

Abstract
2-Ethynylnaphthalene (2EN) is an effective mechanism-based inhibitor of CYP2B4. There are two inhibitory components, (1) irreversible inactivation of CYP2B4 (a typical time-dependent inactivation), and (2) a reversible component. The reversible component was unusual in that the degree of inhibition was not simply a characteristic of the enzyme-inhibitor interaction, but dependent on the size of the substrate molecule used to monitor residual activity. The effect of 2EN on the metabolism of seven CYP2B4 substrates showed that it was not an effective reversible inhibitor of substrates containing a single aromatic ring; substrates with two fused rings were competitively inhibited by 2EN; and larger substrates were non-competitively inhibited. Energy-based docking studies demonstrated that, with increasing substrate size, the energy of 2EN and substrate co-binding in the active site became unfavorable precisely at the point where 2EN became a competitive inhibitor. Hierarchical docking revealed potential allosteric inhibition sites separate from the substrate binding site.
Cytochrome P450 refers to a superfamily of enzymes that catalyze the oxidation of a wide variety of exogenous and endogenous chemicals. The enzyme system most commonly supports oxygen insertion into a substrate molecule, generating a hydroxylated product; however, the initial monooxygenation can lead to a wide variety of reactions such as dealkylation, oxidative deamination, sulfoxidation, and epoxidation (1).
The broad substrate selectivity of the P450 enzymes is due not only to the multiplicity of P450 enzymes, but also due to the characteristics of the active site. The active site for several of the P450 enzymes has been shown to be relatively large and capable of binding and metabolizing substrates of diverse chemical size and structure. A consequence of the large active site is its ability to accommodate multiple substrate/effector molecules. This effect is most commonly associated with CYP3A4 (2;3), where the presence of multiple compounds within the active site has been shown to alter the kinetics to exhibit cooperativity (4;5), and both substrate and product inhibition (2;6). The binding of multiple substrate/inhibitor molecules has also been documented for CYP2C9 (4), CYPERYF (7;8), and P450 cam (9). The presence of active sites on other P450 enzymes that are sufficiently large to bind multiple ligands is clearly possible and likely, based on the relative size of the ligands as compared to the active sites of these non-specific enzymes.
2-Ethynylnaphthalene (2EN) is a selective mechanism-based inhibitor of CYP2B4. CYP2B4 catalyzes the conversion of 2EN to the highly reactive intermediate, 2-naphthylacetic acid, which covalently modifies the apoprotein and results in its inactivation (10;11). In addition to its ability to inhibit CYP2B4-mediated reactions, 2EN could also act as a reversible inhibitor of both CYP1A1 and CYP1A2 (12). Although earlier studies reported that 2EN could act as a mechanism-based inhibitor of CYP1A proteins (13), the binding associated with these complexes is not nearly as tight as that observed between 2EN and CYP2B enzymes (12).
Previously, our laboratory reported on the inhibition of CYP2B4 by 2EN, where both the irreversible and reversible components were characterized (14). This was accomplished by examining the residual metabolism, for seven different CYP2B4 substrates before and after inactivation with 2EN. This inhibitor was effective at inactivating CYP2B4, leading to an inactivation of greater than 80% when preincubated with 1 μM 2EN for 10 min. 2EN also reversibly inhibited CYP2B4 activities; however, the characteristics of the inhibitory response were dependent on the substrate employed. Examination of the reversible component showed that 2EN was a more effective reversible inhibitor with larger substrates, which is not consistent with classical theory of enzyme inhibition. The goal of this report is to further examine the reversible inhibition of CYP2B4 by 2EN as a function of the substrate employed. The results are consistent with the presence of multiple 2EN binding sites on the CYP2B4 molecule, located at or near the substrate binding site, with interplay among these sites leading to the complex inhibition patterns.
EXPERIMENTAL PROCEDURES
Materials
7-ethoxycoumarin (7-EC), 7-hydroxycoumarin (7-HC), 7-pentoxyresorufin (7-PR), 7-benzyloxyresorufin (7-BR), resorufin, were purchased from Sigma-Aldrich (St. Louis, MO). Benzphetamine (BZP) was a gift from Upjohn (Kalamazoo, MI). 7-ethoxy-4-trifluoromethylcoumarin (7-EFC), and 7-hydroxy-4-trifluoromethylcoumarin (7-HFC) were obtained from Molecular Probes (Eugene, OR). p-Nitroanisole (PNA) was provided by Acros Organics (Belgium). Testosterone (TS) and its metabolites were from Steraloids Inc. (Newport, RI). 2-Ethynylnaphthalene (2EN) was synthesized as described (13;15), and its purity was confirmed by GC-MS, NMR, and by TLC using a reference standard for comparison (gift from Maryam Foroozesh, Xavier University, New Orleans, LA).
Enzymes
Cytochrome P450 2B4 (CYP2B4) was expressed in Escherichia coli C41 and purified according to standard procedures (16). NADPH-cytochrome P450 reductase was purified from phenobarbital-treated rabbits as described previously (17). Recombinant rabbit NADPH cytochrome P450 reductase (plasmid: pSC-CPR, provided by Dr. Lucy Waskell (Univ. Michigan); constructed from plasmid pCWori-rabbit reductase and plasmid pOR263-rat reductase, utilizing a T7 promoter) was expressed in E. coli C41, solubilized and purified as described previously (18-20). Both preparations of NADPH-cytochrome P450 reductase showed similar enzyme activities.
Preparation of reconstituted systems
CYP2B4 and NADPH-cytochrome P450 reductase were reconstituted with sonicated dilauroylphosphatidylcholine (DLPC) as described (21). Briefly, DLPC was prepared at a stock concentration of 8 mM in 50 mM potassium phosphate buffer, pH 7.25, containing 20% glycerol, 0.1 M NaCl, and 5 mM EDTA. The DLPC stock suspension was sonicated for approximately 30 min using a bath sonicator, until clarification. The sonicated DLPC was combined with reductase and P450 and preincubated for 2 hr at room temperature. The concentrations of DLPC, reductase, and CYP2B4 during the preincubation were 1.4 mM, 17 μM, and 8.7 μM, respectively. These reconstituted systems were then combined with other assay components to examine their catalytic characteristics as described below.
Determination of the type of reversible inhibition of CYP2B4 with each substrate by 2EN
The reconstituted systems containing 0.05 μM CYP2B4, 0.1 μM rabbit cytochrome P450 reductase and 8 μM DLPC were incubated with each substrate (PNA, 7-EC, 7-EFC, 7-PR, 7-BR, or TS), and 2EN at concentrations ranging from 0-1 μM, unless otherwise indicated in 100 mM potassium phosphate buffer (pH 7.25) at 37°C. The buffer used for BZP metabolism was 10 mM potassium phosphate, pH 7.25. The reaction was initiated by the addition of NADPH to a final concentration of 0.5 mM. Lineweaver-Burk plots were used to determine the type of inhibition caused by 2EN. Kinetic constants were calculated using non-linear regression analysis (GraphPad Prism, San Diego, CA).
Product formation for each of the substrates was measured as described previously (14), using standard fluorescence assays for 7EC, 7EFC, 7PR, 7BR, and BZP assays (22-27). PNA metabolism was determined by monitoring the absorbance at 405 nm resulting from the formation of p-nitrophenol (28). TS metabolism was monitored using a standard HPLC assay (29).
In the absence of 2EN, plots of product formation versus time were linear for at least 20 min. In the presence of 2EN (both substrate and 2EN were added simultaneously), linearity was observed for at least 10 min. After this time the rate of product formation decreased, and product formation began to deviate from linearity, consistent with the mechanism-based inactivation of CYP2B4. All reaction rates were determined using the linear portion of the curve, within 10 min after initiation of the reaction.
Hierarchical Docking
Complimentary to experimental studies, we explored the configuration space available to 2EN and the substrates in and near the heme binding site of CYP2B4 via docking with density functional theoretical (DFT) parameterization of the ligand, inhibitor, and heme charges. While the energies of docked ligand configurations are, in general, not good predictors of ligand binding affinities (30), the lowest energy, docked ligand configurations generally are consistent with structure based analysis (31;32). In this guise, Autodock (Molecular Graphics Laboratory, The Scripps Research Institute) (33) scored amongst the top performing docking approaches in providing ca. 80% of poses consistent with crystallographic orientations (31;32). The initial coordinates for the docking studies were those of the crystal structure of 4-(4-chlorophenyl)imidazole bound CYP2B4 (1SUO) with the inhibitor and waters removed (34).
The docking procedure employed for initial exploration of multiple ligand/inhibitor binding was analogous to one that was explored by a number of groups whose aim was to probe the structural basis of heterotropic/homotropic activation or inhibition (35;36). In this approach, two ligands were docked in a sequential fashion. The first ligand was docked using a pre-evaluated interaction grid based on interactions with atoms in the protein alone. The second ligand was then docked employing an interaction grid including interactions with the protein and bound configurations of the 1st ligand. In this way, the energy landscape for binding of the 2nd ligand includes interactions with the 1st ligand. It is plausible that binding multiple ligands to P450 isozymes involves such a sequential process. Our refinement of this approach, referred to as hierarchical docking, searches for co-bound configurations of inhibitor and substrate by first determining the unique sets of clustered configurations of one of the two ligands, using an RMS criteria of 0.5 Å, followed by docking the 2nd ligand to a representative structure for clusters of the 1st docked ligand. This was performed for ascending mean energies of the docking clusters, hence the hierarchical nature of the process. The hierarchical docking procedure was accomplished by first docking 2EN followed by one of the substrates and vice versa. The conclusions as to whether 2EN and the substrates could co-bind to a common cavity were not dependent on order of docking of 2EN and the substrate. The results are relevant to our prior finding that the reversible inhibition by 2EN of substrate metabolism was highly correlated with substrate size and architecture (14).
Autodock3 Protocol
In the present study the Lamarkian algorithm in Autodock3 (37) was used employing two different grid spacings. In the first a 120×120 grid with 0.375 Å was employed to identify plausible binding sites throughout the protein. Refined sampling proximate to the heme binding site was then performed employing a 120×120 grid with 0.175 Å spacing. In general, similar docked configurations were obtained in the binding site regions with both grids, however, the less dense grid enabled exploration of binding configurations remote from the binding site. In both instances the grid was centered on the compound I oxyferryl oxygen, the form of the heme employed in these docking studies, however, trial runs with ferric heme representation revealed only very minor changes in the docking profiles. Lamarkian and local search parameters were as previously described (38). A 0.5 Å RMS criterion was employed to identify unique clusters of docked configurations.
The low energy docked configurations were analyzed to examine the propensity of substrate and one or more 2EN inhibitor molecules to bind to CYP2B4 from consideration of the number of co-bound configurations.
Properties of low energy substrate conformations
Geometric properties of low energy docked configurations of each of the ligands docked in the binding site were evaluated from output of MOPAC7 calculations with use of the program GRAPHA. The correlation of the percentage of reversible inhibition with Sterimol parameters (39), molecular volumes, and the molecular weight were examined via linear least squares.
Binding Site Cavity Volumes
Potential binding site cavity volumes were obtained employing Internal Coordinate Mechanics (ICM: Molsoft, Co, San Diego, CA) employing the Gauss theorem. While this approach is most accurate for enclosed spaces, the spaces partially enclosed near the surface of CYP2B4 identified with this algorithm also correlated with low-energy interaction sites for docked ligands.
Ligand and Protein Charge Parameterization for Docking Studies
Potential derived charges for 2EN, PNA, 7EC, 7EFC, 7BR, TS, BZP and 7PR were obtained from Restrained Electrostatic Potential (RESP)(40) fits to 6-31G** DFT optimized geometries of the ligands computed using Jaguar 5.5 (Schrodinger, Inc., Portand, OR). Atomic charges for protein atoms employed in docking studies were taken from the AMBER 4.1 (41;42) polar-H representation of amino acids. The latter charges are appropriate for use with the radially screened continuum dielectric treatment in Autodock3 (37). Heme charges were derived from a single point calculation at the DFT optimized geometry obtained using a LACVP**(Fe)/6-31G**(C,N,O,S,H) basis set description and a B3LYP hybrid functional, which incorporates exchange interactions from both computed Hartree-Fock exchange integrals, the Becke exchange functional, and electron correlation from the (local) Vosko, Wilk, and Nusair (VWN) and (non-local)Lee-Yang-Parr (LYP) functionals. The heme charge parameterization has been shown to be adequate for docking and molecular dynamics predictions of binding modes of drugs consistent with their principal metabolites (38) and for prediction of binding free energies of substrates and inhibitors via free-energy simulations (43). Recent rederivation of the potential charges in a QMMM (quantum mechanics-molecular mechanics) calculation of the heme of CYP2B4 in the compound I form with proximal Cys,(436), Leu(437), Ile(435), Gly(438), the protoporphyrin IX and methoxyflurane were found to be consistent with the present charge set.
RESULTS AND DISCUSSION
Examination of the reversible inhibition of CYP2B4-dependent activities by 2EN
As mentioned in our previous report (14), 2EN can serve as both a reversible and an irreversible inhibitor of CYP2B4-mediated metabolism. Although the irreversible inactivation is similar to that expected for typical mechanism-based inhibitors, the reversible component was unusual. The ability of 1 μM 2EN to reversibly inhibit substrate metabolism could be divided into three groups (14): (a) little to no inhibition (PNA), (b) a moderate degree of inhibition (13%-30% for 7EC, 7EFC and BZP), and (c) significant reversible inhibition (50%-71% for 7BR, 7PR, and TS). The ability of 2EN to inhibit metabolism of these substrates was not related to the affinity of the substrates for the active site, but did appear to be related to the molecular size of the substrate, particularly the length of the molecule. The goal of the current study was to further examine the reversible component of this inhibitory response, and to provide a structural explanation for the unusual kinetic behavior.
In an effort to more completely characterize the ability of 2EN to inhibit each of the CYP2B4-dependent activities, metabolism of each of the substrates was examined by varying the substrate concentration both in the absence and presence of different concentrations of the inhibitor 2EN. Because of the focus on the reversible component, the reconstituted systems were not preincubated with 2EN – both inhibitor and substrate were added simultaneously. This experimental design allows for the determination of the type of inhibition, the Km for each substrate, and the for 2EN with each substrate.
The results showing the determination of the reversible inhibition by 2EN are shown in Figure 1 and Table 1. The data were plotted using the Lineweaver-Burk analysis, which was used to illustrate the type of inhibition. The results demonstrate that the apparent mechanism of inhibition by 2EN was dependent on the substrate employed. Metabolism of the small substrate PNA was not inhibited by 2EN at inhibitor concentrations below 1 μM (vide infra). As the size of the substrate molecule was increased (7-EC and 7-EFC), inhibition by 2EN was observed (Fig. 1A). The type of inhibition for these substrates appeared to be competitive in nature with values of 0.55 μM and 0.25 μM for 7-EC and 7-EFC, respectively. Interestingly, the larger substrates (7-PR, 7-BR and TS) exhibited a different mechanism of inhibition, appearing to be largely noncompetitive (Figure 1C and 1D). The values for inhibition of these substrates were 0.2, 0.5, 0.4 μM for 7-PR, 7-BR and TS, respectively (Table 1). The more flexible BZP molecule exhibited a mixed type of inhibitory response (Fig. 1B), with a value of 0.75 μM.
Figure 1. Determination of the type of reversible inhibition of CYP2B4-dependent 7-EC (A), BZP (B), 7-PR (C), and 7-BR (D) metabolism by 2EN.




The data points represent, no inhibitor (■), 0.25 μM (△), 0.5 μM (▼), 0.75 μM (◊), 1.0 μM 2EN (●); as for BZP, the legend is: no inhibitor (■), 1 μM (△), 2 μM (▼), 3 μM (◊), 4 μM (●). The y-axes are expressed as the reciprocal of the rates (pmol product/min/nmol P450) except for bzp which is expressed as nmol product/min/nmol P450. The assays were performed as described in Experimental Procedures.
Table 1.
Summary of the type and kinetic constants for reversible inhibition of CYP2B4 by 2EN
| SUBSTRATE | Km (μM) | (μM) | TYPE OF REVERSIBLE
INHIBITION |
|---|---|---|---|
| PNA | 15 | ------ | No significant inhibition |
| 7-EC | 115 | 0.55 | Competitive |
| 7-EFC | 5.7 | 0.25 | Competitive |
| BZP | 13 | 0.75 | Mixed |
| 7-PR | 0.64 | 0.20 | Noncompetitive |
| TS | 100 | 0.40 | Noncompetitive |
| 7-BR | 0.27 | 0.50 | Noncompetitive |
The inhibitory responses described for 2EN with the different substrates were described at 2EN concentrations below 1 μM. However, at higher 2EN concentrations there was a dramatic increase in the inhibitory potential as well as the pattern of inhibition for some of the substrates. This effect was even observed with the substrate PNA, which was not inhibited below 1 μM. At these higher inhibitor concentrations, PNA demethylation was shown to be significantly inhibited (Figure 2). 7-EFC, which was competitively inhibited at low 2EN concentrations, also exhibited a more complex inhibition pattern as the 2EN concentration exceeded 1 μM (Figure 2). A similar response was observed with the substrate TS (not shown). This alteration in the inhibitory pattern as the 2EN concentration was increased could be explained by the potential for 2EN molecules to bind to a site(s) apart from the active site where 2EN binding could modulate the conformation in the substrate binding site. This could lead to a modification of substrate binding affinity in a non-competitive/allosteric manner. Additionally, a component of this inhibitory process, particularly the response observed at 2EN concentrations above 1 μM, could be due to the stacking of multiple 2EN molecules within the substrate binding site. Such conditions would be expected to alter the manner by which each of the substrates interacts in a productive manner in the active site altering the Km and kcat (both binding free energy and metabolic step activation energy modulation).
Figure 2. Reversible inhibition of CYP2B4-dependent (A) PNA and (B) 7-EFC metabolism by 2EN at higher concentrations.


The data points represent, no inhibitor (■), 1 μM (△), 2 μM (▼), 3 μM (◊), 4 μM 2EN (●) for PNA; no inhibitor (■), 0.5 μM (△), 1 μM (▼), 1.5 μM (◊), 2 μM 2EN (●) for 7-EFC. The inset of B shows competitive inhibition of CYP2B4-dependent 7-EFC deethylation by 2EN at or below 1 μM – no inhibitor (■), 0.5 μM (△), 1 μM (▼). The y-axis for PNA is expressed as the reciprocal of the rates (pmol product/min/nmol P450); 7EFC is expressed as nmol product/min/nmol P450. The data points at the highest 2EN concentrations (at the lowest substrate concentrations) were missing due to not being detectable.
Binding Site Volumes and Substrate/Inhibitor Size
Internal Coordinate Mechanics (ICM: Molsoft Inc, San Diego, CA) binding site locator was used to examine binding site and closed cavity volumes near the substrate/inhibitor binding site of CYP2B4. Figure 3 shows volumes above or near the distal side of the heme identified using this approach. The white enclosure in this figure corresponds to the region above the heme encompassing approximately 594 Å3. We will refer to this region as the primary binding site throughout the manuscript in that it is the largest site and the one where substrate metabolism occurs. The next two largest regions that could plausibly be binding sites are more distant from the heme and are shown in grey (330 Å3) and gold (275 Å3). Additionally, there is a site on the surface of CYP2B4 that is capable of binding 2EN which will be discussed subsequently. The computed volumes of all of the ligands (Table 2) are less than one-half the estimated CYP2B4 primary binding site volume. Indeed the sums of the volume of 2EN plus that of each of the substrate volumes are smaller than the cavity volume (594 Å3). The volumes of 2EN summed with each of the substrates are: PNA (267 Å3), 7EC (298 Å3), 7EFC (327 Å3), 7PR (395 Å3), 7BR (387 Å3), BP (417 Å3), and 2EN (293 Å3). Were binding site volume of each of the ligands in this study the only determinant of the ability to bind to these binding sites, each of them could co-bind in the CYP2B4 primary binding site with 2EN. Clearly the length and width parameters and contour of the molecule play a vital role in determining whether substrate and inhibitor may both fit within the contour of the buried substrate binding site on the distal side of the heme.
Figure 3. Potential substrate binding sites identified from the crystal structure of CYP2B4.
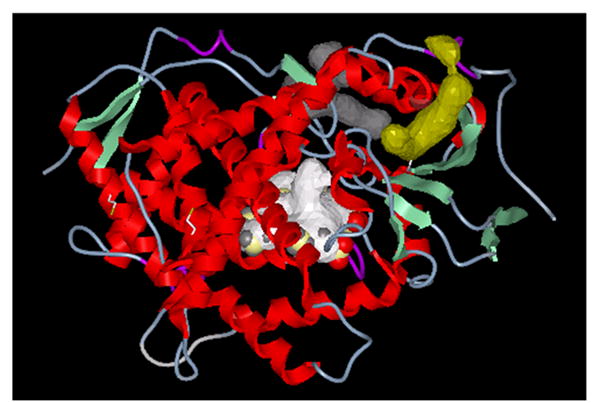
Three potential substrate binding sites were derived from the CYP2B4 crystal structure (34). The white enclosure represents the distal binding site above the heme (Volume=594 Å3). A second potential binding site with a volume of 330 Å3 is separated from the primary binding site and is shown in grey. A site on the surface of the CYP2B4 molecule, yet still in the vicinity of the heme binding region is shown in gold and occupies a volume of about 275 Å3. Each of these enclosures is of sufficient size to bind the inhibitor 2EN.
Table 2.
Correlation of the percentage reversible inhibition with simple properties related to molecular size, % of docked configurations present in the heme binding site both with and without a low energy docked 2EN configuration
| LIGAND | MW | VOLUME a | L b | B4 c | NUMBER OF BOUND CONFIGURATIONS OF SUBSTRATE IN THE PRIMARY BINDING SITE | NUMBER OF CONFIGURATIONS OF SUBSTRATE CO-BOUND WITH 2EN IN THE BINDING SITE | TYPE OF REVERSIBLE INHIBITION |
|---|---|---|---|---|---|---|---|
| PNA | 153.1 | 121.1 | 10.8 | 3.8 | 41 | 15 | No significant inhibition |
| 7EC | 190.2 | 151.2 | 12.2 | 4.2 | 43 | 1 | Competitive |
| 7EFC | 258.2 | 180.6 | 12.3 | 4.3 | 32 | 1 | Competitive |
| BZP | 239.4 | 271.1 | 14.5 | 5.8 | 9 | 0 | Mixed |
| 7PR | 383.3 | 248.4 | 19.1 | 4.3 | 7 | 0 | Noncompetitive |
| TS | 288.4 | 297.6 | 13.7 | 4.7 | 6 | 0 | Noncompetitive |
| 7BR | 303.3 | 240.5 | 17.9 | 4.8 | 2 | 0 | Noncompetitive |
| 2EN | 152.2 | 146.8 | 12.1 | 4.1 | 60 | 10 | ----- |
All length parameters are in units of Angstroms and volume in Å3
The longest dimension of the substrate/inhibitor mapped from points on the Connolly surface
The longest transverse distance perpendicular to the L-axis to a point on the Connolly surface
Figure 4A shows a low energy docked configuration of BZP with exposure of the metabolized N-CH3 group proximate to the oxyferryl heme. One may see that when docking ligands of increasing size in this study, beginning with BZP one begins to fill most of the accessible space in the binding site cavity with even a single ligand present. In contrast, Figure 4B shows that two 2EN inhibitors readily fit as does PNA and 1-2EN (vide infra).
Figure 4. Docking of benzphetamine and 2EN molecules in the primary common cavity site of CYP2B4.
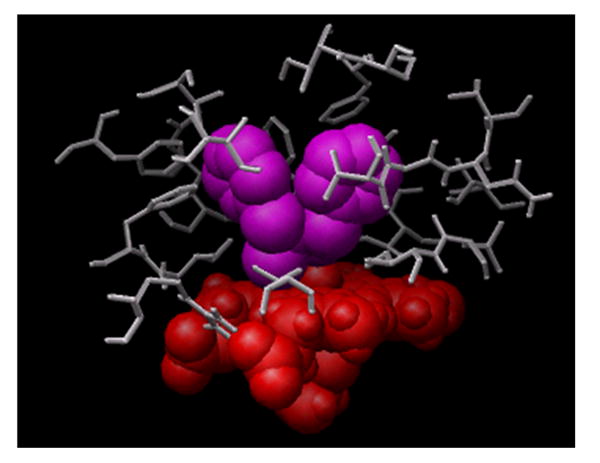
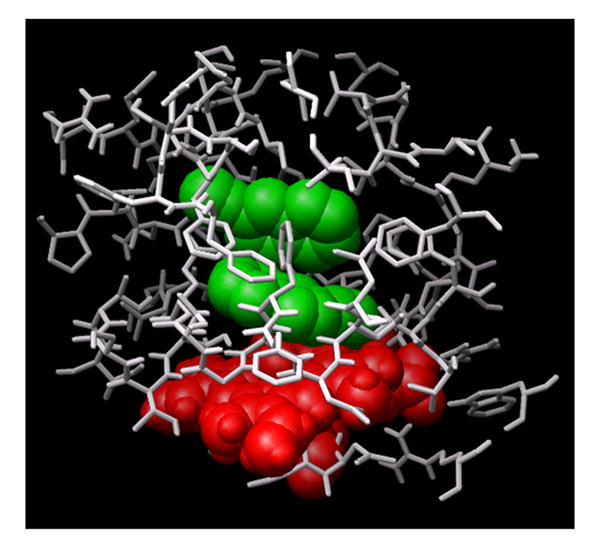
(A) The docked configuration of a single BZP (shown in magenta) nearly fills the primary binding site. (B) In contrast, the smaller size and architecture of 2EN (shown in green) readily accommodates low-energy configurations of two-2EN ligands above the heme.
Hierarchical Docking Probes of Modes of Substrate/Inhibitor Binding
Each of the substrates was docked into CYP2B4 employing a pre-evaluated interaction grid encompassing the entire CYP2B4 enzyme with the grid center at the heme site. Low energy docked configurations are, in principle, possible in both buried and surface sites. Figure 5A shows the number of low-energy docked configurations that were found in the buried binding site above the heme, as opposed to remote separated or surface binding sites. The smaller substrates, such as PNA and 7EC, have numerous low energy docked configurations, with the most configurational diversity. Larger substrates BZP, 7PR, 7BR, and TS fit more tightly into the active site of CYP2B4 and the number of the low-energy configurations found in the distal heme site is fewer for these ligands than for the small PNA, 7EC, and 2EN ligands (Table 2).
Figure 5. Estimates of the relative number of binding configurations for substrates and inhibitors within the primary binding site.


(A) The percentage of substrate bound configurations in the binding site above the heme as opposed to other cavities correlated with substrate volume. (B) Percentage of co-docked low-energy configurations of two ligands in the primary binding site on the distal site of the heme. Above a certain substrate volume, co-occupation of 2EN and substrate in the heme binding site is no longer possible---indicating a size threshold for the transition to lack of inhibition to a competitive interaction.
Figure 5A shows that the number of the docked ligand configurations obtained in the primary binding site, as opposed to more remote sites, was inversely proportional to the size of the substrate (r2 = 0.80). This illustrates that while numerous low energy configurations exist for small substrates in the primary binding site, as size and branching increases, substrate access to the site becomes more restricted. This is a reflection of the ease of fit as a function of substrate size, but does not preclude the primary binding site above the heme being the lowest free energy binding site for the larger substrates.
Hierarchical docking was next used to probe the possible configurations for multiple-ligand binding, including both multiple substrate and substrate/inhibitor binding. In this approach the inhibitor was docked into CYP2B4, and the docked configurations were energy ordered and compared via root mean square (RMS) fits to determine unique energy ordered clusters of docked conformations similar to one another in energy and disposition in the CYP2B4. Next, representative configurations from each of the docked inhibitor clusters in CYP2B4 were used for docking of substrates. This is done by computing an interaction grid including CYP2B4 and the docked configuration of 2EN. This build-up/hierarchy of co-bound configurations served as an efficient method of quickly scoring co-bound configuration sampling prior to additional characterization.
Figure 5B shows that the size of the substrate and inhibitor had a profound effect on the number of primary binding site configurations involving multiple substrate or substrate and inhibitor bound in a common cavity. In this figure, for the purposes of comparison, we have normalized the number of co-bound configurations to that found for 2 co-bound PNA molecules. Figure 5B shows a sharp decline in the number of low energy unique co-bound configurations with increasing substrate/inhibitor size. There were numerous co-bound configurations of 2-PNAs in the heme binding site, fewer for 2EN+PNA, and fewer still for two 2ENs. One notes that with the 2EN inhibitor present in the common cavity, a PNA (or a second 2EN) readily “fits” into the heme binding site. Once the substrate size is as large as 7EC or 7EFC, the volume of 2EN virtually precludes its co-binding with substrate. This interaction pattern would be expected to produce “competitive” inhibition, that is, mutually exclusive binding of 2EN and substrate in the CYP2B4 common cavity. In marked contrast, a large percentage of the co-docked PNA and 2EN ligands revealed occupancy in the primary binding site. These results are consistent with an inhibitory pattern with 2EN that is dependent on the size of the substrate molecule. When the substrate molecule is small (e.g. PNA), both 2EN and PNA can be co-bound in the common cavity of the active site in many different configurations, leading to little or no inhibition (at 2EN concentrations below 1 μM). However, as the size of the substrate molecule is increased, co-binding in the common cavity is no longer possible. Mutually exclusive binding in the common cavity would be expected to produce a simple competitive response.
Docking studies using the X-ray structure of CYP2B4 show that there are three likely sites for 2EN binding (Figure 6). Within the vicinity of the heme, 2EN can be oriented in two configurations, the common cavity configuration (primary binding site) which has been discussed above, and a separated cavity (Fig. 6). Additional sites exist more distal to the heme, along the surface of the CYP2B4 molecule and preliminary molecular dynamics investigations indicate the surface sites of mean residence times of several hundred picoseconds (not shown). Each of these binding sites may participate in the reversible inhibition of CYP2B4 by 2EN, with the contribution from each site being dependent on the substrate present.
Figure 6. A hierarchical docking result showing a low energy configuration of PNA (magenta) and 2EN (green).
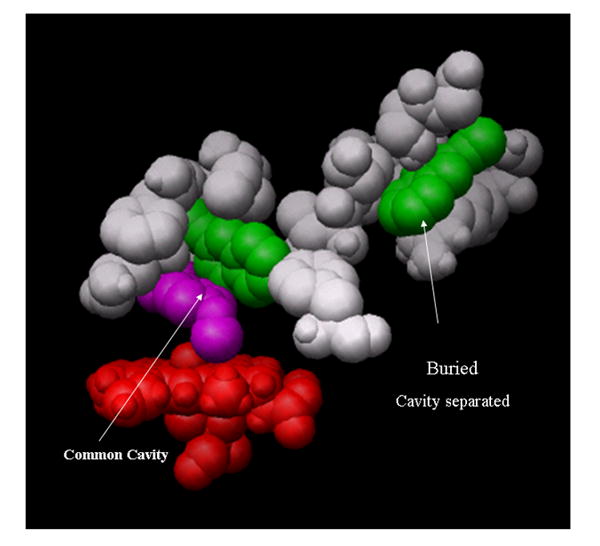
A single 2EN and the PNA are in a “common-cavity” arrangement above the heme (red) shown in the common cavity. A second 2EN is shown in a “separate cavity” adjoining the heme binding site. The residues within 2.8 Å of 2EN and PNA in the closed cavity arrangement are PNA [E301,V367,I363,V477,G478] and 2EN[I101,V104, I114,I209,F297,E301,V367,V477]. The residues within 2.8 Å of 2EN and PNA in the separated cavity configuration are: PNA[V104,F115,F297,E301,I363,V367] and 2EN[H172,F203,F206,S207,S210, T306,L362,G478,N479,V480,I486]. Residues have been removed for clear visualization of the occupancy; both of these sites are buried.
Based on these studies, two types of 2EN-substrate co-bound configurations were observed. In the ‘common cavity’ configuration, both 2EN and substrate simultaneously occupy the common cavity above the heme as shown by the white enclosure in Figure 3. In the ‘cavity separated’ co-bound configurations, the inhibitor is bound in the gold region (Fig. 3) adjoining the binding pocket. The substrate and inhibitor could ‘communicate’ via intervening residues. Figure 6 illustrates PNA co-binding with two-2EN molecules where 2EN is bound in both configurations. The 1st 2EN (green) is present with the PNA (magenta) above the oxyferryl heme with the 2nd 2EN in an adjacent binding pocket. Residues have been removed about the 2nd-cavity (separated) and it is in fact a partially buried site, in contrast to the “surface” 2EN interaction site that were also found as a surface sites.
CYP2B4 residues identified as being in close contact (within 3 Å) to the bound 2EN and PNA are indicated in the legend to Figure 6. Strobel and coworkers (44) have performed a careful assessment of the effect of specific active site residues on the mechanism based inactivation of CYP2B4 and CYP2B5 by 2EN by site-directed mutagenesis. Consistent with our findings (Figure 6), mutations at sequence positions 363 and 367 in CYP2B4 and CYP2B5 significantly modulated the susceptibility to mechanism based inhibition and indicated contact of these residues with 2EN (44), Clearly mutation of these residues alter the dynamics of 2EN in the binding pocket which influences its reversible and mechanism based inhibition.
While there were multiple configurations allowing co-binding of PNA and 2EN in the primary site, there were much fewer with 7EC and 2EN, and none for the larger substrates. Consequently, each of the larger substrates would be expected to exhibit competitive inhibition, rather than the mixed and noncompetitive inhibition patterns actually observed with the larger substrates. It is plausible that the separated cavity, and surface-interaction 2EN inhibitor binding modes that are responsible for the non-competitive and mixed inhibition found with the larger substrates. Interaction with the separated or surface sites could lead to conformational changes in the common cavity that could modulate substrate binding free energies. Joint computational/experimental studies are in progress to address this point.
As previously reported (14), the reversible component of the 2EN inhibition correlated well with the size of the substrate molecules; the bulk of this correlation was due to the effective length of the ligand as captured by the Sterimol L parameter with little correlation with molecular width (Sterimol B4). The numerical results shown in figure 5B and the example in figure 6A illustrate that a small substrate and 2EN may co-bind such that there is little detectable inhibition. Ongoing studies examining the binding free energies of substrate and inhibitor support this concept. With moderately sized substrates, however, one cannot simultaneously bind 2EN and a substrate in the binding site above the heme. At low inhibitor concentrations this results in competitive inhibition. Additional sites in the ‘separated’ and ‘surface’ cavities also may be occupied by 2EN. Binding to these sites may result in conformational compaction of the substrate binding site (14).
In conclusion, both reversible inhibition and mechanism-based inactivation components of CYP2B4 with various substrates by 2EN were observed. The mechanism-based inactivation of CYP2B4 by 2EN was substrate-independent (14). In contrast, the reversible inhibition of CYP2B4 by 2EN was substrate dependent and the degree of reversible inhibition was related to the length or molecular weight of substrate, but not related to the affinity of each substrate with CYP2B4 (14). Examination of the reversible inhibition of CYP2B4-dependent metabolism for seven different substrates by 2EN showed not only that the Ki but also the type of inhibition was dependent on the substrate. Inhibition ranged from no inhibition to competitive, mixed and finally non-competitive as the size of the substrate molecule was increased. Finally, hierarchical docking studies demonstrated that the CYP2B4 active site is sufficiently large to accommodate the simultaneous binding of 2EN inhibitor and small substrates. With larger substrates there may be components of both competitive inhibition due to competing for the distal-heme binding site and non-competitive inhibition when 2EN binds to adjacent binding sites close to the substrate binding site above the heme. These models provide an explanation that is consistent with the experimental data showing multiple mechanisms of reversible inhibition of CYP2B4 by the small 2EN molecule that are dependent on the physical characteristics of the substrate used.
Acknowledgments
This work was supported by US Public Health Service Research Grants from the National Institute of Environmental Health Sciences (R01 ES004344 – WLB), and the National Institutes of Health (R43-DC-6925 – DH), and support from the Stanley S. Scott Cancer Center (JRR). We would like to thank Dr. Maryam Foroozesh (Xavier University of Louisiana, New Orleans) for supplying us with 2-ethynylnaphthalene. We would also like to thank Mr. George Cawley for his assistance in purifying CYP2B4 and NADPH-cytochrome P450 reductase.
The abbreviations used are
- 2EN
2-ethynylnaphthalene
- PNA
p-nitroanisole
- 7-EC
7-ethoxycoumarin
- 7-EFC
7-ethoxy-4-trifluoromethylcoumarin
- BZP
benzphetamine
- 7-PR
7-pentoxyresorufin
- 7-BR
7-benzyloxyresorufin
- TS
testosterone
- DLPC
dilauroylphosphatidylcholine
- NADPH
nicotinamide adenine dinucleotide phosphate reduced tetrasodium salt
- DMSO
dimethylsulfoxide
- 7-HFC
7-hydroxy-4-trifluoromethylcoumarin
- 7-HC
7-hydroxycoumarin
- CYP2B4
cytochrome P450 2B4
the dissociation constant for reversible inhibition by 2EN
- DFT
density functional theory
- RMS
root mean squared
- RESP
restrained electrostatic potential
- B3LYP
Becke-3-parameter exchange-correlation hybrid functional with Lee-Yang-Parr Correlation
- MOPAC
Molecular Orbital Package
- QMMM
quantum mechanics-molecular mechanism
- LACVP
Los Alamos effective core potential
Reference List
- 1.White RE, Coon MJ. Annu Rev Biochem. 1980;49:315–356. doi: 10.1146/annurev.bi.49.070180.001531. [DOI] [PubMed] [Google Scholar]
- 2.Korzekwa KR, Krishnamachary N, Shou M, Ogai A, Parise RA, Rettie AE, Gonzalez FJ, Tracy TS. Biochem. 1998;37:4137–4147. doi: 10.1021/bi9715627. [DOI] [PubMed] [Google Scholar]
- 3.Shou M, Dai R, Cui D, Korzekwa KR, Baillie TA, Rushmore TH. J Biol Chem. 2001;276:2256–2262. doi: 10.1074/jbc.M008799200. [DOI] [PubMed] [Google Scholar]
- 4.Hutzler JM, Wienkers LC, Wahlstrom JL, Carlson TJ, Tracy TS. Arch Biochem Biophys. 2003;410:16–24. doi: 10.1016/s0003-9861(02)00665-3. [DOI] [PubMed] [Google Scholar]
- 5.Ueng YF, Kuwabara T, Chun YJ, Guengerich FP. Biochem. 1997;36:370–381. doi: 10.1021/bi962359z. [DOI] [PubMed] [Google Scholar]
- 6.Wang RW, Newton DJ, Scheri TD, Lu AY. Drug Metab Dispos. 1997;25:502–507. [PubMed] [Google Scholar]
- 7.Yoon MY, Campbell AP, Atkins WM. Drug Metab Rev. 2004;36:219–230. doi: 10.1081/dmr-120033998. [DOI] [PubMed] [Google Scholar]
- 8.Xiang H, Tschirret-Guth RA, Ortiz de Montellano PR. J Biol Chem. 2000;275:35999–36006. doi: 10.1074/jbc.M005811200. [DOI] [PubMed] [Google Scholar]
- 9.Banci L, Bertini I, Marconi S, Pierattelli R, Sligar SG. J Am Chem Soc. 1994;116:4866. [Google Scholar]
- 10.Roberts ES, Hopkins NE, Zaluzec EJ, Gage DA, Alworth WL, Hollenberg PF. Biochem. 1994;33:3766–3771. doi: 10.1021/bi00178a037. [DOI] [PubMed] [Google Scholar]
- 11.Roberts ES, Hopkins NE, Foroozesh M, Alworth WL, Halpert JR, Hollenberg PF. Drug Metab Dispos. 1997;25:1242–1248. [PubMed] [Google Scholar]
- 12.Foroozesh M, Primrose G, Guo Z, Bell LC, Alworth WL, Guengerich FP. Chem Res Toxicol. 1997;10:91–102. doi: 10.1021/tx960064g. [DOI] [PubMed] [Google Scholar]
- 13.Hammons GJ, Alworth WL, Hopkins NE, Guengerich FP, Kadlubar FF. Chem Res Toxicol. 1989;2:367–374. doi: 10.1021/tx00012a003. [DOI] [PubMed] [Google Scholar]
- 14.Cheng D, Reed JR, Harris D, Backes WL. Arch Biochem Biophys. 2007;462:28–37. doi: 10.1016/j.abb.2007.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yun CH, Hammons GJ, Jones G, Martin MV, Hopkins NE, Alworth WL, Guengerich FP. Biochem. 1992;31:10556–10563. doi: 10.1021/bi00158a019. [DOI] [PubMed] [Google Scholar]
- 16.Saribas AS, Gruenke L, Waskell L. Protein Expression and Purification. 2001;21:303–309. doi: 10.1006/prep.2000.1377. [DOI] [PubMed] [Google Scholar]
- 17.Backes WL, Reker-Backes CE. J Biol Chem. 1988;263:247–253. [PubMed] [Google Scholar]
- 18.Yasukochi Y, Masters BS. J Biol Chem. 1976;251:5337–5344. [PubMed] [Google Scholar]
- 19.Shen AL, Porter TD, Wilson TE, Kasper CB. J Biol Chem. 1989;264:7584–7589. [PubMed] [Google Scholar]
- 20.Kelley RW, Reed JR, Backes WL. Biochem. 2005;44:2632–2641. doi: 10.1021/bi0477900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Causey KM, Eyer CS, Backes WL. Mol Pharmacol. 1990;38:134–142. [PubMed] [Google Scholar]
- 22.Roberts ES, Ballou DP, Hopkins NE, Alworth WL, Hollenberg PF. Arch Biochem Biophys. 1995;323:303–312. doi: 10.1006/abbi.1995.9960. [DOI] [PubMed] [Google Scholar]
- 23.Hayashi S, Omata Y, Sakamoto H, Hara T, Noguchi M. Protein Expr Purif. 2003;29:1–7. doi: 10.1016/s1046-5928(03)00023-8. [DOI] [PubMed] [Google Scholar]
- 24.de Andrade JB, Bispo MS, Reboucas MV, Carvall MLSM, Pinheire HLC. Am Lab. 1996;28:56–58. [Google Scholar]
- 25.Hanna IH, Reed JR, Guengerich FP, Hollenberg PF. Arch Biochem Biophys. 2000;376:206–216. doi: 10.1006/abbi.2000.1708. [DOI] [PubMed] [Google Scholar]
- 26.Beebe LE, Roberts ES, Fornwald LW, Hollenberg PF, Alworth WL. Biochem Pharmacol. 1996;52:1507–1513. doi: 10.1016/s0006-2952(96)00525-4. [DOI] [PubMed] [Google Scholar]
- 27.Burke MD, Thompson S, Elcombe JR, Halpert J, Haaparanta T, Mayer RT. Biochem Pharmacol. 1985;34:3337–3345. doi: 10.1016/0006-2952(85)90355-7. [DOI] [PubMed] [Google Scholar]
- 28.Netter KJ, Seidel G. J Pharmacol Exp Ther. 1964;146:61–65. [PubMed] [Google Scholar]
- 29.Wood AW, Ryan DE, Thomas PE, Levin W. J Biol Chem. 1983;258:8839–8847. [PubMed] [Google Scholar]
- 30.Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS. J Med Chem. 2006;49:5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- 31.Ferrara P, Gohlke H, Price DJ, Klebe G, Brooks CL., III J Med Chem. 2004;47:3032–3047. doi: 10.1021/jm030489h. [DOI] [PubMed] [Google Scholar]
- 32.Krovat EM, Steindl T, Langer T. Current Computer-Aided Design. 2005;1:93–102. [Google Scholar]
- 33.Goodsell DS, Morris GM, Olson AJ. Journal of Molecular Recognition. 1996;9:1–5. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 34.Scott EE, White MA, He YA, Johnson EF, Stout CD, Halpert JR. J Biol Chem. 2004;279:27294–27301. doi: 10.1074/jbc.M403349200. [DOI] [PubMed] [Google Scholar]
- 35.Muralidhara BK, Negi SS, Halpert JR. J Am Chem Soc. 2007;129:2015–2024. doi: 10.1021/ja066303w. [DOI] [PubMed] [Google Scholar]
- 36.Locuson CW, Gannett PM, Ayscue R, Tracy TS. J Med Chem. 2007;50:1158–1165. doi: 10.1021/jm060706p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 38.Park JY, Harris D. J Med Chem. 2003;46:1645–1660. doi: 10.1021/jm020538a. [DOI] [PubMed] [Google Scholar]
- 39.Verloop A. The STERIMOL Approach to Drug Design. Marcel Dekker; New York: 1987. [Google Scholar]
- 40.Bayly C, Cieplak P, Cornell W, Kollman P. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 41.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 42.Case DA, Pearlman DA, Caldwell JW, Cheatham TE, III, Ross WS, Simmerling CL, Darden TA, Merz KM, Stanton RV, Cheng AL, Vincent JJ, Crowley M, Tsui V, Radmer RJ, Duan Y, Pitera J, Massova I, Seibel GL, Singh UC, Weiner PK, Kollman PA. Univ California. San Francisco; San Francisco: 1997. [Google Scholar]
- 43.Harris DL, Park JY, Gruenke L, Waskell L. Proteins. 2004;57:643. doi: 10.1002/prot.20062. [DOI] [PubMed] [Google Scholar]
- 44.Strobel SM, Szklarz GD, He Y, Foroozesh M, Alworth WL, Roberts ES, Hollenberg PF, Halpert JR. J Pharmacol Exp Ther. 1999;290:445–451. [PubMed] [Google Scholar]