

Abstract
R2 elements are non-long terminal repeat (non-LTR) retrotransposons with a single open reading-frame encoding reverse transcriptase, DNA endonuclease and nucleic acid binding domains. The elements are specialized for insertion into the 28S ribosomal RNA (rRNA) genes of many animal phyla. The R2-encoded activities initiate retrotransposition by sequence-specific cleavage of the 28S gene target site and the utilization of the released DNA 3′ end to prime reverse transcription (target primed reverse transcription). The activity of the R2 polymerase on RNA templates has been shown to differ from retroviral reverse transcriptases (RTs) in a number of properties. In this report we demonstrate that the R2-RT is capable of efficiently utilizing single-stranded DNA (ssDNA) as a template. The processivity of the enzyme on ssDNA templates is higher than its processivity on RNA templates. This finding suggests that R2-RT is also capable of synthesizing the second DNA strand during retrotransposition. However, R2-RT lacks the RNAse H activity that is typically used by retroviral and LTR-retrotransposon RTs to remove the RNA strand before the first DNA strand is used as template. Remarkably, R2-RT can displace RNA strands that are annealed to ssDNA templates with essentially no loss of processivity. Such strand displacement activity is highly unusual for a DNA polymerase. Thus the single R2 protein contains all the activities needed to make a double-stranded DNA product from an RNA transcript. Finally, during these studies we found an unexpected property of the highly sequence specific R2 endonuclease domain. The endonuclease can non-specifically cleave ssDNA at a junction with dsDNA. This activity suggests that second-strand cleavage of the target site may not be sequence specific, but rather is specified by a single-stranded region generated when the first DNA strand is used to prime reverse transcription.
Keywords: reverse transcriptase, retrotransposons, processivity, strand displacement activity, RNase H
INTRODUCTION
A large fraction of the DNA in most eukaryotic genomes originated from the reverse transcription of RNA sequences. These insertions include several classes of retrotransposons as well as reverse transcripts derived from cellular RNAs (SINEs and processed pseudogenes).1 Two major mechanisms for the integration of reverse transcripts into a genome have been characterized to date. One mechanism is utilized by long-terminal repeat (LTR) retrotransposons and retroviruses. In this mechanism an element encoded reverse transcriptase (RT) typically utilizes tRNA annealed to its RNA template to prime cDNA synthesis in RNP particles in the cytoplasm of the cell. While the first DNA strand is being synthesized the RNA template is destroyed by an RNase H activity located in a separate domain of the RT. Specific remnants of the RNA primes the synthesis of the second DNA strand again by the RT. Finally, the resulting double-stranded DNA is inserted into the genome by an integrase also encoded by the element.2,3
The second mechanism for inserting reverse transcripts into a genome is catalyzed by the non-LTR retrotransposons (or LINEs). This mechanism differs from the first mechanism in that the priming of cDNA synthesis occurs directly at the insertion site. In the non-LTR mechanism an element-encoded endonuclease cleaves the chromosomal target site, and the released 3′ OH is utilized by the element-encoded RT to prime cDNA synthesis. The formation of this first DNA strand is referred to as target primed reverse transcription (TPRT).4 Because the reverse transcriptase of many elements has minimal specificity for its own RNA, this TPRT mechanism appears to be the origin of insertions derived from the reverse transcription of cellular RNAs.5,6 Unfortunately many questions remain concerning the steps used to complete integration after this initial TPRT reaction. While the simplest models suggest second-strand synthesis occurs by a DNA-directed DNA polymerase activity of the RT, such activity has not been extensively characterized for non-LTR retrotransposon RTs. Furthermore, most elements do not appear to encode an RNase H domain, thus it is not clear how the RNA is removed from the first DNA strand to allow second-strand synthesis.
Much of our current understanding of the TPRT mechanism is based upon in vitro studies of the protein encoded by the R2 retrotransposons.4,7,8 These elements specifically insert into the large ribosomal subunit gene, 26S or 28S rRNA, in many groups of organisms.9,10 In addition to its ability to conduct TPRT, previous studies have revealed several properties of R2 reverse transcriptase that differentiates it from the RTs encoded by LTR-elements and retroviruses. First, R2-RT can efficiently synthesize cDNA using RNA primers that are not annealed to the RNA template (i.e. in the absence of complementarity between the primer and template).11 Second, the R2 polymerase is able to jump from the 5′-end of one RNA template to the 3′-end of a second RNA template in the absence of sequence identity between the two templates (end-to-end template jumping).12 Finally, R2-RT can reverse transcribe RNA even if a complementary RNA strand is annealed to the template RNA.13
In this report we characterize the DNA-directed DNA polymerase activity of the R2 polymerase. We show that the R2 polymerase can efficiently utilize DNA templates even when these templates are annealed with a complementary RNA or DNA strand. Thus the R2 enzyme contains both activities needed to make a double-stranded DNA product from an RNA transcript.
RESULTS
R2 reverse transcriptase has high processivity on DNA templates
We first compared the ability of the R2 reverse transcriptase (R2-RT) to use single-stranded (ss) DNA versus RNA templates. The ssDNA template was a 320 nt sequence obtained by restriction digestion of the bacteriophage M13mp18, while the 320 nt RNA template was obtained by in vitro transcription of this same sequence (see Materials and Methods). The assays were designed to measure the processivity of the enzyme (i.e. the length of product that can be formed without the enzyme dissociating from the template). The R2 protein was preincubated with the RNA or DNA template containing an annealed 5′ end-labeled DNA primer (Figure 1a), and polymerization started by the addition of dNTPs together with a “trap” [(heparin and poly(A)/oligo(dT)], which prevents the reassociation of the polymerase with the template once it dissociates.13 The products of the single round extension reactions were separated on 6% denaturing polyacrylamide gels (Figure 1b). As evidence for the effectiveness of the trap no initiation of DNA synthesis was observed if the trap was added prior to the enzyme (Figure 1b, control lane). The R2 protein efficiently used both the RNA and DNA templates with the R2 protein showing greater processivity on the ssDNA template based on the reduced levels of products less than 320 nt in length.
Figure 1.
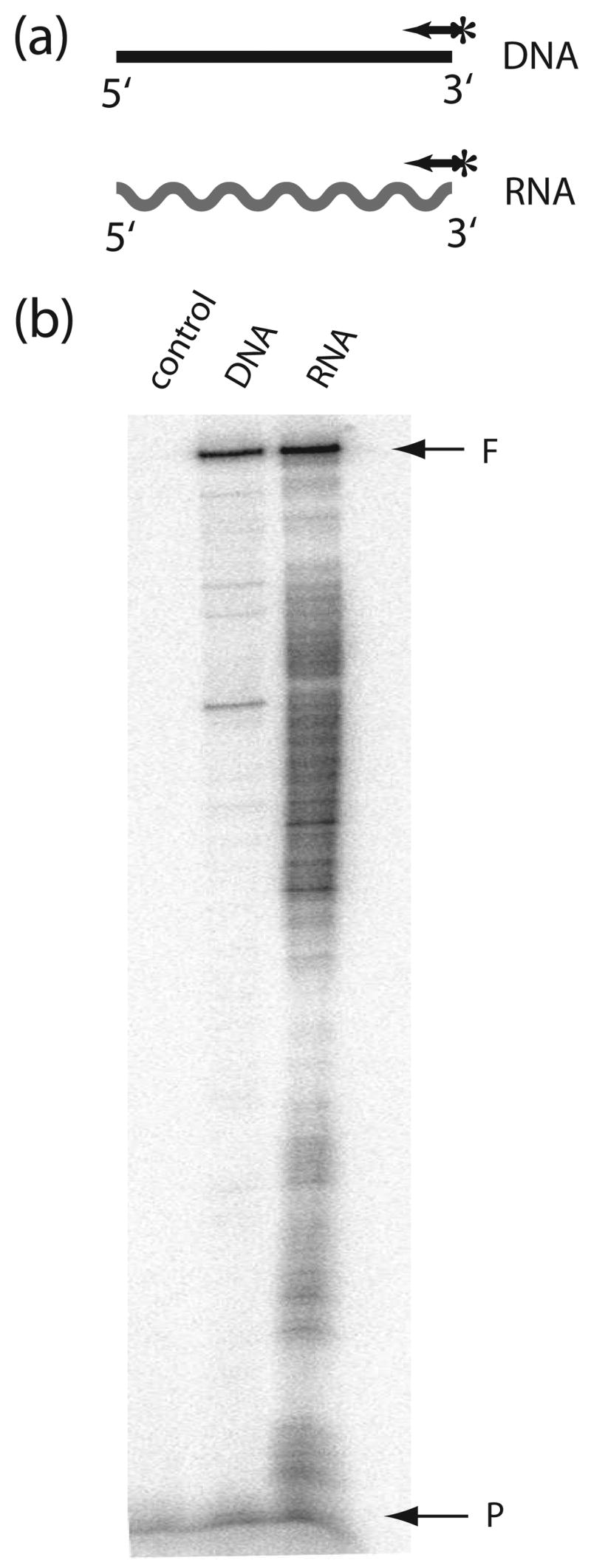
The R2 polymerase can utilize both RNA and DNA templates. (a) Schematic diagrams of templates used in the experiment. DNA synthesis on a 320 nt ssDNA template (horizontal line) and a 320 nt RNA template of the same sequence (wavy line) was primed by a 5′-end labeled primer (arrow with asterisk) annealed to each template. (b) PhosphorImager scan of the extension products separated on a 6% denaturing polyacrylamide gel. Templates with annealed primers were preincubated with R2 protein for 10 min at room temperature. Reactions were started by the addition of dNTPs and a reinitiation trap [20 μg heparin and 300 ng poly(rA)/poly(dT)13–18]. Incubations were for 20 minutes at 37°C. In the control reaction the trap was added prior to the addition of the R2 protein to the template. F, full-length extension products; P, non-extended primer.
To quantify the processivity of R2-RT the probability of premature termination per nucleotide were calculated for RNA and DNA templates. The fraction of the total extension products greater than various lengths were determined from PhosphoImager scans like that in Figure 1. The curve fitted to those values is described by the function exp(−Poff × n) where n is the number of incorporated nucleotides, and Poff is the probability of premature termination per nucleotide. Thus the lower the -Poff value for an enzyme the higher the processivity (see ref. 13 for a more detailed discussion). The -Poff values for R2-RT on the DNA and RNA templates in Figure 1 was 1.5 × 10−3 and 4.8 × 10−3, respectively (Table 1, template B). Because the processivity of a polymerase can depend upon the sequence of the template, the processivity of R2-RT was also determined on other RNA and DNA templates (Table 1). The mean -Poff value for R2-RT on ssDNA was consistently lower than the mean -Poff value for RNA templates. For comparison, the processivity of the Avian Moloney Virus (AMV) RT was also tested on most of the templates. As previously reported14 the processivity of AMV RT was also lower on ssDNA compared to RNA templates. On all templates the R2 polymerase had 2–3 fold higher processivity than the retroviral enzyme.
Table 1.
Processivity of the R2 polymerase on DNA and RNA templates.
| DNA
|
RNA
|
||||
|---|---|---|---|---|---|
| A | B | B | C | D | |
| R2 RT | 0.7 ± 0.1 | 1.5 ± 0.1 | 4.8 ± 0.1 | 2.6 ± 0.1 | 4.0 ± 0.3 |
| AMV RT | 2.5 ± 0.2 | 4.4 ± 0.2 | - | 5.4 ± 0.4 | 9.3 ± 0.7 |
Numbers presented are the -Poff value calculated as the average fraction of the enzyme that dissociates from the RNA or DNA template during the incorporation of each nucleotide (see reference 13 for a more extensive discussion). Lower -Poff values indicate increased processivity. All values are the averages calculated from three separate experiments with the standard deviation. All values should be multiplied by 10−3. Templates B are the 320 nt DNA or RNA templates used in Figure 1. Template A is a HhaI-digested M13mp18 fragment annealed with the universal M13/pUC sequencing primer (Fermentas), and RNA templates C and D are described in a previous report.13
R2-RT does not have RNase H activity
After utilizing an RNA template for reverse transcription retroviral RTs degrade the RNA template with an RNase H domain located immediately C-terminal to the reverse transcriptase domain.15 Analysis of the protein domains encoded by non-LTR retrotransposons suggests that only a few of the most recently evolved lineages of elements encode a domain with sequence similarity to known RNase H domains.16,17 R2 elements represent one of the oldest lineages of non-LTR retrotransposons and lack such RNase H domains. To rule out possible RNase H activity derived from a previously uncharacterized domain as well as possible contaminating RNase H activity in our protein preparation it was important to confirm that our purified R2 protein preparations lacked RNase H activity.
To this end an internally labeled 100 nt RNA transcript was annealed with a 30 nt DNA oligonucleotide (Figure 2a) and incubated with the R2 protein for 20 minutes at 37°C (Figure 2b, lane 2). Control reactions included incubation of the substrate with the reaction buffer without protein (lane 3) and incubation of the substrate with the reverse transcriptase from Moloney Murine Leukemia Virus (MMLV), an enzyme with well characterized RNase H activity (lane 1). MMLV RT cleaved the substrate to fragments 35 nt or shorter in length. No cleavage products were observed for the reactions containing the R2 protein, indicating that at least under the conditions of the assays used in this report, the R2 protein had no RNase H activity.
Figure 2.
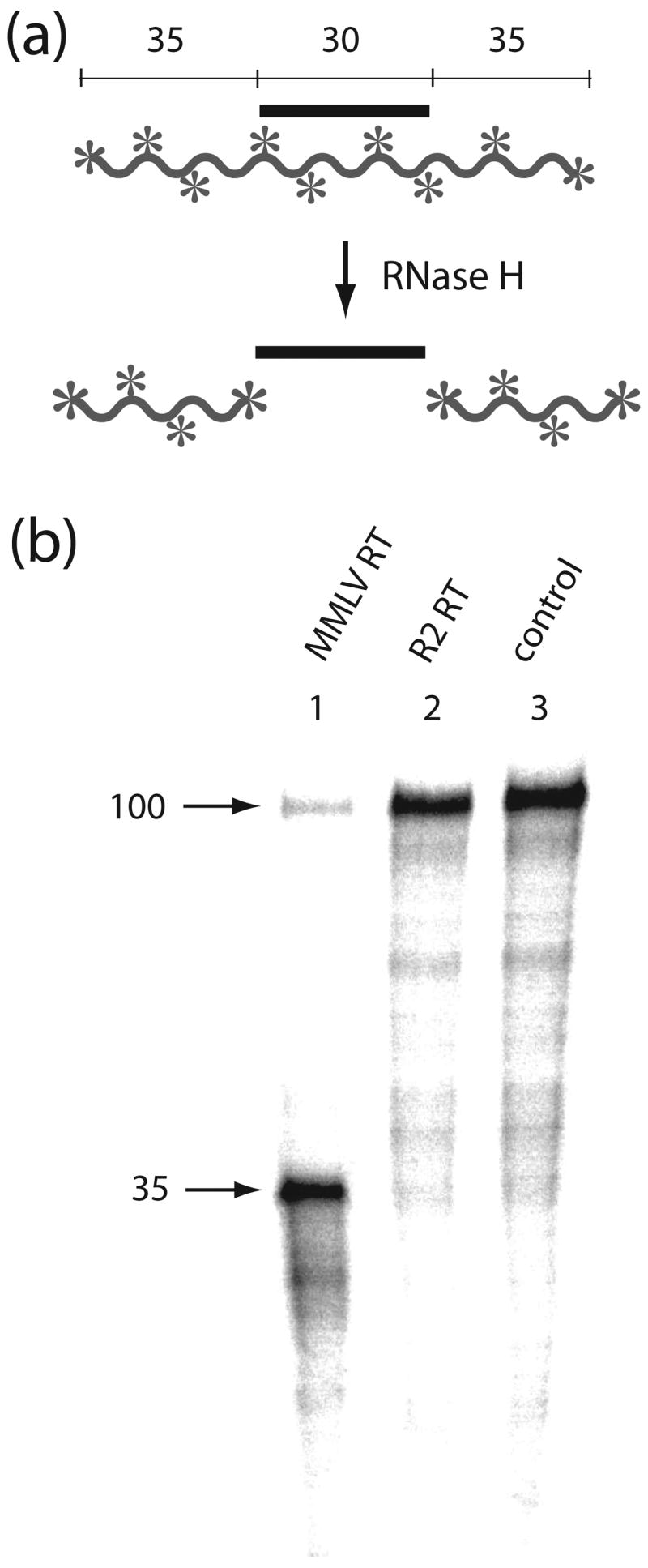
The R2 protein does not have RNase H activity. (a) Schematic diagram of the RNase H assay. In the presence of RNase H an internally 32P-labeled 100 nt RNA (waved line with asterisks) annealed with a centrally located 30 nt DNA fragment (straight line) will be cleaved to 35 nt fragments. (b) PhosphorImager scan of the products of an RNase H assay using retroviral (MMLV) RT and R2-RT. The RNA:DNA substrates were incubated for 20 min at 37°C in the presence of either MMLV RT, which has RNase H activity (lane 1), the R2 protein, which does not have RNase H activity (lane 2), or with buffer (lane 3).
R2 can displace RNA or DNA annealed to a DNA template
The high processivity of the R2 polymerase on ssDNA templates suggests that like retroviral RTs, R2-RT also performs second-strand DNA synthesis during retrotransposition. However, if the R2 protein does not contain RNase H activity, how is the RNA removed from the first strand of synthesized DNA (cDNA) in order for it to be used as a template? One possibility is that the R2 protein can displace the annealed RNA strand as the cDNA strand is used as template. While retroviral RTs have only weak RNA displacement activity14,18, we have shown that the R2 polymerase can displace RNA strands annealed to RNA templates.13
To determine whether the R2-RT can also displace an RNA strand annealed to a DNA template, the experiments diagrammed in Figure 3a was conducted. A 340 nt complementary RNA strand was annealed to the 5′ end of a 560 nt DNA template. Extension reactions on this blocked template were compared to that on the 560 nt ssDNA without the RNA block. Extension reactions were again initiated with a 5′ end labeled primer annealed to the 3′ end of the DNA template. To insure that the complementary RNA was annealed to all DNA templates, control reactions were conducted with T4 DNA polymerase. Like most DNA polymerases T4 DNA polymerase does not have strand displacement activity, and thus can be used to test the efficiency of the RNA block. As shown in Figure 3b, T4 DNA polymerase efficiently used the ssDNA template, giving rise to full-length 560 nt extension products, but few extension products greater than 220 nts in length were generated on the DNA:RNA duplex. Thus most DNA templates in the assay had an annealed RNA strand blocking polymerization. In the case of the R2 protein, while processivity was lower than that of the T4 DNA polymerase, the presence of the RNA block had minimal effects on the polymerization reaction catalyzed by the R2 polymerase. Only minor increases in extension products were detected a short distance before and within the duplex region (bands from 180 to 240 nt).
Figure 3.

R2-RT can displace RNA annealed to a DNA template. (a) Schematic diagram of the displacement assays. Polymerization on a 560 nt ssDNA template (horizontal line) was compared to polymerization on the same DNA template annealed to a 340 nt complementary RNA strand (wavy line). Polymerization was primed by a 5′-end labeled primer (arrow with asterisk) annealed to the 3′-end of the template. (b) PhosphorImager scans of the products from the displacement reactions. Reactions were conducted at 37°C as in Figure 1 but without the trap. As a control, displacement reactions were also conducted with T4 DNA polymerase. The vertical line next to the gel indicates those extension products requiring RNA displacement. T4 DNA polymerase does not have strand displacement activity, thus on the annealed template, polymerization is blocked at the beginning of the double-strand region. The RNA block has little affect on R2-RT extension. A 100 nt DNA ladder is shown at left as size markers. F, full-length extension products; B, beginning of the RNA block.
We next compared the ability of the R2 protein at various temperatures to displace DNA or RNA annealed to a DNA template. DNA:DNA duplexes are less stable than DNA:RNA duplexes, and the stability of both duplexes is decreased as the temperature of the polymerization reaction is increased (see Table 2 for the calculated stability of the duplexes used in these assays). To the same DNA template-primer complex used for the experiments in Figure 1 a 100 nt ssDNA (DNA block) or 100 nt RNA (RNA block) was hybridized to the 5′-end of this 320 nt ssDNA template, and the extension reactions were again initiated with a 5′-end labeled DNA primer (Figure 4a).
Table 2.
Efficiency of the R2 polymerase strand displacement synthesis at different temperatures.
| 25°C | 37°C | 42°C | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Δ G0 | %FL | -Poff | Δ G0 | %FL | -Poff | Δ G0 | %FL | -Poff | |
| DNA alone | -- | 71 | 3.2 ± 0.2 | -- | 77 | 1.5 ± 0.1 | -- | 77 | 1.6 ± 0.1 |
| DNA:DNA | −162 | 71 | 4.0 ± 0.1 | −135 | 84 | 1.5 ± 0.1 | −124 | 81 | 1.5 ± 0.1 |
| DNA:RNA | −178 | 65 | 3.1 ± 0.1 | −153 | 78 | 1.6 ± 0.1 | −143 | 79 | 1.6 ± 0.1 |
G0 describes the Gibbs free energy (kcal/mol) calculated for the complexes shown in Figure 4a using the predictions of nucleic acid hybridization thermodynamics in HyTher (http://ozone3.chem.wayne.edu/cgi-bin/login/login/loginVerify.cgi). % FL (full-length) is the percentage of the extension products that are full-length in the reaction of Figure 4b. Full-length products include the 320 nt band (F) and the end-to-end template jump band (J) seen in some reactions, because these jumps only occur with the polymerase reaches the end of the first template (see ref. 12 and Figure 5c). -Poff values define the fraction of the enzyme that dissociates from the RT complex after incorporation of a single nucleotide as described in Table 1. All values are the mean and standard deviation of three determinations and should be multiplied by 10−3.
Figure 4.

R2 polymerase displacement of DNA and RNA strands at different temperatures. (a) Schematic diagram of the primer extension reactions. Polymerization on a 320 nt ssDNA template (DNA template) was compared to polymerization on the same template annealed to a 100 nt complementary DNA (DNA:DNA template) or RNA (RNA:DNA template). Polymerization was primed by a 5′-end labeled primer (arrow with asterisk) annealed to the 3′-end of the template. (b) PhosphorImager scans of the products from the displacement reactions. Reactions were conducted as in Figure 4 at 25°C, 37°C and 42°C. Control displacement reactions with T4 DNA polymerase were also conducted as in Figure 4. The R2 protein extensions on the DNA:DNA template also generated ~420 nt products (band J). These products were generated by the R2 polymerase jumping from the 5′ end of the original template onto the 3′ end of the excess 100 nt ssDNA present in the reaction (see Figure 5 and text). F, full-length extension products; B, beginning of the RNA or DNA block; vertical line at right, those extension products requiring strand displacement.
Full-length extension products can be seen in all lanes in Figure 4b indicating the R2 polymerase was able to displace both DNA and RNA strands at 25°C, 37°C and 42°C. The absence of extension products corresponding to the beginning of the RNA block suggests that R2-RT does not even pause at this junction. While extension products do accumulate at the beginning of the DNA block, as will be discussed below, these products are derived from the R2 endonuclease cleaving the block junction. To quantify the relative efficiencies of the R2 polymerase to displace an RNA or DNA strand at the different temperatures, the level of extensions that terminated within the region of the duplex was compared to the level of products that was full-length. These percent full-length extensions (%FL) and calculated -Poff values are shown in Table 2. At each temperature the ability of the polymerase to displace the weaker DNA:DNA complex was only slightly greater than that of the DNA:RNA complex. Decreasing the temperature to 25°C, which greatly increased the stability of the DNA:DNA and DNA:RNA complexes, only resulted in a two-fold decrease in the processivity of the enzyme. However, this reduction in processivity on the annealed templates at the lowest temperature mirrored a similar decrease with the ssDNA template alone, thus appears to reflect a general decrease in processivity of the R2 polymerase at lower temperatures, rather than a decreased ability of the enzyme to displace RNA or DNA strands. These results suggest the presence of an annealed RNA or DNA strand has little affect on the ability of the R2 protein to use DNA as a template.
End-to-end template jumps from DNA templates
Distinct extension products greater than 400 nt in length (band J) were also generated in the DNA:DNA template assays at the higher temperatures (Figure 4b). This longer product is also present at a lower level in the DNA:RNA assays. We have previously shown that the R2 protein can efficiently jump from the 5′ end of an RNA template to the 3′ end of a second RNA template in the absence of initial sequence complementarity.12 The jumps typically occur by the polymerase adding non-templated nucleotides (predominately purines) to the newly synthesized DNA strand, and then using microhomologies in these extensions to anneal to sequences near the 3′ end of a second RNA strand. The lengths of the longer extension products in Figure 4 suggested they were derived from jumps to the 100 nt DNA blocking strands in the assays rather than to a second 320 nt ssDNA template. An excess of the 100 nt DNA block was present in the reactions to insure all of the 320 nt DNA template contained a DNA block.
To confirm that the longer products are indeed end-to-end jumps to the blocking DNA strand, additional extension reactions were conducted with the original 100 nt block (100 block lane) as well as a ssDNA block that started at the 5′ end of the template but only extending 60 nt (60 block lane). As shown in Figure 5a, ~420 nt extension products were obtained with the 100 nt block and 380 nt products with the 60 nt block. To show that the R2-RT could template jumps to other ssDNA templates in the reactions, the 60 nt block extensions was repeated with and without a 5-fold excess of an unrelated 100 nt ssDNA. As shown in Figure 5b the major ~380 nt extension product (lane 2) is replaced with a more abundant 430 nt product in the presence of the excess 100 nt sequence (lane 3). Also visible in lane 3 is an ~530 nt extension product suggesting some of the extension reactions had undergone two consecutive template jumps.
Figure 5.

The R2 polymerase can conduct end-to-end template jumps after strand displacement reactions. (a) PhosphoImager scans of the products from extensions reactions similar to those conducted at 37°C in Figure 4. Lane 1, 320 nt DNA template in the absence of a DNA block; lane 2, 320 nt DNA template in the presence of the 100 nt DNA block as diagramed in Figure 4a; and lane 3, 320 nt DNA template in the presence of a 60 nt DNA block. Products of the template jumps onto the excess 100 nt and 60 nt DNA molecules in the reactions appear as 420 nt and 380 nt bands, respectively. Vertical lines indicate those extension products requiring strand displacement. (b) PhospoImager scan of the products from extensions reactions as in (a): lane 1, absence of a DNA block; lane 2, presence of 60 nt DNA block; and lane 3, presence of the 60 nt DNA block and a 100 nt DNA template. The molar ratio of the 100 nt template to 60 nt block was 5:1. Extension products derived from template jumps are indicated with a J. (c) Sequences at the junctions of the template jumps from the 5′ end of 320 nt DNA template to the 3′ end of 100 nt DNA template. Shown at the top are the sequences at the ends of the DNA templates. Shown below are the junction sequences of 16 PCR amplified and cloned products from the 420 nt J band in panel b, lane 3. In parenthesis are numbers of clones obtained with each junction sequence.
To determine if the end-to-end template jumps from DNA to DNA are also associated with the addition of non-templated purine nucleotides, the junction between the 320 nt DNA template and the 100 nt ssDNA was PCR amplified, cloned and individual products sequenced (Figure 5c.) Of the 16 junctions sequenced, 14 were a jump from the last nucleotide at the 5′ end of the 320 nt template to the first nucleotide at the 3′ end of the 100 nt template with the addition of an extra G. Of the two remaining clones, one had an additional A at the junction, and one jump occurred to a location 26 bp into the 100 nt ssDNA. Thus the end-to-end template jumps involving DNA templates that are catalyzed by the R2 polymerase appear similar to those previously described involving RNA templates.12
DNA cleavage activity at the junction between a DNA:DNA helix and ssDNA
A second feature that can be noted in Figure 4b, as well as in Figures 5a and b, are that extension products from 220 to 240 nt in length were generated by the R2 polymerase on the DNA:DNA templates with the 100 nt block and ~ 260 nt with the 60 nt block. While the lengths of these products are near the beginning of the duplex region of the templates, their accumulation did not appear consistent with the R2 protein occasionally being unable to displace the annealed DNA strand. First these truncation products were less frequent at 25°C, even though the DNA:DNA duplex at this lower temperature would be more stable. Second, the truncation products did not accumulate on the DNA:RNA template at any temperature even though this duplex is more stable than the DNA:DNA duplex.
The single protein encoded by the R2 ORF contains an endonuclease domain responsible for the specific cleavage of the R2 target site within the 28S gene.19 While this endonuclease has been shown to be highly sequence specific for its target site on double-stranded DNA substrates7,8, it appeared the R2 endonuclease might exhibit non-sequence specific cleavage of the partially ssDNA templates used in the extension reaction. We therefore repeated the extension reactions with R2 protein in which the active site of the endonuclease had been mutated. All previously characterized reverse transcriptase and DNA and RNA binding properties of the R2 protein are unaffected by this mutation.8,20 Figure 6 presents a comparison between strand displacement reactions carried out by the native R2 protein (R2 EN+) and the endonuclease mutant (R2 EN−). Truncation products observed at the beginning of the helical region on the DNA:DNA template with the R2 EN+ protein, were not observed with the R2 EN− protein, suggesting that the products generated with the R2 EN+ protein was a result of endonuclease activity.
Figure 6.

Effects of an R2 endonuclease mutation on the displacement reactions. The templates and the conditions of the reactions were identical to those conducted at 37°C in Figure 4. R2 EN+, wild type R2 protein with a functional endonuclease domain; EN−, R2 protein with an amino acid substitution eliminating endonuclease activity8. All DNA binding and RT activities of this mutant are similar to wild-type.8,20 The EN− mutation has similar levels of strand displacement activity to the EN+ protein, but does not show an accumulation of extension products near the beginning of the DNA:DNA block (~ 220–240 nt). F, B and vertical lines are as described in Figure 4.
To directly assay this R2 endonuclease activity near the junction of ssDNA and a DNA:DNA duplex, DNA cleavage assays were conducted with a 100 nt DNA substrate annealed with a centrally located 30 nt DNA oligonucleotide (Figure 7a). The cleavage assays were conducted under the same conditions as the extension reactions, except that the 5′-end label was on the 100 nt DNA template rather than the DNA primer. Specific cleavage products ~35 and ~65 nt in length were observed with the EN+ protein but not with the EN− protein (Figure 6b, lanes 1 and 3). Thus the R2 endonuclease is able to cleave ssDNA extending from both ends of a double-stranded region. Similar cleavage by the R2 endonuclease at the junction of the annealed DNA strand used in the extension assays in Figures 4 and 5 would explain the observed 220–260 nt termination products. The presence of multiple termination products in the extension reactions with the 100 nt block suggests that additional cleavage of the DNA substrate can occur within the helical region during a polymerization reaction (Figures 4 and 6). These additional cleavages did not occur with the 60 nt block (Figure 5). Cleavage of helical junctions by the R2 endonuclease requires both strands of the helix to be DNA, because cleavages were not detected at the junctions of RNA annealed to longer DNA strands (Figure 4 and 6), or of DNA strands annealed to longer RNA sequences (data not shown).
Figure 7.

Nonspecific endonuclease activity of the R2 protein. (a) Schematic diagrams of the DNA substrates used in the experiment. A 30 nt (1) or 20 nt (2) complementary oligonucleotide was annealed to a 5′-end labeled 100 nt ssDNA. DNA fragments generated by cleavage at the junctions of the duplex regions are indicated below each substrate. (b) PhosphorImager scans of the gel products of the R2 endonuclease cleavage reactions. Substrates were incubated for 20 min at 37°C in the R2 extension buffer with wild type R2 (EN+, lanes 1 and 2) or endonuclease-deficient (EN−, lanes 3 and 4) protein. Lanes 1 and 3, cleavage reactions with substrate 1; lanes 2 and 4, cleavage reactions with substrate 2; lane C, control reaction in which substrate 1 is incubated in the reaction buffer without protein. The three lanes at left contain size standards including 5′-end labeled oligonucleotides 30 and 100 nt in length.
Finally, because all extension assays used in this report were primed by a DNA oligonucleotide annealed to the template DNA cleavage reactions were conducted with a 100 nt 5′-end labeled DNA substrate containing the standard 20 nt primer annealed to its 3′ end (Figure 7a). The conditions of the reaction were identical to the extension reactions. No cleavage products were detected (Figure 7b, lane 2). Thus in the various extension assays in this report there was no reduction in yield of products due to cleavage of the template at its junction with the oligonucleotide primer.
DISCUSSION
Current models of non-LTR retrotransposition suggest that their RTs synthesize both DNA strands during a retrotransposition reaction.1,4,20,21 However, these models are based more on analogy to the mechanism of retroviral retrotransposition than to known properties of the non-LTR RTs. Cost and co-workers observed second-strand DNA synthesis by the human L1 element; but the products were synthesized at only low levels such that detection required PCR amplification followed by Southern blotting.22 More recently the ability of L1 RT to utilize ssDNA templates was demonstrated at levels high enough to be directly detect using labeled dNTPs in the extension reactions, but no detailed characterization of the activity was conducted.23 This report is the first to extensively characterize the DNA-dependent DNA polymerase activity of the RT encoded by a non-LTR retrotransposon. The processivity of R2-RT on DNA templates was 3.5 greater than its processivity on an RNA template of the same sequence. The greater processivity of R2-RT on DNA substrates compared to RNA substrates is similar to that exhibited by retroviral RTs (Table 1). While it has been suggested that the greater processivity of retroviral RT on DNA templates is a result of the less stable secondary and tertiary structures of ssDNA compared to RNA,14 R2-RT appears minimally affected by RNA secondary structures13, thus such an explanation is unlikely to account for the greater processivity of R2-RT on ssDNA substrates.
If R2-RT is able to synthesize the second DNA strand during a retrotransposition reaction, then the question becomes how is the RNA bound to the first synthesized DNA strand removed to permit second-strand DNA synthesis. We show here that even though R2-RT does not have RNase H activity, an annealed RNA strand is not an impediment to second-strand synthesis. The R2 protein can efficiently displace such RNA strands as it uses DNA strands as template. Indeed, the processivity of R2-RT on DNA templates is minimally affected by the presence of an annealed RNA or DNA strand (Table 2). The absence of RNase H activity has also been reported for the RT encoded by the human L1 element,23 and inspection of the protein sequences encoded by all non-LTR retrotransposon suggests that only a few lineages encode RNase H domains.16,17 Thus it appears likely that the ability to displace RNA from DNA templates will be found as a common property of many non-LTR retrotransposon RTs.
While efficient RNA displacement is not needed by retroviral enzymes to complete the synthesis of the second DNA strand, because of their RNase H activity, retroviral polymerases must be able to extend through DNA:DNA helixes the length of their LTRs (at least several 100 nt). The efficiency of retroviral RT displacement synthesis through DNA:DNA helixes are about one-third their efficiency on ssDNA templates.24,25 It has been proposed that the finger subdomains of retroviral RTs are involved in DNA:DNA displacement synthesis through interactions with either the template or non-template strands.25–27 Mutations in these domains have been found that influence strand-displacement activity without affecting the processivity of the enzyme.26,27 Non-LTR retrotransposon RTs have more extended fingers regions compared to retroviral RTs.28,29 These extended fingers regions may enable greater binding to single-stranded templates which could permit more extensive displacement synthesis.
Alternatively, while most cellular DNA polymerases have minimal strand displacement activity, relying instead on accessory proteins to provide single-stranded templates,32,33 a few bacteriophage DNA polymerases are able to catalyze strand displacement DNA synthesis.34–36 Crystallographic analysis of one of these polymerases, φ29, suggests that the displacement of helical regions occurs when the bound template DNA passes through a narrow tunnel before entering the polymerase active site that is only able to accommodate ssDNA.34,37 Thus an alternative model for the extensive finger region domains of R2-RT are that they form a similar constriction that is responsible for the strand displacement activity of the polymerase.
Based on their sequence relationships the RT domains of non-LTR retrotransposons are most similar to those of mobile group II introns.16 The mobility of these introns involves specific cleavage of the target site by reverse splicing of the intron RNA into one strand of the DNA with cleavage of the second strand catalyzed by an element-encoded endonuclease.30 As with non-LTR retrotransposons, reverse transcription of the intron RNA (first-strand DNA synthesis) is primed by the 3′ end of the DNA strand generated by the endonuclease, a process also referred to a TPRT.30 However, unlike R2-RT, group II intron RTs are unable to use DNA as substrates suggesting that they must rely upon cellular encoded polymerases for the synthesis of the second DNA strand.31 Clearly the RNA- and DNA-polymerizing abilities of the R2 protein differes from that of any previously characterized polymerase.
The data in this report adds support to our previously suggested model of R2 retrotransposition.38,39 This model, shown in Figure 7, assumes an ordered series of cleavage and polymerization reactions carried out by two R2 protein subunits. The R2 protein is able to bind two alternative RNA sequences: the 250 nt 3′ untranslated region of the R2 transcript or a 300 nt region near the 5′ end of the transcript. Binding of these RNA ends promotes the binding of the R2 protein either upstream or downstream of the DNA insertion site.20,38,39 The subunit bound to the 3′ end of the R2 transcript, binds the DNA upstream of the insertion site and is responsible for the first two steps of the integration reaction: cleavage of the bottom DNA strand (step 1) and the synthesis of cDNA primed from the 3′-end released by the cleavage (step 2). The protein subunit bound to the 5′ end of the R2 transcript binds downstream of the insertion site and is responsible for the next two steps of the integration reaction: cleavage of the top strand (step 3), and the synthesis of the second DNA strand primed from the released 3′-end of the top strand (step 4).20,39 Although this last step of the integration reaction has not been observed in vitro, the results in this report demonstrate that the R2 protein has the DNA-directed polymerase and strand-displacement activities needed to carry out this final step. Once initiated, second-strand synthesis will be highly efficient, because the processivity of the R2 polymerase in synthesizing the second DNA strand while displacing RNA is even higher than its processivity in reverse transcribing RNA to make the first DNA strand (compare -Poff values in Tables 1 and 2). Our results also suggest that the displacement activity may enable the polymerase to proceed past the R2 sequences into the flanking chromosomal sequences during both first- and second-strand synthesis.
Finally in this report we detected a previously unrecognized cleavage activity of the R2 protein: the R2 endonuclease can cleave ssDNA at its junction with duplex DNA (Figure 7). We propose this activity may be related to the second-strand cleavage activity of the R2 protein (step 3 in Figure 8). When viewed from the 3′ end of the element, the location of all R2 insertions within the 28S rRNA gene are identical in all animal taxa.9,10 This precision is determined by the highly sequence-specific cleavage of the first DNA strand.4,7,8 On the other hand, the location of R2 insertions when viewed from their 5′ end varies between elements from the same or different species. This variation is proposed to result from second-strand cleavage at different position either upstream of downstream of the first stand cleavage site.28 Cleavage upstream results in small target site deletions, while cleavage downstream results in target site duplications. This variability suggests the R2 endonuclease does not recognize a specific sequence, but simply cleaves the second (top) strand at locations near the first (bottom) strand cleavage site. We propose that binding of the R2 proteins and the utilization of the nicked bottom strand to prime reverse transcription (step 2 in Figure 7) results in local denaturation of the DNA at the insertion site. The target site would thus contain a ssDNA:dsDNA junction which could be cleaved by the endonuclease. Further work on this ssDNA cleavage activity and its potential use to prime second-strand DNA synthesis is needed, as it represents the single remaining step needed in a complete in vitro reconstitution of R2 retrotransposition.
Figure 8.

Current model of R2 retrotransposition. The proposed mechanism involves an ordered series of cleavage and polymerization reactions carried out by two R2 protein subunits (light and dark gray ovals). Each R2 subunit binds an RNA segment from either the 3′ end or near the 5′ end of the R2 transcript (wavy lines). Binding of this RNA determines whether the protein will bind upstream or downstream of the DNA target site and thus conducts the first two or the last two steps of the integration. The endonuclease of the subunit bound upstream of the target site (darker oval binding the 3′ end of the transcript) nicks the bottom strand (step 1). The RT of this upstream subunit uses the 3′ DNA end released by the cleavage to prime reverse transcription of the R2 transcript (step 2). This step has been termed target primed reverse transcription (TPRT). When the synthesis of the first strand is completed, the second subunit which binds downstream of the target site (lighter oval binding the 5′ end of the transcript) cleaves the top DNA strand (step 3). The RT of this downstream subunit uses the 3′ DNA end released by this cleavage to prime second-strand DNA synthesis (step 4). See references 20 and 41 for a detailed description of the evidence for this model.
MATERIALS AND METHODS
DNA and RNA Templates
The 560 nt and 320 nt single stranded DNA (ssDNA) templates were derived from the ssDNA bacteriophage M13mp18 vector (Invitrogen). To generate the DNA, oligonucleotides complementary to regions containing the BglII and PvuII restriction endonuclease recognition sites at positions 6934 and 6376 (5′-GCTATTTTTGAGAGATCTACAAAGGCTATC-3′ and 5′-GCTATTACGCCAGCTGGCGAAAGGGG-3′) or 6054 and 6376 (5′-CCTGTCGTGCCAGCTGCATTAATGAATC-3′) were annealed to the vector DNA. The complex was then cleaved with BglII and PvuII or PvuII only, respectively, and the digestion products purified using the Qiaquick purification kit (Qiagen).
RNA templates and blocks were generated in vitro by transcription using the T7 RNA polymerase (AmpliScribe™ T7-Flash™ Transcription Kit, Epicentre). The DNA templates used for RNA synthesis were obtained by PCR amplification of M13mp18 using the primers 5′-GCGTAATACGACTCACTATAGCTGGCACGACAGGTTTCCCG-3′ and 5′-CTGGCGAAAGGGGGATGTGC-3′ for the 320 nt RNA template, primers 5′-GCGTAATACGACTCACTATAGACCAATAGGAACGCCATC-3′ and 5′-CTGGCGTAATAGCGAAGAGGCC-3′ for the 340 nt RNA block, and primers 5′-GCGTAATACGACTCACTATAGGCATAAAGTGTAAAGCCTGGGG-3′ and 5′-CTGGCACGACAGGTTTCCCG-3′ for the 100 nt block. All PCR products were purified from agarose gels before RNA synthesis. The internally [α-32P]-labeled 100 nt RNA used in the RNase H assay was synthesized as above with the addition of [α-32P]-labeled UTP in the polymerization reaction. The 100 nt DNA block (5′-GCATAAAGTGTAAAGCCTGGGGTG CCTAATGAGTGAGCTAACTCACATTAATTGCGTTGCGCTCACTGCCCGCTTTCCAGT CGGGAAACCTGTCGTGCCA-3′), the 60 nt block (same 3′ end sequence as the 100 nt DNA block but is 40 nt shorter on its 5′ end), and the 100 nt ssDNA used to monitor template jumping (5′-TGCGCGTCACTAATTAGATGACGAGGCATTTGGCTACCTTAAGAGAGTCATAGTT ACTCCCGCCGTTTACCCGCGCTTGCTTGAATTTCTTCACGTTGAC-3′) were obtained by chemical synthesis of the oligonucleotide (Integrated DNA Technologies, Inc.).
Protein purification
R2 protein of Bombyx mori was expressed in Escherichia coli JM109/pR260 and purified as previously described.4,20 Enzymatic activity was equilibrated between preparation by measuring the efficiency of cDNA synthesis on a poly(rA)/poly(dT)13–18 template with α-32P-labeled dTTP.
Processivity assays
The R2 polymerase processivity assays were performed in 30 μl volumes. To the 3′-end of either the 320 nt ssDNA or RNA template, a [γ32P] 5′-end labeled DNA primer SD-1 (5′-CTGGCGAAAGGGGGATGTGC-3′) was first annealed to prime the reaction. 300 fmol of the DNA or RNA template-primer complex was preincubated for 10 min at room temperature with approximately 40 fmol of the R2 protein in 50 mM Tris HCl (pH 7.5), 0.2 M NaCl, 10 mM MgCl2, 2.5 mM DTT, 0.01% Triton X-100 (standard R2 reaction buffer). Reactions were started by the simultaneous addition of 20 μg of heparin, 300 ng of poly(rA)/poly(dT)13–18 and each dNTP to a final concentration of 0.5 mM. In the control reaction, the heparin and poly(rA)/poly(dT) was added in the beginning of the preincubation (before the R2 protein was added) to prevent the initiation of DNA synthesis. Extension reactions were for 20 min at 37°C and stopped by the addition of 3 volumes of 95% ethanol containing 0.3 M sodium acetate (pH 5.3) and 0.1% SDS. After precipitation, the products were dissolved in 15 μl of 7 M urea loading dye, separated on a denaturing 6% polyacrylamide-7 M urea gels and the relative intensity of the different length extension products quantified using a PhosphorImager.
RNase H activity assay
The RNA:DNA hybrid was prepared by incubating the internally labeled 100 nt RNA and the complementary 30 nt DNA (5′-GTGAGCGCAACGCAATTAATGTGAGTTAGC-3′) at 95°C for 5 min and slow cooling for 2 hours. The reaction mixture was in a total volume of 10 μl and contained the above described extension buffer, 3 pmol RNA:DNA hybrid and approximately 40 fmol of the R2 protein. The incubations were for 20 min. at 37°C and stopped by addition of 3 volumes of 7 M urea loading buffer. The products were analyzed on a denaturing 8% polyacrylamide-7 M urea gels. The reaction with MMLV reverse transcriptase (Promega) was conducted under the same conditions with 100 units of enzyme.
Strand displacement synthesis assays
The DNA:DNA, DNA:RNA, or ssDNA templates were prepared in 30 μl volumes by heating 300 fmol of the template with the appropriate 5′-end labeled DNA primer SD-2 (5′-TACAAAGGCTATCAGGTC-3′) or SD-1 and the DNA or RNA block (1:10 and 1:5 molar ratio to the template, respectively) in the R2 reaction buffer at 95°C for 5 min, and slow cooling for 90 minutes. The complexes were preincubated either with 40 fmol of the R2 protein or 5 units of T4 DNA polymerase (Fermentas) for 10 min at room temperature. Reactions were started by the addition of dNTPs to the final concentration of 0.5 mM for each nucleotide, incubated at 25°C, 37°C or 42°C and stopped after 20 min. by addition of 3 volumes of 95% ethanol containing 0.3 M sodium acetate (pH 5.3) and 0.1% SDS.
Sequencing the junctions of the template jumps
The band corresponding to the jump product was excised from a polyacrylamide gel, eluted with 10% sodium acetate (pH 5.2) for several hours at room temperature and ethanol precipitated. The extracted DNA was used as template for PCR amplification using the primers 5′-CTGGCGAAAGGGGGATGTGC-3′ and 5′-TGCGCGTCACTAATTAGATGA-3′. The amplified PCR products from four separate PCR reactions were then cloned into the TA cloning vector pCR2.1 (Invitrogen, Carlsbad, CA), followed by transformation into DH5α (Invitrogen) and plated on 2XYT/Amp/X-gal/IPTG plates for blue-white selection. Sixteen white colonies confirmed to contain the appropriate sized insertion by EcoR I digestion were sequenced using TOPO Rev primer 5′-CAGTGAATTGTAATACGACTCA-3′.
Nuclease activity assay
The DNA:DNA hybrids were prepared by annealing of 5′-end labeled 100 nt ssDNA with complementary 30 nt ssDNA (5′-GTGAGCGCAACGCAATTAATGTGAGTTAGC-3′) or 20 nt ssDNA (5′-CTGGCACGACAGGTTTCCCG-3′) in the R2 reaction buffer. Components were placed at 95°C for 5 min and slowly cooled for 2 hours. Cleavage reactions were in a total volume of 10μl and contained R2 reaction buffer, 3 pmol DNA:DNA hybrid and 40 fmol of the R2 protein (EN+, wild type or EN−, the single-amino-acid mutation D966A R2 protein8,20). The samples were incubated for 20 min. at 37°C and stopped by addition of 3 volumes of 7 M urea loading buffer. Products were separated by electrophoresis in an 8% denaturing polyacrylamide-7 M urea gel and analyzed by PhosphorImager.
Acknowledgments
This work was supported by National Institutes of Health Public Health Services Grant GM42790. We thank S. M. Christensen for discussions and providing the single-amino-acid mutation D966A R2 protein (R2 EN−), and W. D. Burke with assistance in sequencing the end-to-end jumping products. We also thank D. Eickbush for comments on the manuscript.
Abbreviations
- RT
reverse transcriptase
- LTR
long terminal repeat
- TPRT
target primed reverse transcription
- EN
endonuclease
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ostertag EM, Kazazian HH., Jr Biology of mammalian L1 retrotransposons. Annu Rev Genet. 2001;35:501–538. doi: 10.1146/annurev.genet.35.102401.091032. [DOI] [PubMed] [Google Scholar]
- 2.Whitcomb JM, Hughes SH. Retroviral reverse transcription and integration: progress and problems. Annu Rev Cell Biol. 1992;8:275–306. doi: 10.1146/annurev.cb.08.110192.001423. [DOI] [PubMed] [Google Scholar]
- 3.Voytas DF, Boeke JD. Ty1 and Ty5 of Sacharomyces cerevisiae. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. American Society for Microbiology; Washington DC: 2002. pp. 631–662. [Google Scholar]
- 4.Luan DD, Korman MH, Jakubczak JL, Eickbush TH. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell. 1993;72:595–605. doi: 10.1016/0092-8674(93)90078-5. [DOI] [PubMed] [Google Scholar]
- 5.Dewannieux M, Esnault C, Heidmann T. LINE-mediated retrotransposition of marked Alu sequences. Nat Genet. 2003;35:41–48. doi: 10.1038/ng1223. [DOI] [PubMed] [Google Scholar]
- 6.Dewannieux M, Heidmann T. LINEs, SINEs and processed pseudogenes: parasitic strategies for genome modeling. Cytogenet Genome Res. 2005;110:35–48. doi: 10.1159/000084936. [DOI] [PubMed] [Google Scholar]
- 7.Luan DD, Eickbush TH. RNA template requirements for target DNA-primed reverse transcription by the R2 retrotransposable element. Mol Cell Biol. 1995;15:3882–3891. doi: 10.1128/mcb.15.7.3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang J, Malik HS, Eickbush TH. Identification of the endonuclease domain encoded by R2 and other site-specific, non-long terminal repeat retrotransposable elements. Proc Natl Acad Sci U S A. 1999;96:7847–7852. doi: 10.1073/pnas.96.14.7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eickbush TH. In: Mobile DNA II. Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. American Society of Microbiology Press; Washington DC: 2002. pp. 813–835. [Google Scholar]
- 10.Kojima KK, Kuma K, Toh H, Fujiwara H. Identification of rDNA-specific non-LTR retrotransposons in Cnidaria. Mol Biol Evol. 2006;23:1984–1993. doi: 10.1093/molbev/msl067. [DOI] [PubMed] [Google Scholar]
- 11.Bibillo A, Eickbush TH. The reverse transcriptase of the R2 non-LTR retrotransposon: continuous synthesis of cDNA on non-continuous RNA templates. J Mol Biol. 2002;316:459–473. doi: 10.1006/jmbi.2001.5369. [DOI] [PubMed] [Google Scholar]
- 12.Bibillo A, Eickbush TH. End-to-end template jumping by the reverse transcriptase encoded by the R2 retrotransposon. J Biol Chem. 2004;279:14945–14953. doi: 10.1074/jbc.M310450200. [DOI] [PubMed] [Google Scholar]
- 13.Bibillo A, Eickbush TH. High processivity of the reverse transcriptase from a non-long terminal repeat retrotransposon. J Biol Chem. 2002;277:34836–34845. doi: 10.1074/jbc.M204345200. [DOI] [PubMed] [Google Scholar]
- 14.Lanciault C, Champoux JJ. Single unpaired nucleotides facilitate HIV-1 reverse transcriptase displacement synthesis through duplex RNA. J Biol Chem. 2004;279:32252–32261. doi: 10.1074/jbc.M404117200. [DOI] [PubMed] [Google Scholar]
- 15.Wisniewski M, Balakrishnan M, Palaniappan C, Fay PJ, Bambara RA. Unique progressive cleavage mechanism of HIV reverse transcriptase RNase H. Proc Natl Acad Sci U S A. 2000;97:11978–11983. doi: 10.1073/pnas.210392297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malik HS, Burke WD, Eickbush TH. The age and evolution of non-LTR retrotransposable elements. Mol Biol Evol. 1999;16:793–805. doi: 10.1093/oxfordjournals.molbev.a026164. [DOI] [PubMed] [Google Scholar]
- 17.Malik HS, Eickbush TH. Phylogenetic analysis of ribonuclease H domains suggests a late, chimeric origin of LTR retrotransposable elements and retroviruses. Genome Res. 2001;11:1187–1197. doi: 10.1101/gr.185101. [DOI] [PubMed] [Google Scholar]
- 18.Kelleher CD, Champoux JJ. Characterization of RNA strand displacement synthesis by Moloney Murine Leukemia Virus reverse transcriptase. J Biol Chem. 1998;273:9976–9986. doi: 10.1074/jbc.273.16.9976. [DOI] [PubMed] [Google Scholar]
- 19.Xiong YE, Eickbush TH. Functional expression of a sequence-specific endonuclease encoded by the retrotransposon R2Bm. Cell. 1988;55:235–246. doi: 10.1016/0092-8674(88)90046-3. [DOI] [PubMed] [Google Scholar]
- 20.Christensen SM, Eickbush TH. R2 target-primed reverse transcription: ordered cleavage and polymerization steps by protein subunits asymmetrically bound to the target DNA. Mol Cell Biol. 2005;25:6617–6628. doi: 10.1128/MCB.25.15.6617-6628.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moran JV, Holmes SE, Nass PT, DeBarardinis RJ, Boeke JD, Kazazian HH., Jr High frequency retrotransposition in cultured mammalian cells. Cell. 1996;87:917–927. doi: 10.1016/s0092-8674(00)81998-4. [DOI] [PubMed] [Google Scholar]
- 22.Cost GJ, Feng Q, Jacquier A, Boeke JD. Human L1 element target-primed reverse transcription in vitro. EMBO J. 2002;21:5899–5910. doi: 10.1093/emboj/cdf592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Piskareva O, Schmatchenko V. DNA polymerization by the reverse transcriptase of the human L1 retrotransposon on its own template in vitro. FEBS Lett. 2006;580:661–668. doi: 10.1016/j.febslet.2005.12.077. [DOI] [PubMed] [Google Scholar]
- 24.Whiting SH, Champoux JJ. Properties of strand displacement synthesis by Moloney Murine Leukemia Virus reverse transcriptase: mechanistic implications. J Mol Biol. 1998;278:559–577. doi: 10.1006/jmbi.1998.1720. [DOI] [PubMed] [Google Scholar]
- 25.Winshell J, Paulson BA, Buelow BD, Champoux JJ. Requirements for DNA unpairing during displacement synthesis by HIV-1 reverse transcriptase. J Biol Chem. 2004;279:52924–52933. doi: 10.1074/jbc.M409134200. [DOI] [PubMed] [Google Scholar]
- 26.Kew Y, Olsen L, Japour A, Prasad VR. Insertions into the β3–β4 hairpin loop of HIV-1 reverse transcriptase reveal a role for fingers subdomain in processive polymerization. J Biol Chem. 1998;273:7529–7537. doi: 10.1074/jbc.273.13.7529. [DOI] [PubMed] [Google Scholar]
- 27.Fisher TS, Darden T, Prasad VR. Substitutions at Phe61 in the beta3–beta4 hairpin of HIV-1 reverse transcriptase reveal a role for the Fingers subdomain in strand displacement DNA synthesis. J Mol Biol. 2003;325:443–459. doi: 10.1016/s0022-2836(02)01225-1. [DOI] [PubMed] [Google Scholar]
- 28.Burke WD, Malik HS, Jones JP, Eickbush TH. The domain structure and retrotransposition mechanism of R2 elements are conserved throughout arthropods. Mol Biol Evol. 1999;16:502–511. doi: 10.1093/oxfordjournals.molbev.a026132. [DOI] [PubMed] [Google Scholar]
- 29.Blocker FJ, Mohr G, Conlan LH, Qi L, Belfort M, Lambowitz AM. Domain structure and three-dimensional model of a group II intron-encoded reverse transcriptase. RNA. 2005;11:14–28. doi: 10.1261/rna.7181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zimmerly S, Guo H, Perlman PS, Lambowitz AM. Group II intron mobility occurs by target DNA-primed reverse transcription. Cell. 1995;82:545–554. doi: 10.1016/0092-8674(95)90027-6. [DOI] [PubMed] [Google Scholar]
- 31.Smith D, Zhong J, Matsuura M, Lambowitz AM, Belfort M. Recruitment of host functions suggests a repair pathway for late steps in group II intron retrohoming. Genes Dev. 2005;19:2477–2487. doi: 10.1101/gad.1345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor AF, Smith GR. RecBCD enzyme is a DNA helicase with fast and slow motors of opposite polarity. Nature. 2003;423:889–893. doi: 10.1038/nature01674. [DOI] [PubMed] [Google Scholar]
- 33.Dekker J, Kanellopoulos PN, Loonstra AK, van Oosterhout JA, Leonard K, Tucker PA, van der Vliet PC. Multimerization of the adenovirus DNA-binding protein is the driving force for ATP-independent DNA unwinding during strand displacement synthesis. EMBO J. 1997;16:1455–1463. doi: 10.1093/emboj/16.6.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamtekar S, Berman AJ, Wang J, Lazaro JM, de Vega M, Blanco L, Salas M, Steitz TA. Insights into strand displacement and processivity from the crystal structure of the protein-primed DNA polymerase of bacteriophage phi29. Mol Cell. 2004;16:609–618. doi: 10.1016/j.molcel.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 35.Zhu W, Ito J. Purification and characterization of PRD1 DNA polymerase. Biochem Biophys Acta. 1994;1219:267–276. doi: 10.1016/0167-4781(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 36.Fujimura RK, Roop BC. Characterization of DNA polymerase induced by bacteriophage T5 with DNA containing single strand breaks. J Biol Chem. 1976;251:2168–2174. [PubMed] [Google Scholar]
- 37.Blanco L, Bernad A, Lazaro JM, Martin G, Garmendia C, Salas M. Highly efficient DNA synthesis by the phage phi 29 DNA polymerase. Symmetrical mode of DNA replication. J Biol Chem. 1989;264:8935–8940. [PubMed] [Google Scholar]
- 38.Christensen SM, Bibillo A, Eickbush TH. Role of the Bombyx mori R2 element N-terminal domain in the target-primed reverse transcription (TPRT) reaction. Nucleic Acids Res. 2005;33:6461–6468. doi: 10.1093/nar/gki957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Christensen SM, Ye J, Eickbush TH. RNA from the 5′end of the R2 retrotransposon controls R2 protein binding to and cleavage of its DNA target site. Proc Natl Acad Sci U S A. 2006;103:17602–17607. doi: 10.1073/pnas.0605476103. [DOI] [PMC free article] [PubMed] [Google Scholar]