

Abstract
ATP-sensitive K+ (KATP) channels are present in the sarcolemma of cardiac myocytes where they link membrane excitability with the cellular bioenergetic state. These channels are in vivo composed of Kir6.2, a pore-forming subunit, SUR2A, a regulatory subunit, and at least four accessory proteins. In the present study, real-time RT-PCR has demonstrated that of all six sarcolemmal KATP channel-forming proteins, SUR2A was probably the least expressed protein. We have generated mice where the SUR2A was under the control of a cytomegalovirus promoter, a promoter that is more efficient than the native promoter. These mice had an increase in SUR2A mRNA/protein levels in the heart whereas levels of mRNAs of other channel-forming proteins were not affected at all. Imunoprecipitation/Western blot and patch clamp electrophysiology has shown an increase in KATP channel numbers in the sarcolemma of transgenic mice. Cardiomyocytes from transgenic mice responded to hypoxia with shortening of action membrane potential and were significantly more resistant to this insult than cardiomyocytes from the wild-type. The size of myocardial infarction in response to ischemia-reperfusion was much smaller in hearts from transgenic mice compared to those in wild-type. We conclude that overexpression of SUR2A generates cardiac phenotype resistant to hypoxia/ischemia/reperfusion injury due at least in part to increase in levels of sarcolemmal KATP channels.
Keywords: SUR2A, KATP channel, cardioprotection, hypoxia, heart
Throughout a lifetime, the heart is subjected to a variety of metabolic stresses, including ischemia/hypoxia. Work carried out over the last 15 years has demonstrated that powerful cardioprotective signaling pathways exist in cardiomyocytes that, when activated, enable cardiac cells to function under adverse conditions (1, 2).
Sarcolemmal ATP-sensitive K+ (KATP) channels, ion channels that couple metabolic status of the cell with membrane excitability (3), have been implicated in cardioprotective signaling (4, 5). These channels are selectively permeable to K+ ions and are closed by high intracellular ATP levels (3). It has been suggested that the opening of KATP channels protects against myocardial infarction, mediates ischemic preconditioning (a phenomenon when brief periods of ischemia/reperfusion protect the heart against sustained ischemia, 2), and promotes survival of cardiomyocytes exposed to different kinds of metabolic stresses (4, 5). Conditions associated with an increase in expression of sarcolemmal KATP channels, such as estrogen treatment and chronic hypoxia, are associated with increased cardiac resistance to stress (6-8). Also, ischemic preconditioning seems to be mediated by recruitment and an increase in number of these channels (9). However, preconditioning, estrogens, and hypoxia activate other signaling pathways as well (1, 10, 11), making it impossible to draw definite conclusions about the contribution of each of these pathways to the cardioprotective outcome.
Nevertheless, up-regulation of sarcolemmal KATP channels would seem a therapeutic strategy against ischemic heart disease that deserves to be tested. This strategy would be particularly attractive considering that KATP channels are silent under physiological conditions and the appearance of adverse effects would be unlikely (3). However, in vivo, cardiac sarcolemmal KATP channels are multiprotein complexes composed of Kir6.2, a pore-forming subunit, SUR2A, a regulatory subunit, and at least four accessory proteins, which include adenylate kinase type 1 (AK), creatine kinase type 1 (CK), the muscle form of lactate dehydrogenase (m-LDH), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (12-16). Four molecules of Kir6.2 and four molecules of SUR2A are required to form the functional channel (17) whereas the stoichiometry for accessory proteins is still unknown. Considering the complexity of the KATP channel protein complex and 1:1 Kir6.2:SUR2A stoichiometry, controlling the expression of these channels would seem to be a difficult task. In the present study, we aimed to find a way to manipulate the number of sarcolemmal KATP channels and to assess the consequence of overexpression of sarcolemmal KATP channels on cardiac resistance to metabolic stress.
MATERIALS AND METHODS
Molecular genetics
Full coding sequence was subcloned by PCR using primers for mouse SUR2A: sense, 5′-GCAGATCTGGC AGGCTGTTGGTAGCTCA-3′, and antisense, 5′-GCCTCGAGCTACTTGTTGGTCAT CACCA-3′. The template for the full SUR2A sequence was kindly provided by Y. Kurachi (Osaka University, Osaka, Japan). PCR was performed using the highest fidelity enzyme PfuUltra™ hotstart DNA polymerase (Stratagene, La Jolla, CA, USA) under the following conditions: hot start for 2 min at 95°C, followed by 30 cycles of 0.5 min at 95°C, 0.5 min at 58°C, and 5 min at 72°C; with a final extension 10 min at 72°C. The PCR product of the SUR2A gene was cloned into the BamH I/XhoI sites of pcDNA3.1 vector (Invitrogen, Paisley, UK), and placed downstream of the human cytomegalovirus immediate-early (CMV) promoter to provide high-concentration expression in mammalian cells (Fig. 1A). The final ≈6.5 kb CMV promoter-SUR2A construct was excised by using BglII and Nsi I (Promega, Southampton, UK). The linearized construct was injected into fertilized eggs from superovulated female derived from CBAxBalb/c crosses (male and female F1's). Founder mice were checked for successful integration of the transgene CMV-SUR2A by PCR using CMV-specific primers (Fig. 1B). Mouse genomic DNA as template was extracted from mouse ears using the Wizard® SV Genomic DNA Purification System (Promega). Primers for CMV promoter of the transgene construct was: Sense, 5′-CTGCTTCGCGATGTACGGGCC-3′; and antisense, 5′-TTCGATAAGCCAGTAAGCAGT-3′, the PCR product was 660 bp. The quality of genomic DNA as a template for the reaction was checked with primers for intron 1 of the endogenous mouse SUR2A gene: sense, 5′-GTAAGCTGTTGATTTTGATCTA-3′; and antisense, 5′-TAAAGAGCAAAGAGTAACAAAT-3′, this PCR product was 260 bp. PCR was performed as above in a thermal Cycler Mastercycler Gradient (Eppendorf, Dorset, UK) using TaqDNA polymerase (Sigma, Dorset, UK). Founder mice were bred between themselves and with C57BL/6J mice. Every mouse was genotyped using the CMV-specific primers (CMV-positive signal was indicative of the presence of CMV-SUR2A construct). It was not possible to determine whether founder mice were homozygous or heterozygous, but each breeding pair, including pairs of founders, produced litters that included both CMV-positive and CMV-negative mice. We specifically used lines producing wild-types and transgenes, which allowed us to do experiments on littermates. Thus, all wild-type mice used in the study as controls were either littermates (in most cases) or brothers/sisters of the transgenic mice. Experiments were designed in such a manner to allow use of the same tissue/cells for different experiments so that results obtained with different methodologies can be matched individually (as an example, real-time RT-PCR experiments were always done on the same tissue as Western blot, cells from same hearts were used for patch clamp and laser confocal experiments, etc.). No significant differences were observed between wild-type and transgenic mice with respect to appearance, body size (adult wt was 32.5±1.7 g and 29.0±1.3 g for wild-type and transgenes, respectively, n=15–16, P>0.05, Fig. 1C), heart morphology and size (heart wt was 233±30 mg and 210±9 mg for wild-type and transgenes, respectively, n=6, P=0.47, Fig. 1D), and health status (no signs of disease/conditions were observed in 390 CMV negative and 364 CMV-positive mice; only one unexpected death was registered in each of the groups).
Figure 1.
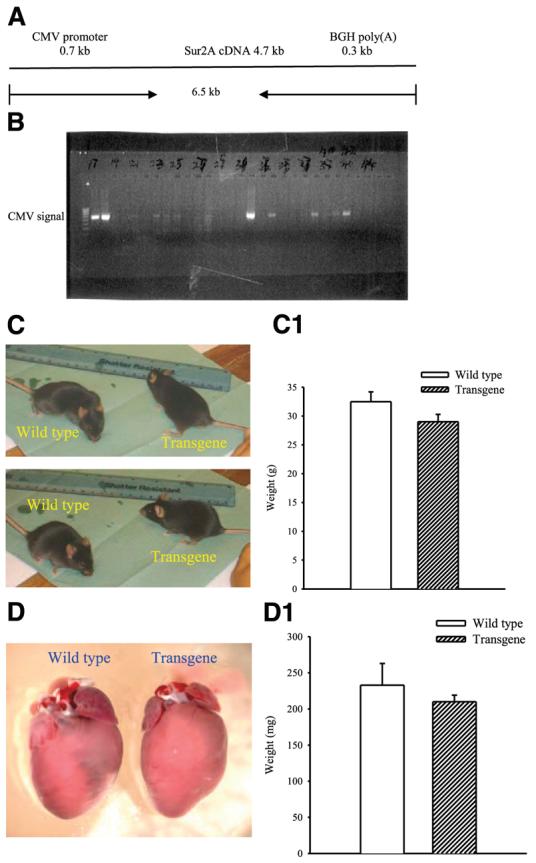
Generation of transgenic mice. A) Schematic representation of the CMV-SUR2A construct used to generate transgenic mice. B) Founder mice were checked for successful integration of the transgene CMV-SUR2A by PCR. PCR products obtained with CMV-specific primers using mouse genomic DNA as template. C, D) Photographs of wild-type and transgenic mice (C) and hearts (D) from these mice. C1 and D1 correspond to panels C and D, respectively. Each bar represents mean ± se (n=6–16).
Real-time RT-PCR
Total RNA was extracted from cardiac ventricular tissue of transgenic and wild-type mice using TRIZOL reagent (Invitrogen, Paisley, UK) according to the manufacturer's recommendations. Extracted RNA was further purified with RNeasy Mini Kit (Qiagen, Crawley, UK) according to the manufacturer's instruction. The specific primers for mouse SUR2A, SUR2A(B), SUR1, Kir6.2, Kir6.1, adenylate kinase type 1 (AK), creatine kinase type 1 (CK), muscle form of lactate dehydrogenase (m-LDH), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were designed as follows. For SUR2A: sense, ACTATGGAGTCCGAGAACTA, antisense, AGGTTTGGACCAGTATCACA; for Kir6.2: sense; ACATGCAGGTGGTGCGCAAG, antisense, AGGGCATCCAGCAGACTGCG; for AK: sense, 5′-ATGAATTCGTGGTGGGCGGACCTGGCTC-3′, antisense, 5′-GCCTCGAGTTAAATGCCACGCTTGTCATAGA-3′; for CK: sense, 5′-ATGAATTCGAGTACCCAGACCTCAGCAA-3′, antisense, 5′-GCCTCGAGCTACTTCTGCGCGGGGATCA-3′; for m-LDH: sense, 5′-GGCAGATCTATGCAGAACAAGATTACAGTTGT, antisense, 5′-GCCTCGAGTTAATTGATTCCATAGAGACCCT-3′; for GAPDH: sense, 5′-GAGAATTCATGGTGAAGGTCGGTGTGAA-3′ , antisense, 5′-GCCTCGAGTTACTCCTTGGAGGCCATGT-3′; for Kir6.1: sense, 5′-GTCACACGCTGGTCATCTTCAC-3, antisense, 5-GGCATTCCTCAGTCATCATTCTCC-3′; for SUR1: sense, 5′-GGAAGGACTCACCACCATC-3′, antisense, 5′-GAGACCATCAAGGCGTAGG-3′; for SUR2(A)B (designed to amplify SUR2B and natively present SUR2A): sense, 5′-GATGCCACTGTCACCGAAG-3′, antisense, 5-TCATCACAATAACCAGGTCTGC-3′. The reverse transcription (RT) reaction was carried out with ImProm-II Reverse Transcriptase (Promega, Southampton, UK). A final vol of 20 μl of RT reaction containing 4 μl of 5× buffer, 3 mM MgCl2, 20 U of RNasin® Ribonuclease inhibitor, 1 U of ImProm-II reverse transcriptase, 0.5 mM each of dATP, dCTP, dGTP, and dTTP, 0.5 μg of oligo(dT), and 1 μg of RNA was incubated at 42°C for 1 h, then inactivated at 70°C for 15 min. The resulting cDNA was used as a template for real-time PCR. A SYBR Green I system was used for the RT-PCR and the 25 μl reaction mixture contained: 12.5 μl of iQ™ SYBR® Green Supermix (2×), 7.5 nM each primers, 9 μl of ddH2O, and 2 μl of cDNA. In principle, the thermal cycling conditions were as follows: an initial denaturation at 95°C for 3 min, followed by 40 cycles of 10 s of denaturing at 95°C, 15 s of annealing at 56°C, and 30 s of extension at 72°C. This protocol was modified for the m-LDH-, GAPDH-, and CK-specific primers changing the extension temperature to 55°C. The real-time PCR was performed in the same wells of a 96-well plate in the iCycler iQ™ Multicolor Real-Time Detection System (Bio-Rad, Hercules, CA, USA). Data were collected after each cycle and displayed graphically (iCycler iQ™ Real-time Detection System Software, version 3.0A, Bio-Rad). Primers were tested for their ability to produce no signal in negative controls by dimer formation, then with regard to the efficiency of the PCR reaction. Efficiency is evaluated by the slope of the regression curve obtained with several dilutions of the cDNA template. Melting curve analysis tested the specificity of primers. Threshold cycle values, PCR efficiency (examined by serially diluting the template cDNA and performing PCR under these conditions), and PCR specificity (by constructing the melting curve) were determined by the same software. Each mouse cDNA sample was measured at three different quantities (and duplicated at each concentration, the corresponding no-RT mRNA sample was included as a negative control (blank).
Immunoprecipitation and Western blot analysis
Sheep anti-SUR2 and anti-Kir6.2 antibodies were used for immunoprecipitation and Western blot in this study (described in detail in refs 6, 12). To obtain the membrane cardiac fraction, mouse ventricular tissue was homogenized in buffer I (TRIS 10 mM, NaH2PO4 20 mM, EDTA 1 mM, PMSF 0.1 mM, pepstatin 10 μg/ml, leupeptin 10 μg/ml, at pH=7.8) and incubated for 20 min (at 4°C). The osmolarity was restored with KCl, NaCl, and sucrose and the mixture obtained was centrifuged at 500 g. The supernatant was diluted in buffer II (imidazole 30 mM, KCl 120 mM, NaCl 30 mM, NaH2PO4 20 mM, sucrose 250 mM, pepstatin 10 μg/ml, leupeptin 10 μg/ml, at pH=6.8) and centrifuged at 7000 g, the pellet removed, and supernatant centrifuged at 30,000 g. The obtained pellet contains the membrane fraction. Ventricular tissue was snap-frozen immediately upon extraction and ground to a powder under liquid nitrogen. The powder was resuspended in 10 ml of tissue buffer (20 mM HEPES, 150 mM NaCl, 1% Triton-X 100, pH 7.5) and homogenized. Protein concentration was determined using the Bradford method. Ten micrograms of the epitope-specific Kir6.2 antibody (Ab) or 40 μg of the epitope-specific SUR2A Ab were prebound to protein-G Sepharose beads and used to immunoprecipitate from 50 μg of membrane fraction protein extract. The pellets of this precipitation were run on SDS-polyacrylamide gels for Western analysis. In a separate series of experiments (to measure levels of total SUR2 protein), immunoprecipitation stage was omitted and total protein extract was run directly on SDS-polyacrylamide gels. Western blot probing was performed using 1/200 and 1/300 dilutions of anti-SUR2 and anti-Kir6.2 Ab, respectively, and detection was achieved using Protein-G HRP and ECL reagents. The band intensities were analyzed using the Quantiscan software (6-9).
Isolation of single cardiomyocytes
Ventricular cardiomyocytes were dissociated from the mouse using an established enzymatic procedure (6-9). In brief, hearts were retrogradely perfused (at 37°C) with medium 199, followed by Ca2+-EGTA-buffered low Ca2+ medium (pCa=7), and finally low Ca2+ medium containing Pronase E (8 mg/100 ml), proteinase K (1.7 mg/100 ml), bovine albumin (0.1 g/100 ml, fraction V), and 200 μM CaCl2. Ventricles were cut into fragments in the low Ca2+ medium enriched with 200 μM CaCl2. Cells were isolated by stirring the tissue (at 37°C) in a solution containing Pronase E and proteinase K supplemented with collagenase (5 mg/10 ml). The first aliquot was removed, filtered through a nylon sieve, centrifuged for 60 s (at 300–400 rpm), and washed. Remaining tissue fragments were re-exposed to collagenase and isolation continued for 2–3 such cycles.
Patch clamp electrophysiology
Measurement of whole cell K+ current has been done as described (6). In brief, cells were superfused with Tyrode solution (in mM: 136.5 NaCl; 5.4 KCl; 1.8 CaCl2; 0.53 MgCl2; 5.5 glucose (Glc); 5.5 HEPES-NaOH; pH 7.4). Fire-polished pipettes (resistance 3–5 MΩ), were filled with: 140 mM KCl, 1 mM MgCl2, 3 μM ATP, 5 mM HEPES-KOH (pH 7.3). Recordings were made at room temperature (22°C). During each experiment, the membrane potential was normally held at −40 mV and the currents evoked by a series of 400 ms depolarizing and hyperpolarizing current steps (−100 mV to +80 mV in 20 mV steps) recorded directly to hard disk using an Axopatch-200B amplifier, Digidata-1321 interface and pClamp8 software (Axon Instruments, Inc., Forster City, CA, USA). The capacitance compensation was adjusted to null the additional whole-cell capacitative current. The slow capacitance component measured by this procedure was used as an approximation of the cell surface area and allowed normalization of current amplitude (i.e., current density). Currents were low-pass filtered at 2 kHz. Pooled data are presented as mean ± se and values of n refer to the number of experiments in each group.
Experimental protocol of cellular hypoxia
Hypoxia of isolated cardiomyocytes has been performed as described (9). Thus, cardiomyocytes were placed into Tyrode's solution (in mM: NaCl 136.5, KCl 5.4, CaCl2 1.8, MgCl2 0.53, Glc 5.5, HEPES-NaOH 5.5, pH 7.4), plated out on glass coverslips, and paced to beat by field stimulation (parameters of the stimulation: 5–20 mV depending on cellular threshold, 5 ms, 1 Hz). Beating cardiomyocytes were perfused with Tyrode solution at a rate of 3 ml/min; under these conditions, the partial pressure of O2 (PO2) in perfusate was 140 mmHg. To induce hypoxia, Tyrode solution was bubbled with 100% argon (PO2=20 mM Hg). Cells exposed to this experimental protocol were used for digital epifluoresecent experiments (9).
Laser confocal microscopy
Sarcolemmal membrane potential was monitored in cells exposed to hypoxia as described above. Cells were loaded with di-8-ANEPPS according to the manufacturer's instruction (Invitrogen, Paisley, UK) and the sarcolemma imaged using laser confocal microscopy in line-scan mode (LSM-510, Zeiss, Gottingem, Germany). Cells were scanned under control conditions, then exposed to hypoxia. Cells were scanned at 5 min after the beginning of hypoxia (a time point when sarcolemmal KATP channels are still closed in cardiomyocytes expressing physiological levels of KATP channels, 9). Fluorescence was detected/imaged at 488 nM excitation wavelength and emission was captured at >505 nM.
Digital epifluorescence imaging
Cell morphology, size parameters, and intracellular concentration of Ca2+ were continuously monitored in cells exposed to the experimental protocol described in the previous section. To measure intracellular Ca2+, cardiomyocytes were loaded with the esterified form of the Ca2+ sensitive fluorescent probe Fura-2 and imaged using a digital epifluorescence imaging system coupled to an inverted microscope (Image Solutions, Standish, UK). A mercury lamp served as a source of light to excite Fura-2 at 340 and 380 nM. Fluorescence emitted at 520 nM was captured, after crossing dichroic mirrors, by an intensified charge coupled device camera, and digitized using an imaging software. The moment of cell death was defined as a point when irreversible cellular hypercontracture associated with the Ca2+ overload occurred (6-9).
Heart collection and ischemia-reperfusion injury
Mice were sacrificed by cervical dislocation (according to UK Home Office procedures), weighed immediately, and the heart was dissected into ice-cold Tyrode's solution (pH 7.4). The aorta was then cannulated and the heart attached to a Langendorff perfusion apparatus where it was perfused with 37°C oxygenated Tyrode's solution for a stabilization period of 70 min. The heart was then subjected to 30 min of ischemia by placing it into degassed Tyrode's and switching off perfusion. A second 30 min reperfusion with oxygenated Tyrode's followed the ischemia. After reperfusion, hearts were snap-frozen in liquid nitrogen and stored at −80°C. The frozen heart was divided into ∼5–6 transverse sections, which were weighed before staining for 1 h in 10% triphenyltetrazolium chloride (TTC) in PBS (both Sigma-Aldrich, Dorset, UK) at 37°C. The stain was fixed in 4% paraformaldehyde (Sigma-Aldrich) for 30 min, then the tissue was photographed and the area of infarcted tissue was measured using Zeiss LSM Image Analysis software.
Statistical analysis
Data are presented as mean ± se, with n representing the number of analyzed mice or cells. Mean values were compared by the Student's t test, Mann-Whitney rank sum test, or χ2 test, where appropriate, using SigmaStat program (Jandel Scientific, Chicago, IL, USA). P < 0.05 was considered statistically significant.
RESULTS
SUR2A is the least-expressed KATP channel-forming protein in the heart
In the present study, we have designed efficient Kir6.2- and SUR2A-specific primers (90.4% and 98.6% of PCR efficiency for SUR2A and Kir6.2, respectively, Fig. 2A) that have produced a single product as indicated by the shape of melting curves (Fig. 2B). Real-time RT-PCR has shown that the concentration of Kir6.2 is much higher in the mouse heart than those for SUR2A (Fig. 2C; average cycle threshold was 21.9±0.3 and 28.4±0.8 at dilution 1 for Kir6.2 and SUR2A, respectively, n=5 for each, P<0.0001). We have also checked the levels of expression of accessory proteins. Primers for AK, CK, m-LDH, and GAPDH were efficient (100%, 98%, 90%, and 100%, respectively, Fig. 2D) and produced a single product (Fig. 2E). In this series of experiments, cycle threshold was 17.6 ± 0.4 for GAPDH, 19.9 ± 0.1 for AK, 20.2 ± 0.3 for CK and 20.2 ± 0.1 for m-LDH (n=4 for each). All these values were significantly lower then the value for SUR2A (23.4±0.2, n=4, P<0.01 compared with any other protein, Fig. 2F), showing that mRNAs of all proteins forming KATP channels are at much higher levels than mRNA of the SUR2A subunit.
Figure 2.
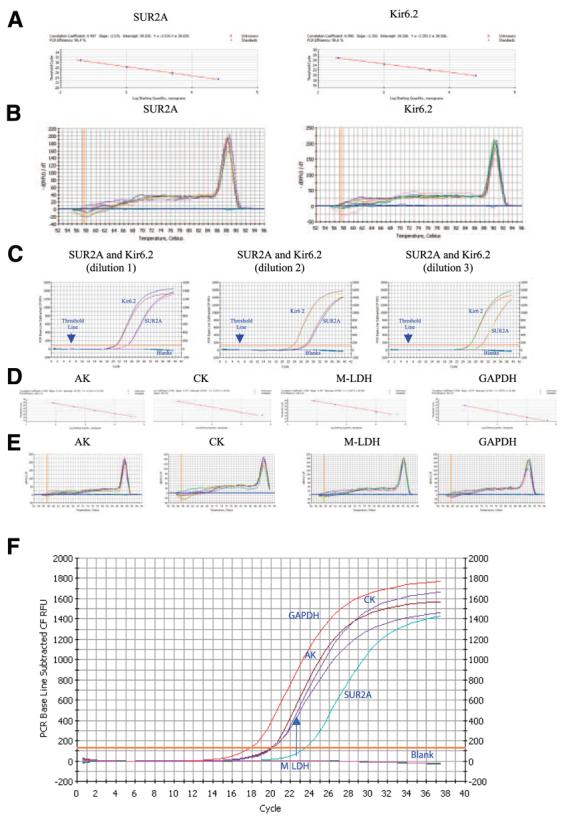
Levels of mRNA of sarcolemmal KATP channel subunits and accessory proteins in the mouse heart. A) Standard curve for the real-time PCR amplification of SUR2A and Kir6.2 cDNA. Shown is a linear regression plot of the cross point (cycle number) vs. the logarithm of cDNA amounts (40 pg, 200 pg, 1 ng, 5 ng, 25 ng). Each point represents an experiment. B) Melting curve analysis of Sur2A and Kir6.2 amplicons. Shown are plots of fluorescence (−dF1/dT) vs. temperature with different amounts of cDNA template (40 pg, 200 pg, 1 ng. 5 ng, 25 ng) done in duplicate. The presence of a single peak is consistent with the formation of a single amplicon and indicates the lack of primer-dimer formation. C) Representative progress curves done in duplicate for the real-time PCR amplification of SUR2A and Kir6.2 cDNA (dilution 1: 25 ng of template; dilution 2: 5 ng of template; dilution 3: 1 ng of template). D) Standard curve for the real-time PCR amplification of adenylate kinase type 1 (AK), creatine kinase type 1 (CK), muscle form of lactate dehydrogenase (m-LDH) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) cDNA. Shown is a linear regression plot of the cross point (cycle number) vs. the logarithm of cDNA amounts (80 pg, 400 pg, 2 ng, 10 ng, 50 ng). Each point represents an experiment. E) Melting curve analysis of AK, CK, m-LDH, and GAPDH amplicons. Shown are plots of fluorescence (−dF1/dT) vs. temperature with different amounts of cDNA template (80 pg, 400 pg, 2 ng, 10 ng, 50 ng) done in duplicate. F) Representative progress curves for the real-time PCR amplification of SUR2A, AK, CK, m-LDH, and GAPDH cDNA (5 ng of template was used).
Transgenic mice overexpress SUR2A without affecting expression of any other KATP channel pore-forming protein
An excess of Kir6.2, AK, CK, m-LDH, and GAPDH mRNAs over SUR2A mRNA suggested that the numbers of sarcolemmal KATP channels in the heart could be controlled solely by the concentration of SUR2A expression. To check this hypothesis, we generated transgenic mice overexpressing the SUR2A subunit. This was achieved using a SUR2 gene construct linked to a CMV promoter; this promoter is more efficient than the indigenous SUR2 promoter, which produces low levels of SUR2A mRNA (8). Real-time RT-PCR has demonstrated that SUR2A mRNA levels were increased in transgenic compared to wild-type mice (Fig. 3; average cycle threshold for SUR2A was 28.4±0.8 and 22.0±0.2 for control and transgenic mice, respectively, n=5–6, P≪0.0001; that an increase in SUR2A mRNA is associated with an increase was confirmed by Western blot, depicted on inset of Fig. 3). On the other hand, no difference was observed between the levels of Kir6.2, AK, CK, m-LDH, and GAPDH mRNA (Fig. 3; average cycle threshold for Kir6.2 was 21.9±0.3 and 21.8±0.5, 23.6±0.2 and 23.4±0.4 for AK, 23.6±0.1 and 23.7±0.3 for CK, 22.1±0.3, and 22.2±0.2 for m-LDH, and 19.1±0.1 and 19.1±0.2 for GAPDH for wild-type and transgenic mice, respectively, n=3–6, P>0.50 for comparison between the groups). We have also checked the expression of proteins that are either proposed to form sarcolemmal KATP, such as Kir6.1 (18) or proteins that do not form sarcolemmal KATP channels but whose mRNA is present in the heart (SUR1 and SUR2B, 18, 19). Primers for Kir6.1, SUR1, and SUR2(A)B were efficient (96.5%, 93.4%, and 100%, respectively) and produced a single product (data not shown). No differences in mRNA levels of Kir6.1, SUR1, and SUR2B plus natively expressed SUR2A were observed between wild-type and transgenic mice (Fig. 3; average cycle threshold for Kir6.1 was 19.9±0.2 vs. 19.9±0.2, 22.8±0.3 vs. 22.7±0.1 for SUR1, and 20.9±0.7 vs. 20.4±0.3 for SUR2(A)B for wild-type and transgenic mice, respectively, n=3–6, P>0.50 for a comparison between the groups).
Figure 3.

Levels of mRNA of sarcolemmal KATP channel subunits and accessory proteins in transgenic and wild-type hearts. Bar graphs showing cycle threshold for the real-time PCR amplification of SUR2A, Kir6.2, AK, CK, m-LDH, GAPDH, Kir6.1, SUR1, and natively expressed SUR2A/SUR2B (SUR2AB, native) cDNA (25 ng of template was used) from wild-type (WT) and transgenic mice. Inset in Fig. 3 (SUR2A): Western blot of total protein extract with anti-SUR2 Ab. Each bar represents mean ± se (n=3–6). *P < 0.01.
Overexpression of the SUR2A subunit generates cardiac phenotype with increased number of sarcolemmal KATP channels
Real-time RT-PCR experiments suggested that the transgenic cardiac phenotype has an increased expression of the SUR2A, but not the Kir6.2, subunit. How this difference affects the levels of assembled sarcolemmal KATP channels was tested by imunoprecipitation/Western blot and patch clamp electrophysiology.
We probed anti-Kir6.2 immunoprecipitate of cardiac membrane fraction with anti-SUR2 Ab and vice versa. Using this strategy, we measured only those Kir6.2 and SUR2A subunits that physically assemble in sarcolemma to form a channel (6-9). Western blot has demonstrated that the levels of both Kir6.2 and SUR2 proteins were increased in sarcolemma in transgenic mice in comparison to wild-type mice (Fig. 4A, A1, B, B1; SUR2A: the band intensity was 1.9±0.5 and 5.5±1.1 in wild-type and transgenic mice, respectively (n=3 for each, P=0.04); Kir6.2: the band intensity was 12.6 ± 1.5 and 24.8 ± 2.8 in wild-type and transgenic mice, respectively (n=3 for each, P=0.02). To determine whether increased levels of physically associated Kir6.2/SUR2A results in increase number of functional sarcolemmal KATP channels we have applied whole-cell configuration of patch clamp electrophysiology. Under physiological conditions, high levels of intracellular ATP keeps KATP channels closed. Therefore, to activate sarcolemmal KATP channels, a pipette solution contained only 3 μM of ATP, a concentration too low to affect channel activity but still able to decrease the probability of the channel rundown. Under these conditions, whole-cell K+ current was significantly larger in transgenic cells then in wild-type cells (at membrane potential of 80 mV current density was 10.1±3.1 pA/pF in wild-type and 20.8±2.1 pA/pF in transgenic cells, P=0.01, n=6–7, Fig. 5A, B). HMR 1098 (100 μM), a selective antagonist of sarcolemmal KATP channels (20), inhibited the whole-cell K+ current, which was also associated with the appearance of N shape of steady-state voltage-current (I-V) relationship (Fig. 5A) due to the strong inward rectification of Ik1 channels and absence of active KATP channels (see also 13-15). HMR 1098-sensitive component of the current was significantly larger in transgenic then in wild-type cells (at membrane potential of 80 mV current density of HMR 1098-sensitive component of the current was 3.2±0.6 pA/pF in wild-type and 10.6±2.7 pA/pF in transgenic cells, P=0.02, n=6, Fig. 5A, C).
Figure 4.
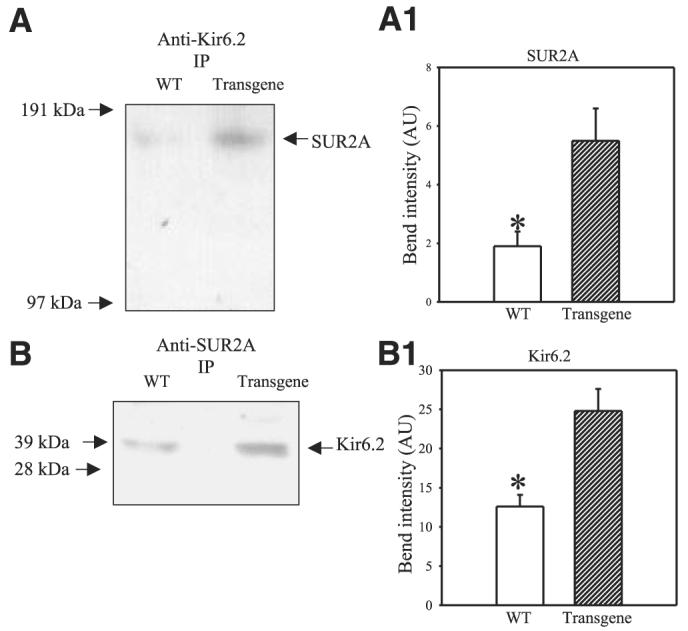
A, B) Western blot with anti-Kir6.2 and anti-SUR2 Ab of anti-Kir6.2 (A) and anti-SUR2A (B) immunoprecipitate pellets (IP) from cardiac membrane fractions obtained from wild-type (WT) and transgenic mice. A1, B1). Graphs corresponding to panels A and B, respectively. Each bar represents mean ± se (n=3 for each). *P < 0.01.
Figure 5.

Whole cell K+ currents in cells from wild-type (WT) and transgenic mice. A) Superimposed membrane currents evoked by identical families of 400 ms voltage pulses (from −100 mV to 80 mV; holding potential was −40 mV) in cells that were first maintained in the absence of HMR 1098, then exposed to 100 μM HMR 1098 for 2 min. Scatter-line graphs on the right correspond to the recordings on the left. Dotted lines correspond to the zero current concentration. B) Current densities at 80 mV in the absence of 100 μM HMR 1098 in cardiac cells from wild-type (WT) and transgenic mice. Each bar represents mean ± se (n=6–7). *P < 0.05.
Cardiomyocytes overexpressing SUR2A acquire resistance to hypoxia
Cardiomyocytes were stimulated to beat and were exposed to hypoxia (as described in ref 9). It has been shown that sarcolemmal KATP channels are not active in first 5 min of hypoxia in single beating cardiomyocytes (9). Here, we measured membrane potential under control conditions and after 5 min of hypoxia in wild-type and transgenic cells. A representative example of action membrane potentials is depicted on Fig. 7. In wild-type, a prolongation of action membrane potential was a response found in all cells tested (Fig. 6). In contrast, in 5 of 6 transgenic cells, shortening of action membrane potential was observed after 5 min of hypoxia, which is a typical finding when sarcolemmal KATP channels are activated (5). Furthermore, cells overexpressing SUR2A were significantly more resistant to hypoxia than control ones (Fig. 7A-C). Only 23.5% of wild-type cells survived 60 min-long hypoxia whereas this number was 90.9% for transgenic cells, n = 17–22, P < 0.001 (Fig. 7C). The average time of survival was 48.2 ± 7.4 min (n=17) for wild-type cells and 85.3 ± 4.9 min (n=18) for transgenic cells (P<0.001).
Figure 7.

Resistance of cardiomyocytes from wild-type and transgenic mice to hypoxia. A) Epifluorescent images of Fura-2-loaded cardiomyocytes exposed to hypoxia. ×40. Time course of Fura-2 ratio (A1) and cell diameters (A2) corresponding to experiments in panel A. B) Average survival time of wild-type and transgenic cardiomyocytes exposed to hypoxia. Bars represent mean ± se (n=17–18). *P < 0.01. C) Percentage of cells from wild-type or transgenic mice that died/survived, n = 17–18. *P < 0.01. Note that cardiomyocytes from transgenic mice are more resistant to hypoxia then cardiomyocytes from wild-type mice.
Figure 6.

The response of cardiac action membrane potential to hypoxia in wild-type and transgenic mice. Original line scans of di-8-ANEPPS-loaded cardiomyocytes and corresponding time courses under depicted conditions. AU = arbitrary units.
Hearts overepressing SUR2A acquire resistance to ischemia-reperfusion injury
Ischemia-reperfusion induced myocardial infarction in wild-type mice that was 21.2 ± 5.1% of the area at risk zone (n=17; Fig. 8A, C). The size of myocardial infarction was dramatically smaller in transgenic mice and it was just 5.3 ± 1.2% (Fig. 8B, C; n=6, P=0.02).
Figure 8.
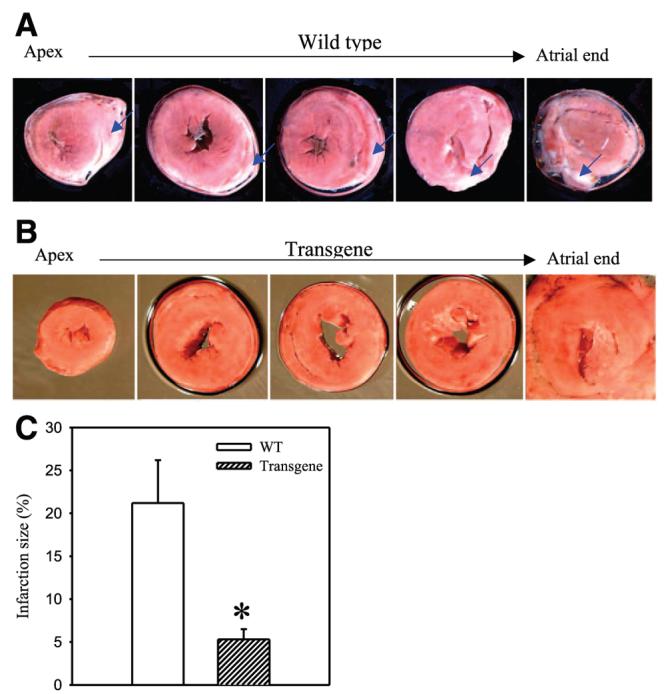
Resistance of wild-type and transgenic hearts to ischemia-reperfusion. A, B) Typical photographs of myocardial slices from wild-type and transgenic mice exposed to ischemia-reperfusion. Infarcted areas are pale/gray (indicated also with the blue arrows) whereas viable myocardium is dark/red. C) Myocardial infarct size expressed as a percentage of area at risk zone (n=6–17). *P < 0.01. Note that hearts from transgenic mice are more resistant to myocardial infarction then hearts from wild-type mice.
DISCUSSION
In the present study we have shown that a sole overexpression of the SUR2A protein generates cardiac phenotype with more sarcolemmal KATP channels and increased resistance to hypoxia and ischemia-reperfusion.
It is generally accepted that cardiac sarcolemmal KATP channels are heteromultimers comprised of pore-forming Kir6.2 and regulatory SUR2A subunits (21). More recent research has suggested that the cardiac sarcolemmal KATP channel protein complex in vivo also contains enzymes AK, CK, m-LDH and GAPDH. These enzymes govern the concentration of KATP channel ligands in the microenvironment surrounding the channel and regulate the activity of KATP channels (12-16). The involvement of at least six proteins in the channel makes it difficult to figure out how expression of such a protein complex can be regulated. The initial idea behind this study was that not all proteins forming KATP channels are expressed at the same concentration and that the protein least expressed is the one whose concentration determines the number of KATP channels. In the present study, real-time RT-PCR has revealed that SUR2A mRNA levels are lower then the concentration of mRNA of any other KATP channel-forming protein in the heart. SUR2A is a regulatory subunit required for KATP channel trafficking and no functional channels can be assembled without this protein (21, 22). The low intracellular concentration of SUR2A could mean that this subunit is comparative low in relation to other KATP channel-forming proteins, suggesting that SUR2A could be a limiting factor in the generation of KATP channels. However, this reasoning had two important limitations. First, mRNA levels do not always match protein levels (for a review, see ref 23), and the conclusions drawn from RNA data were not guaranteed to be completely accurate. Second, AK, CK, m-LDH, and GAPDH are probably not the only accessory proteins of Kir6.2/SUR2A complex (24-26), raising the possibility that some yet unidentified protein might be more important in regulating KATP channel expression than SUR2A. However, it should be pointed out that our previous studies have shown that female gender, young age, estrogen treatment, and chronic hypoxia are associated with an increase in the number of sarcolemmal KATP channels and a sole increase of SUR2A mRNA levels (6-8, 27). Thus, in principle, if the concentration of SUR2A is the limiting factor that regulates the number of sarcolemmal KATP channels, then the up-regulation of this subunit should increase the number of KATP channels. Therefore, we decided to take risks and test our hypothesis by generating mice overexpressing SUR2A. To do that, we used a SUR2A construct where the native SUR2 promoter was replaced with the more efficient CMV promoter. Real-time RT-PCR experiments have demonstrated that transgenic mice had indeed increased expression of SUR2A when compared to the wild-type without any changes in levels of other KATP channel-forming proteins.
Immunoprecipitation, followed by Western blot, as well as whole cell electrophysiology are strategies to measure levels of assembled sarcolemmal KATP channels that we implemented successfully earlier (6-9). Here, this strategy has shown that transgenic mice do have more sarcolemmal KATP channels in the heart in comparison to wild-type. As SUR2A mRNA was the only KATP channel-forming protein increased in transgenic mice, it is reasonable to conclude that the concentration of SUR2A is a limiting factor in determining the number of sarcolemmal KATP channels.
The consequence of an increased number of sarcolemmal KATP channels on cardiac sensitivity to metabolic stress had never been tested before. Estrogens, chronic hypoxia, and preconditioning are associated with an increase in number of sarcolemmal KATP channels and increased resistance to stress (6-9). However, all these treatments activate a number of signaling pathways, making it impossible to tell whether the cardioprotection observed was due solely to increased numbers of sarcolemmal KATP channels (1, 10, 11). A total lack of these channels impairs adaptation to physical stress in mice (28), but it is interesting that the hearts from these mice are not more sensitive to ischemia-reperfusion injury (29). In the present study, we have exposed adult beating cardiomyocytes to genuine hypoxia (hypoxia was induced solely by reducing partial oxygen tension without using additional means to metabolically challenge the cells). This particular experimental model is closer to in vivo conditions than most other cellular models of hypoxia (9, 30, 31). As isolated cardiomyocytes are a pure myocardial preparation free of any neuronal, humoral, and vascular influences, these experiments allowed us to assess the consequences of increased numbers of sarcolemmal KATP channels on cardiac resistance in the absence of any noncardiac confounding factors. It has been shown that long-lasting ischemia/hypoxia induces shortening of action membrane potential, which is mediated by the opening of sarcolemmal KATP channels (5, 32). However, action potential shortening is known to be preceded by an initial prolongation due to the inhibition of transient outward current whereas L-type Ca2+ and steady-state outward current remained unchanged (33). It has been shown that during the first 5 min of hypoxia sarcolemmal KATP channels remain mostly closed in single beating nonpreconditioned cardiomyocytes (9). In the present study, hypoxia was associated with a prolongation of action membrane potential duration in wild-type mice, and this would be expected based on previous findings (9, 33). However, in transgenic mice, shortening of action membrane potential has occurred, which would be in accord with the activation of sarcolemmal KATP channels. It has been reported that preconditioning induces an acute increase in sarcolemmal KATP channel numbers resulting in early opening of these channels. In this respect, the results obtained with the transgenic cells resemble those obtained on preconditioned cells (9). In support of such conclusion is our finding that overexpression of sarcolemmal KATP channels was also associated with increased resistance to hypoxia in cardiomyocytes.
Whether increased resistance of cardiomyocytes to hypoxia transposes into increased resistance of the heart to myocardial infarction was further tested by exposing mice hearts to ischemia-reperfusion challenge. These experiments have shown that the size of myocardial infarction in response to ischemia-reperfusion was much smaller in transgenic mice than in wild-type, suggesting that the increase in number of sarcolemmal KATP channels create a cardiac phenotype with increased resistance to hypoxia/ischemia/reperfusion and myocardial infarction.
The therapy of myocardial infarction at present is based on restitution of blood supply to the heart. However, this strategy does not protect against the reperfusion stage of injury, and activation of endogenous cardioprotective signaling would be theoretically the most efficient way to address this problem (2). Our results imply that up-regulation of sarcolemmal KATP channels is a strategy that deserves to be thoroughly examined. This strategy was impossible to test prior to this study, as it was unknown how to up-regulate sarcolemmal KATP channels. However, we have demonstrated here that regulation of the least expressed KATP channel-forming protein, SUR2A, is sufficient to regulate the number of sarcolemmal KATP channels and cardiac resistance to ischemia. It should also be mentioned that overexpression of SUR2A might have additional beneficial effects on the heart in the context of ischemia/reperfusion. As an example, it has been suggested that SUR2A might be a part of putative mitochondrial KATP channels (19, 34). Therefore, it would probably be worthwhile to test whether mitochondrial events also contribute to ability of this cardiac phenotype to resist an injury. Nevertheless, as the SUR2 promoter has already been cloned (8), therapeutic strategy against ischemic heart disease centered around regulation of SUR2A expression seems to be feasible and technically attainable.
Acknowledgments
We thank Aventis Pharma (Frankfurt, Germany) for providing HMR 1098. This research was supported by grants from BBSRC, British Heart Foundation, MRC, TENOVUS-Scotland, Wellcome Trust, and Anonymous Trust.
REFERENCES
- 1.Kloner RA, Jennings RB. Consequences of brief ischaemia: stunning, preconditioning and their clinical implications: part 2. Circulation. 2001;104:3158–3167. doi: 10.1161/hc5001.100039. [DOI] [PubMed] [Google Scholar]
- 2.Kloner RA, Rezkalla SH. Cardiac protection during acute myocardial infarction: where do we stand in 2004? J. Am. Coll. Cardiol. 2004;44:276–286. doi: 10.1016/j.jacc.2004.03.068. [DOI] [PubMed] [Google Scholar]
- 3.Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305:147–148. doi: 10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- 4.Hanley PJ, Daut J. KATP channels and preconditioning: A re-examination of the role of mitochondrial KATP channels and an overview of alternative mechanisms. J. Mol. Cell. Cardiol. 2005;39:17–50. doi: 10.1016/j.yjmcc.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Kane GC, Liu XK, Yamada S, Olson TM, Terzic A. Cardiac KATP channels in health and disease. J. Mol. Cell. Cardiol. 2005;38:937–943. doi: 10.1016/j.yjmcc.2005.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ranki HJ, Budas GR, Crawford RM, Jovanović A. Gender-specific difference in cardiac ATP-sensitive K+ channels. J. Am. Coll. Cardiol. 2001;38:906–915. doi: 10.1016/s0735-1097(01)01428-0. [DOI] [PubMed] [Google Scholar]
- 7.Ranki HJ, Budas GR, Crawford RM, Davies AM, Jovanović A. 17β-estradiol regulates expression of KATP channels in heart-derived H9c2 cells. J. Am. Coll. Cardiol. 2002;40:367–374. doi: 10.1016/s0735-1097(02)01947-2. [DOI] [PubMed] [Google Scholar]
- 8.Crawford RM, Jovanović S, Budas GR, Davies AM, Lad H, Wenger RH, Robertson KA, Roy DJ, Ranki HJ, Jovanović A. Chronic mild hypoxia protects heart-derived H9c2 cells against acute hypoxia/reoxygenation by regulating expression of the SUR2A subunit of the ATP-sensitive K+ channels. J. Biol. Chem. 2003;278:31444–31455. doi: 10.1074/jbc.M303051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Budas GR, Jovanović S, Crawford RM, Jovanović A. Hypoxia-induced preconditioning in adult stimulated cardiomyocytes is mediated by the opening and trafficking of sarcolemmal KATP channels. FASEB J. 2004;18:1046–1048. doi: 10.1096/fj.04-1602fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Babiker FA, De Windt LJ, van Eickels M, Grohe C, Meyer R, Doevendans PA. Estrogenic hormone action in the heart: regulatory network and function. Cardiovasc. Res. 2002;53:709–719. doi: 10.1016/s0008-6363(01)00526-0. [DOI] [PubMed] [Google Scholar]
- 11.Kolar F, Ostadal B. Molecular mechanisms of cardiac protection by adaptation to chronic hypoxia. Physiol. Res. 2004;53(Suppl 1):S3–S13. [PubMed] [Google Scholar]
- 12.Carrasco AJ, Dzeja PP, Alekseev AE, Pucar D, Zingman LV, Abraham MR, Hodgson D, Bienengraeber M, Puceat M, Janssen E, Wieringa B, Terzic A. Adenylate kinase phosphotransfer communicates cellular energetic signals to ATP-sensitive potassium channels. Proc. Acad. Natl. Sci. USA. 2001;98:7623–7628. doi: 10.1073/pnas.121038198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crawford RM, Ranki HJ, Booting CH, Budas GR, Jovanović A. Creatine kinase is physically associated with the cardiac ATP-sensitive K+ channel in vivo. FASEB J. 2002;16:102–104. doi: 10.1096/fj.01-0466fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crawford RM, Budas GR, Jovanović S, Ranki HJ, Wilson TJ, Davies AM, Jovanović A. M-LDH serves as a sarcolemmal KATP channel subunit essential for cell protection against ischemia. EMBO J. 2002;21:3936–3948. doi: 10.1093/emboj/cdf388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jovanović S, Jovanović A. High glucose regulates the activity of cardiac sarcolemmal KATP channels via 1,3-bisphosphoglycerate: a novel link between cardiac membrane excitability and glucose metabolism. Diabetes. 2005;54:383–393. doi: 10.2337/diabetes.54.2.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jovanović S, Du Q, Crawford RM, Budas GR, Stagljar I, Jovanović A. Glyceraldehyde 3-phosphate dehydrogenase serves as an accessory protein of the cardiac sarcolemmal KATP channel. EMBO Rep. 2005;6:848–852. doi: 10.1038/sj.embor.7400489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shyng S, Nichols CG. Octameric stoichiometry of the KATP channel complex. J. Gen. Physiol. 1997;110:655–664. doi: 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Bever L, Poitry S, Faure C, Norman RI, Roatti A, Baertschi AJ. Pore loop-mutated rat KIR6.1 and KIR6.2 suppress KATP current in rat cardiomyocytes. Am. J. Physiol. 2004;287:H850–H859. doi: 10.1152/ajpheart.00054.2004. [DOI] [PubMed] [Google Scholar]
- 19.Singh H, Hudman D, Lawrence CL, Rainbow RD, Lodwick D, Norman RI. Distribution of Kir6.0 and SUR2 ATP-sensitive potassium channel subunits in isolated ventricular myocytes. J. Mol. Cell. Cardiol. 2003;35:445–459. doi: 10.1016/s0022-2828(03)00041-5. [DOI] [PubMed] [Google Scholar]
- 20.Gogelein H, Ruetten H, Albus U, Englert HC, Busch AE. Effects of the cardioselective KATP channel blocker HMR 1098 on cardiac function in isolated perfused working rat hearts and in anesthetized rats during ischemia and reperfusion. Naunyn Schmiedeberg's Arch. Pharmacol. 2001;364:33–41. doi: 10.1007/s002100000391. [DOI] [PubMed] [Google Scholar]
- 21.Inagaki N, Gonoi T, Clement JP, Wang CZ, Aguilar-Bryan L, Bryan J, Seino S. A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron. 1996;16:1011–1017. doi: 10.1016/s0896-6273(00)80124-5. [DOI] [PubMed] [Google Scholar]
- 22.Schwappach B, Zerangue N, Jan YN, Jan LY. Molecular basis for K(ATP) assembly: transmembrane interactions mediate association of a K+ channel with an ABC transporter. Neuron. 2000;26:155–167. doi: 10.1016/s0896-6273(00)81146-0. [DOI] [PubMed] [Google Scholar]
- 23.Greenbaum D, Colangelo C, Williams K, Gerstein M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003;4:117. doi: 10.1186/gb-2003-4-9-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang Y, Leung YM, Manning-Fox JE, Xia F, Xie H, Sheu L, Tsushima RG, Light PE, Giasano HY. Syntaxin-1A inhibits cardiac KATP channels by its actions on nucleotide binding folds 1 and 2 of sulfonylurea receptor 2A. J. Biol. Chem. 2004;279:47125–47131. doi: 10.1074/jbc.M404954200. [DOI] [PubMed] [Google Scholar]
- 25.Shibasaki T, Sunaga Y, Seino S. Integration of ATP, cAMP, and Ca2+ signals in granule exocytosis. Diabetes. 2004;53(Suppl 3):S59–S62. doi: 10.2337/diabetes.53.suppl_3.s59. [DOI] [PubMed] [Google Scholar]
- 26.Dhar-Chowdhury P, Maddison DH, Han SY, Jankowska D, Parachuru L, Morrissey A, Srivastava S, Liu W, Malester B, Yoshida H, Coetzee WA. The glycolytic enzymes, glyceraldehyde-3-phosphate dehydrogenase, triose-phosphate isomerase, and pyruvate kinase are components of the KATP channel macromolecular complex and regulate its function. J. Biol. Chem. 2005:38464–38470. doi: 10.1074/jbc.M508744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ranki HJ, Crawford RM, Budas GR, Jovanović A. Ageing is associated with decrease in number of sarcolemmal ATP-sensitive K+ channels in a gender-dependent manner. Mech. Ageing Dev. 2002;123:695–705. doi: 10.1016/s0047-6374(01)00415-8. [DOI] [PubMed] [Google Scholar]
- 28.Zingman LV, Hodgson DM, Bast PH, Kane GC, Perez-Terzic C, Gumina RJ, Pucar D, Bienengraeber M, Dzeja PP, Miki T, Seino S, Alekseev AE, Terzic A. Kir6.2 is required for adaptation to stress. Proc. Natl. Acad. Sci. USA. 2002;99:13278–13283. doi: 10.1073/pnas.212315199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suzuki M, Sasaki N, Miki T, Sakamoto N, Ohmoto-Sekine Y, Tamagawa M, Seino S, Marban E, Nakaya H. Role of sarcolemmal K(ATP) channels in cardioprotection against ischemia/reperfusion injury in mice. J. Clin. Invest. 2002;109:509–516. doi: 10.1172/JCI14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mora A, Davies AM, Bertrand L, Sharif I, Budas GR, Jovanović S, Mouton V, Kahn RC, Lucocq JM, Gray GA, Jovanović A, Alessi DR. Deficiency of PDK1 in heart results in heart failure and increased sensitivity to hypoxia. EMBO J. 2003;22:4666–4676. doi: 10.1093/emboj/cdg469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jovanović S, Jovanović N, Jovanović A. High glucose protects single beating adult cardiomyocytes against hypoxia. Biochem. Biophys. Res. Commun. 2006;341:57–66. doi: 10.1016/j.bbrc.2005.12.147. [DOI] [PubMed] [Google Scholar]
- 32.Billman GE. Role of ATP-sensitive potassium channel in extracellular potassium accumulation and cardiac arrhythmias during myocardial ischaemia. Cardiovasc. Res. 1994;26:762–769. doi: 10.1093/cvr/28.6.762. [DOI] [PubMed] [Google Scholar]
- 33.Verkerk AO, Veldkamp MW, van Ginneken AC, Bouman LN. Biphasic response of action potential duration to metabolic inhibition in rabbit and human ventricular myocytes: role of transient outward current and ATP-regulated potassium current. J. Mol. Cell. Cardiol. 1996;28:2443–2456. doi: 10.1006/jmcc.1996.0237. [DOI] [PubMed] [Google Scholar]
- 34.Lacza Z, Snipes JA, Miller AW, Szabo C, Grover G, Busija DW. Heart mitochondria contain functional ATP-dependent K+ channels. J. Mol. Cell. Cardiol. 2003;35:1339–1347. doi: 10.1016/s0022-2828(03)00249-9. [DOI] [PubMed] [Google Scholar]