). p22-FLIP is generated by procaspase-8 cleavage of both c-FLIP isoforms, c-FLIPL and c-FLIPS. Furthermore, p22-FLIP is a strong activator of NF-κB, acting directly at the level of the IKK complex by binding to IKKγ. In addition to its role as an activator of NF- κB, p22-FLIP can block apoptosis by directly binding to the DISC.
Figure 7.

The dual function of p22-FLIP in the cell. p22-FLIP induces NF-κB by interacting with IKKγ in the IKK complex (right side). In addition, p22-FLIP can block death receptor–mediated apoptosis by binding to the DISC via DED interactions and inhibiting procaspase-8 activation (left side).
p22-FLIP is generated by cleavage of c-FLIPL/S at Asp198. The resulting NH2-terminal fragment, p22-FLIP, contains two tandem DEDs and has a high structural homology to v-FLIP of the Kaposi's sarcoma-associated herpesvirus and other v-FLIPs (14) that are also characterized by the presence of two DEDs followed by a short COOH terminus.
We argue that p22-FLIP is the final cleavage product of c-FLIP that serves as a mediator of NF-κB activation. This result is in contrast to previous studies, where it was suggested that p43-FLIP is a cleavage product of c-FLIP that mediates NF-κB activation (18). We have clearly shown that upon zVAD-fmk treatment, p43-FLIP–mediated activation of NF-κB is decreased, whereas the p22-FLIP–mediated NF-κB response remained unaltered. These results indicate that p43-FLIP requires further processing.
Importantly, our study demonstrates a new NF-κB–activating pathway initiated by procaspase-8. We show that independently of death receptor stimulation, procaspase-8 generates the p22-FLIP cleavage product, which leads to the induction of NF-κB (Fig. 7). Recently, procaspase-8 was reported to play a prominent role in NF-κB induction via its involvement in the MALT1–Bcl-10 adaptor complex that is formed upon TCR stimulation (26). Our findings further elucidate the involvement of procaspase-8 in NF-κB induction.
Interestingly, we could also show that the active mature caspase-8 heterotetramer p102-p182 cleaves c-FLIPL in vitro to p43-FLIP and p12-FLIP, but not to p22-FLIP. Thus, we observe that procaspase-8 and caspase-8 heterotetramer cleave c-FLIP in two different ways. This is consistent with reports upon different substrate specificities of the caspase-8 proform and active caspase-8 (7). Of note, our study is the first one demonstrating two different caspase-8 specificities with respect to c-FLIP cleavage.
In addition, we have observed in primary cells that upon increase of the ratio of c-FLIP to procaspase-8, the amount of p22-FLIP was substantially increased. It is likely that procaspase-8 constitutively cleaves c-FLIP to p22-FLIP, forming dimers with c-FLIP. The formation of such dimers between procaspase-8 and c-FLIP in the cytosol was described previously (6, 23). Thus, the ratio of procaspase-8 to c-FLIP in a particular cell type would be the crucial factor defining the amount of generated p22-FLIP and, correspondingly, the potential to induce NF-κB.
Our findings provide a molecular mechanism for how c-FLIP and procaspase-8 contribute to the activation and proliferation of primary lymphocytes. It was reported that caspase activity is essential for T cell activation, as it was shown in experiments with caspase inhibitors (27, 28). Analysis of caspase-8 and c-FLIP conditional knockout mice has demonstrated that those mice show severe defects in T cell activation and proliferation (29). Caspase-8 was reported to be essential for antigen-induced NF-κB activation in T, B, and NK cells (26, 30, 31). We show that upon silencing of c-FLIP in primary cells, the proliferation of these cells is impaired. This demonstrates the importance of our new p22-FLIP–mediated NF-κB–activating pathway in primary lymphocytes.
In conclusion, we described a new NF-κB–activating pathway, which is mediated by two well-described apoptotic DED-containing proteins: procaspase-8 and c-FLIP via their cleavage product, p22-FLIP. The balance between DED-containing proteins may provide sensitive signaling check points that cells use for signaling cross-talk and switching between apoptosis-resistant and -sensitive phenotypes and, thus, between life and death.
MATERIALS AND METHODS
Cell lines.
The T cell lines HUT78, CEM, H9, Jurkat (clone A3), and Jurkat (clone J16); the B lymphoblastoid cell lines SKW6.4, Raji, and BJAB; and the pre–B cell line BoeR were maintained in RPMI 1640, 10 mM Hepes, 50 μg/ml gentamycin, and 10% fetal calf serum (all from Life Technologies) in 5% CO2.
Antibodies and reagents.
Anti-FADD mAb (IgG1) was purchased from Transduction Laboratories. Anti-FLAG mAb was purchased from Sigma-Aldrich. Anti-CD95 polyclonal antibody C20 was purchased from Santa Cruz Biotechnology, Inc. Anti–caspase-8 mAb C15 (mouse IgG2b) recognizes the p18 subunit of caspase-8 (32). Anti-IKKα/β and anti-IKKγ antibodies were purchased from Santa Cruz Biotechnology, Inc., and anti-ERK1 mAb was purchased from BD Biosciences. Anti–APO-1 is an agonistic mAb (IgG3, κ) recognizing an epitope on the extracellular part of CD95 (APO-1/Fas; reference 33). FLAG-TRAIL was obtained from H. Walczak (DKFZ, Heidelberg, Germany). Horseradish peroxidase–conjugated goat anti–mouse IgG1, IgG2a, and IgG2b were from SouthernBiotech. All chemicals used were of analytical grade and purchased from Merck or Sigma-Aldrich. Plasmids encoding c-FLIPL and c-FLIPS have been described previously (2). The plasmid encoding FLAG-c-FLIPL has also been described (3). NF-κB reporter plasmid was provided by M. Li-Weber (DKFZ, Heidelberg, Germany). Constructs of FLAG-IKKα, FLAG-IKKβ, DN-FLAG-IKKα (K44A), and DN-FLAG-IKKβ (K44A) were provided by H. Nakano (Juntendo University, Tokyo, Japan).
EMSA.
Soluble nuclear proteins were prepared and used for EMSA as described previously (34). For each reaction, 10–20 fmol of 32P-labeled oligonucleotides comprising an NF-κB binding site (5′-TCAGAGGGGACTTTCCGAGAGGCG-3′) or NF-Y binding site (5′-CACCTTTTAACCAATCAGAAAAAT-3′) were used.
Cloning of p22-FLIP and D198A-FLAG-c-FLIPL.
p22-FLIP was cloned into the pEF4 expression vector (Invitrogen) using the PCR and the following primers: sense (encoding the KpnI restriction site): 5′-ggggtaccccATGTCTGCTGAAGTCATCC-3′ and antisense (encoding the XbaI restriction site): 5′-gctctagagcctaATCCTTGAGACTCTTTTGG-3′.
The D198A-FLAG-c-FLIPL mutant was cloned via overlap-PCR into the pEF4 expression vector (Invitrogen). The NH2-terminal part was amplified using the following: forward primer 1: 5′-ggggtaccccATGGACTACAAGGACGACGACAAGGGGATGTCTGCTGAAGTCATCCATCAGG-3′; reverse primer 1: 5′-CCTGAAAGTTATTTGAAGGTGCCTTGAGACTCTTTTGG-3′. The COOH-terminal part was amplified using the following: reverse primer 2: 5′-gctctagagcCTATGTGTAGGAGAGGATAAG-3′; forward primer 2: 5′-CCAAAAGAGTCTCAAGGCACCTTCAAATAACTTCAGG-3′. The overlap-PCR was performed using the forward and reverse primer 2.
CD95 DISC analysis.
Composition of the CD95 DISC was determined as follows. 5 × 107 cells were treated either with 1 μg/ml anti–APO-1 (IgG3) for 5 min at 37°C or left untreated, washed twice in 1× PBS, and lysed in lysis buffer (30 mM Tris/HCl, pH 7.5, 150 mM NaCl, 2 mM EDTA, 1 mM phenylmethylsulfonyl fluoride [Sigma-Aldrich], protease inhibitor cocktail [Roche], 1% Triton X-100 [Serva], and 10% glycerol). If pretreated with zVAD-fmk, cells were preincubated for 30 min at 37°C with the indicated concentrations of zVAD-fmk before stimulation. CD95 was immunoprecipitated with anti–APO-1 and protein A–Sepharose beads (Sigma-Aldrich) overnight at 4°C. Beads were washed five times with 20 volumes of lysis buffer. Immunoprecipitates were used for in vitro assay or analyzed using SDS-PAGE gels (35). Gels were transferred to Hybond nitrocellulose membrane (GE Healthcare), blocked with 5% nonfat dry milk in PBS/Tween (PBS plus 0.05% Tween 20) for 1 h, washed with PBS/Tween, and incubated with the primary antibody in PBS/Tween overnight at 4°C. Blots were developed with a chemoluminescence method according to the manufacturer's protocol (PerkinElmer).
Immunoprecipitations.
For c-FLIP immunoprecipitation, 5 × 107 cells were lysed in a volume of 1 ml for 30 min at 0°C, followed by the addition of 100 μl NF6 hybridoma supernatant together with 30 μl protein A–Sepharose. For FLAG immunoprecipitation, 2 × 106 cells were transfected using the calcium phosphate method 1 d before lysis. Immunoprecipitation was performed by using 4 μg anti-FLAG mAb together with 30 μl protein A–Sepharose. For IKKγ immunoprecipitation, 107 primary human T cells were lysed with or without stimulation, and the immunoprecipitation was performed by using 2 μg anti-IKKγ mAb together with 30 μl protein A– Sepharose. Immunoprecipitations were performed for 1 h at room temperature or overnight at 4°C. Beads were then washed five times with 20 volumes of lysis buffer and subjected to Western blot analysis as described above.
In vitro c-FLIP cleavage assays.
Lysates of the indicated cell lines were prepared as described above and incubated with in vitro–translated [35S]-labeled c-FLIPL, FLAG-c-FLIPL, D198A-FLAG-c-FLIPL, or c-FLIPS (TNT, T7-coupled reticulocyte lysate system; Promega) overnight at 4°C (36). The procaspase-8 cleavage assay was performed as follows: 5 × 107 HUT78 cells were lysed, and procaspase-8 was immunoprecipitated using 100 μl anti–caspase-8 C15 hybridoma together with 30 μl protein A–Sepharose. Beads with bound procaspase-8 were incubated in reaction buffer (50 mM Hepes, pH 7.4, 100 mM NaCl, 0.1% CHAPS, 10 mM dithiothreitol, and 10% sucrose) for 1 h at 37°C together with in vitro–translated c-FLIPL. The recombinant caspase-8 cleavage assay was performed as follows: 5 or 50 ng/ml recombinant caspase-8 was incubated in reaction buffer for 1 h at 37°C together with in vitro–translated c-FLIPL. Reactions were separated on 12% SDS-PAGE gels, blotted, and subjected to autoradiography.
Transfection of BJAB and BoeR cells.
Stable transfection of BJAB as well as transient transfection of BoeR cells was performed using expression vectors or the empty vector by electroporation (950 μF, 200 V). Selection pressure was added 48 h after transfection (100 μg/ml zeocin) for 2 wk (BJAB cells) or 10 d (BoeR cells). Expression was controlled by Western blot using anti-FLIP mAb NF6. The p22-FLIP–expressing BJABs as well as the empty vector–transfected control cells were subcloned.
Cytotoxicity assay.
To assay apoptosis, 5 × 105 cells were incubated in 48-well plates with or without the indicated amounts of anti–APO-1, LZ- CD95L, or FLAG-TRAIL for 16 h at 37°C. Cell death was measured by FSC/SSC via flow cytometry, and specific cell death was calculated as follows: (percentage of experimental cell death − percentage of spontaneous cell death)/(100 − percentage of spontaneous cell death) × 100.
NF-κB activation assay.
The day before transfection, 24-well titer plates were seeded with 0.5 × 105 293T cells. The cells were transfected using the calcium phosphate method with various expression vectors together with 500 ng of the NF-κB–driven luciferase reporter plasmid. Cells were washed with PBS 16 h after transfection and lysed for 20 min at room temperature in 50 μl lysis buffer (passive lysis buffer; Promega), followed by centrifugation (10,000 g) for 20 min to sediment insoluble materials. A total of 5 μl of cell lysates was mixed with 50 μl of the luciferase assay mixture (470 μM Beetle Luciferin [Promega], 1.07 mM (MgCO3)4Mg(OH)2 × 5 H2O, 20 mM N-Tris-(hydroxymethyl)-methylglycine, 2.67 mM MgSO4, 100 μM EDTA, 33.3 mM DTT, 270 μM CoA(OAc), and 530 μM ATP), and relative light units were measured with a Berthold duoluminomat (Bad Wildbad).
Preparation and activation of primary human lymphocytes and DCs.
Human peripheral T and B cells were prepared as described previously (37). For activation, resting primary human T cells (day 0) were cultured at 2 × 106 cells/ml with 1 μg/ml PHA for 16 h (day 1), and primary human B cells (day 0) were cultured at 2 × 106 cells/ml with 2 μg/ml PHA. After preparing lymphocytes, the primary human monocytes were isolated using cell adhesion onto cell culture flasks. Leukocytes were resuspended in 20–30 ml RPMI 1640 with 10% FCS, and 2-ml aliquots were seeded into six-well titer plates. After incubation for 1 h, adherent cells were washed with PBS. Monocytes were differentiated into immature DCs by adding 1% human AB serum, 1% donor plasma, 1,000 U/ml GM-CSF (Schering), and 500 U/ml IL-4 (Immunotools) for 3 d. Cytokines were renewed after 3 d for an additional 3 d, and immature DCs were stimulated with 500 ng/ml LPS for 16 h.
CHX experiments.
For CHX treatments, cells were incubated with 10 μg/ml CHX for the indicated periods of time. For assaying apoptosis in a cytotoxicity assay, 106 cells were pretreated with 10 μg/ml CHX for 4 h, washed, and incubated with the indicated concentrations of 1 μg/ml anti– APO-1 or 50 ng/ml FLAG-TRAIL for 16 h at 37°C in 24-well plates. Cell death was measured by FSC/SSC via flow cytometry, and specific cell death was calculated as follows: (percentage of experimental cell death − percentage of spontaneous cell death)/(100 − percentage of spontaneous cell death) × 100.
Annexin V and propidium iodide staining.
To detect phosphatidylserine exposure by flow cytometry, the T and B cell lines were washed once with PBS, incubated for 10 min on ice in 400 μl binding buffer (2.5 mM Hepes–NaOH, pH 7.4, 35 mM NaCl, and 0.625 mM CaCl2) with 1 μl annexin V–FITC (Qbiogene) or 10 μg/ml propidium iodide (Invitrogen), and analyzed via flow cytometry.
siRNA-mediated knockdown of c-FLIP and proliferation assays.
Primary human T cells were transfected by HiPerfect (QIAGEN) with a negative control or siRNA oligonucleotids specific for human CFLAR (Hs_CFLAR_1 HP; QIAGEN). For the lipofection, 0.2 μg (75 nM) siRNA was used, and transfected cells were rested for 48 h before further analysis. For proliferation assays, 105 cells were seeded into a 96-well titer plate and stimulated with either 1 μg/ml PHA or 0.5 μg/ml each of anti-CD3/CD28 for up to 4 d. Proliferation was measured with a scintillation counter after tritiated thymidine ([3H]TdR) incorporation during the final 15–18 h of the culture.
Online supplemental material.
Fig. S1 shows the living status of tumor cell lines used, and Fig. S2 shows that the resistance of BoeR cells toward CD95- or TRAIL-induced apoptosis is mediated by c-FLIP. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20051556/DC1.
Supplemental Material
Acknowledgments
We thank Ralf Marienfeld for providing the FLAG-IKKγ plasmid, Hiroyasu Nakano for the FLAG-IKKα and FLAG-IKK-β plasmids, and Peter Angel for a MEKK1 plasmid. We thank Tobias Haas and Henning Walczak for providing FLAG-TRAIL and Min Li-Weber for advice. We thank Heiko Weyd for isolation and differentiation of primary human immature DCs. We would like to thank Sherryl Sundell for critically reading the manuscript; Karsten Gülow, Dagmar Riess, and Rüdiger Arnold for their critical comments; Heidi Sauter for excellent secretarial work; and the Wilhelm Sander Stiftung, SFB 405, and Tumorzentrum Heidelberg/Mannheim for financial support.
The authors have no conflicting financial interests.
Abbreviations used: CHX, cycloheximide; DED, death effector domain; DISC, death-inducing signaling complex; EMSA, electrophoretic mobility shift assay; IKK, IκB kinase complex; siRNA, small interfering RNA.
References
- 1.Krueger, A., S. Baumann, P.H. Krammer, and S. Kirchhoff. 2001. FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol. Cell. Biol. 21:8247–8254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Golks, A., D. Brenner, C. Fritsch, P.H. Krammer, and I.N. Lavrik. 2005. c-FLIPR, a new regulator of death receptor-induced apoptosis. J. Biol. Chem. 280:14507–14513. [DOI] [PubMed] [Google Scholar]
- 3.Krueger, A., I. Schmitz, S. Baumann, P.H. Krammer, and S. Kirchhoff. 2001. Cellular flice-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the cd95 death-inducing signaling complex. J. Biol. Chem. 276:20633–20640. [DOI] [PubMed] [Google Scholar]
- 4.Scaffidi, C., I. Schmitz, P.H. Krammer, and M.E. Peter. 1999. The role of c-FLIP in modulation of CD95-induced apoptosis. J. Biol. Chem. 274:1541–1548. [DOI] [PubMed] [Google Scholar]
- 5.Thome, M., P. Schneider, K. Hofmann, H. Fickenscher, E. Meinl, F. Neipel, C. Mattmann, K. Burns, J.L. Bodmer, M. Schröter, et al. 1997. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 386:517–521. [DOI] [PubMed] [Google Scholar]
- 6.Micheau, O., M. Thome, P. Schneider, N. Holler, J. Tschopp, D.W. Nicholson, C. Briand, and M.G. Grutter. 2002. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J. Biol. Chem. 277:45162–45171. [DOI] [PubMed] [Google Scholar]
- 7.Chang, D.W., Z. Xing, V.L. Capacio, M.E. Peter, and X. Yang. 2003. Interdimer processing mechanism of procaspase-8 activation. EMBO J. 22:4132–4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rasper, D.M., J.P. Vaillancourt, S. Hadano, V.M. Houtzager, I. Seiden, S.L. Keen, P. Tawa, S. Xanthoudakis, J. Nasir, D. Martindale, et al. 1998. Cell death attenuation by ‘Usurpin’, a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ. 5:271–288. [DOI] [PubMed] [Google Scholar]
- 9.Yeh, W.C., J.L. Pompa, M.E. McCurrach, H.B. Shu, A.J. Elia, A. Shahinian, M. Ng, A. Wakeham, W. Khoo, K. Mitchell, et al. 1998. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science. 279:1954–1958. [DOI] [PubMed] [Google Scholar]
- 10.Yeh, W.C., A. Itie, A.J. Elia, M. Ng, H.B. Shu, A. Wakeham, C. Mirtsos, N. Suzuki, M. Bonnard, D.V. Goeddel, and T.W. Mak. 2000. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity. 12:633–642. [DOI] [PubMed] [Google Scholar]
- 11.Varfolomeev, E.E., M. Schuchmann, V. Luria, N. Chiannilkulchai, J.S. Beckmann, I.L. Mett, D. Rebrikov, V.M. Brodianski, O.C. Kemper, O. Kollet, et al. 1998. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 9:267–276. [DOI] [PubMed] [Google Scholar]
- 12.Kataoka, T., R.C. Budd, N. Holler, M. Thome, F. Martinon, M. Irmler, K. Burns, M. Hahne, N. Kennedy, M. Kovacsovics, and J. Tschopp. 2000. The caspase-8 inhibitor FLIP promotes activation of NF-kappaB and Erk signaling pathways. Curr. Biol. 10:640–648. [DOI] [PubMed] [Google Scholar]
- 13.Hu, W.H., H. Johnson, and H.B. Shu. 2000. Activation of NF-kappaB by FADD, Casper, and caspase-8. J. Biol. Chem. 275:10838–10844. [DOI] [PubMed] [Google Scholar]
- 14.Thome, M., and J. Tschopp. 2001. Regulation of lymphocyte proliferation and death by FLIP. Nat. Rev. Immunol. 1:50–58. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh, S., and M. Karin. 2002. Missing pieces in the NF-kappaB puzzle. Cell. 109:S81–S96. [DOI] [PubMed] [Google Scholar]
- 16.Hayden, M.S., and S. Ghosh. 2004. Signaling to NF-kappaB. Genes Dev. 18:2195–2224. [DOI] [PubMed] [Google Scholar]
- 17.Chaudhary, P.M., M.T. Eby, A. Jasmin, A. Kumar, L. Liu, and L. Hood. 2000. Activation of the NF-kappaB pathway by caspase 8 and its homologs. Oncogene. 19:4451–4460. [DOI] [PubMed] [Google Scholar]
- 18.Kataoka, T., and J. Tschopp. 2004. N-terminal fragment of c-FLIP(L) processed by caspase 8 specifically interacts with TRAF2 and induces activation of the NF-kappaB signaling pathway. Mol. Cell. Biol. 24:2627–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hur, G.M., J. Lewis, Q. Yang, Y. Lin, H. Nakano, S. Nedospasov, and Z.G. Liu. 2003. The death domain kinase RIP has an essential role in DNA damage-induced NF-kappa B activation. Genes Dev. 17:873–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu, Z.G. 2005. Molecular mechanism of TNF signaling and beyond. Cell Res. 15:24–27. [DOI] [PubMed] [Google Scholar]
- 21.Peter, M.E., and P.H. Krammer. 2003. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 10:26–35. [DOI] [PubMed] [Google Scholar]
- 22.Boatright, K.M., M. Renatus, F.L. Scott, S. Sperandio, H. Shin, I.M. Pedersen, J.E. Ricci, W.A. Edris, D.P. Sutherlin, D.R. Green, and G.S. Salvesen. 2003. A unified model for apical caspase activation. Mol. Cell. 11:529–541. [DOI] [PubMed] [Google Scholar]
- 23.Boatright, K.M., C. Deis, J.B. Denault, D.P. Sutherlin, and G.S. Salvesen. 2004. Activation of caspases-8 and -10 by FLIP(L). Biochem. J. 382:651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmitz, I., H. Weyd, A. Krueger, S. Baumann, S.C. Fas, P.H. Krammer, and S. Kirchhoff. 2004. Resistance of short term activated T cells to CD95-mediated apoptosis correlates with de novo protein synthesis of c-FLIPshort. J. Immunol. 172:2194–2200. [DOI] [PubMed] [Google Scholar]
- 25.Fulda, S., E. Meyer, and K.M. Debatin. 2000. Metabolic inhibitors sensitize for CD95 (APO-1/Fas)-induced apoptosis by down-regulating Fas-associated death domain-like interleukin 1-converting enzyme inhibitory protein expression. Cancer Res. 60:3947–3956. [PubMed] [Google Scholar]
- 26.Su, H., N. Bidere, L. Zheng, A. Cubre, K. Sakai, J. Dale, L. Salmena, R. Hakem, S. Straus, and M. Lenardo. 2005. Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science. 307:1465–1468. [DOI] [PubMed] [Google Scholar]
- 27.Kennedy, N.J., T. Kataoka, J. Tschopp, and R.C. Budd. 1999. Caspase activation is required for T cell proliferation. J. Exp. Med. 190:1891–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Misra, R.S., D.M. Jelley-Gibbs, J.Q. Russell, G. Huston, S.L. Swain, and R.C. Budd. 2005. Effector CD4+ T cells generate intermediate caspase activity and cleavage of caspase-8 substrates. J. Immunol. 174:3999–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chau, H., V. Wong, N.J. Chen, H.L. Huang, W.J. Lin, C. Mirtsos, A.R. Elford, M. Bonnard, A. Wakeham, A.I. You-Ten, et al. 2005. Cellular FLICE-inhibitory protein is required for T cell survival and cycling. J. Exp. Med. 202:405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chun, H.J., L. Zheng, M. Ahmad, J. Wang, C.K. Speirs, R.M. Siegel, J.K. Dale, J. Puck, J. Davis, C.G. Hall, et al. 2002. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature. 419:395–399. [DOI] [PubMed] [Google Scholar]
- 31.Dohrman, A., T. Kataoka, S. Cuenin, J.Q. Russell, J. Tschopp, and R.C. Budd. 2005. Cellular FLIP (long form) regulates CD8+ T cell activation through caspase-8-dependent NF-kappa B activation. J. Immunol. 174:5270–5278. [DOI] [PubMed] [Google Scholar]
- 32.Scaffidi, C., J.P. Medema, P.H. Krammer, and M.E. Peter. 1997. FLICE is predominantly expressed as two functionally active isoforms, caspase-8/a and caspase-8/b. J. Biol. Chem. 272:26953–26958. [DOI] [PubMed] [Google Scholar]
- 33.Trauth, B.C., C. Klas, A.M. Peters, S. Matzku, P. Moller, W. Falk, K.M. Debatin, and P.H. Krammer. 1989. Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science. 245:301–305. [DOI] [PubMed] [Google Scholar]
- 34.Brenner, D., A. Golks, F. Kiefer, P.H. Krammer, and R. Arnold. 2005. Activation or suppression of NFkappaB by HPK1 determines sensitivity to activation-induced cell death. EMBO J. 24:4279–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227:680–685. [DOI] [PubMed] [Google Scholar]
- 36.Medema, J.P., C. Scaffidi, F.C. Kischkel, A. Shevchenko, M. Mann, P.H. Krammer, and M.E. Peter. 1997. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J. 16:2794–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klas, C., K.M. Debatin, R.R. Jonker, and P.H. Krammer. 1993. Activation interferes with the APO-1 pathway in mature human T cells. Int. Immunol. 5:625–630. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}