

Abstract
Patients with Familial adenomatous polyposis coli (FAP) may rarely develop hepatocellular adenoma. Here we report the case of a 37 years old FAP woman presenting a hepatocellular adenoma after oestroprogestative oral contraception use. In this steatotic adenoma, we identified an inactivating biallelic mutation of HNF1α. In addition to the known germline APC mutation Q1062fs, we did not find an inactivation of the second APC allele nor an activation of the β-catenin target genes GLUL and GPR49. Our findings contrast with two hepatocellular adenoma cases related to FAP, for which a biallelic inactivation of the APC gene was previously described. Altogether, these results suggest that benign hepatocellular carcinogenesis may be dependent or independent of the Wnt/β-catenin pathway in patients with FAP.
Keywords: Hepatocellular adenoma, Familial adenosis polyposis coli, Hepatocyte Nuclear Factor 1, β-catenin, gene mutation
Keywords: Adenomatous Polyposis Coli; complications; genetics; Adult; Alleles; Carcinoma, Hepatocellular; complications; genetics; pathology; Diagnosis, Differential; Female; Gene Silencing; Genes, APC; Genetic Predisposition to Disease; Germ-Line Mutation; Hepatocyte Nuclear Factor 1-alpha; genetics; Humans; Immunohistochemistry; Liver Neoplasms; complications; genetics; pathology; Reverse Transcriptase Polymerase Chain Reaction
Introduction
Hepatocellular adenomas (HCA) are benign liver tumors, most frequently occurring in women using oral contraception (1). The other risk factors for HCA are glycogen storage diseases and androgen therapy. However, HCA are rare tumors, their estimated incidence in France being approximately one case per 100,000 women.
These tumors result from a benign proliferation of hepatocytes that destroy the normal architecture of the liver. They are usually hyper-vascularised and typical adenomas correspond to a proliferation of benign hepatocytes, intermingled with numerous thin-walled vessels, without portal tracts (2).
Molecular analysis of HCA revealed that half of the adenoma cases are mutated for the TCF1 gene, encoding HNF1α (3). These mutations are inactivating and both alleles are mutated in tumors. Patients with an inherited mutation in one allele of HNF1α may develop maturity onset diabetes of the young type 3 (MODY3, OMIM#600496) and familial liver adenomatosis, when the second allele is inactivated in hepatocytes by somatic mutation or chromosome deletion (3–5). Mutations of CTNNB1, activating the β-catenin was also found in 15% of the HCA cases (6). Recently, we established a molecular and pathological classification of hepatocellular adenomas and showed strong genotype-phenotype correlations in 96 analysed cases (7). We also showed that adenomas with β-catenin activation had a higher risk of malignant transformation.
HCA is also a rare extracolonic tumor developed in patients presenting familial adenomatous polyposis coli (FAP). This syndrome is characterized by an early onset of multiple colonic adenomatous polyps that progress to colon carcinomas (OMIM: #175100 (8, 9)). In addition to colorectal tumors, FAP patients may develop extracolonic tumors, mainly desmoid tumors, adenomas and carcinomas of the upper gastrointestinal tract. In colorectal tumors associated to FAP, a biallelic inactivation of the APC gene is consistently found and inactivation of the APC gene in tumors leads to the β-catenin accumulation and therefore to the activation of the Wnt/wingless pathway. A biallelic inactivation of the APC gene was also recently described in extracolonic associated tumors and particularly in two cases of hepatocellular adenoma developed in FAP patients (10, 11).
We tested an HCA found in a FAP woman for particular genetic alterations. We looked for mutations in TCF1/HNF1α, CTNNB1, encoding β-catenin, and TP53 genes. Activation of the β-catenin pathway was qualified both by immunohistochemistry to detect β-catenin and Glutamine Synthetase, and by quantification of β-catenin target genes GLUL, encoding the Glutamine Synthetase and GPR49, encoding an orphan nuclear receptor, using quantitative RT-PCR.
Material and Methods
Patient and samples
A 22 years old woman was treated for FAP in 1983 by a total colectomy and ileo-rectal anastomosis for FAP. Pathology showed multiple adenomatous polyps with low grade dysplasia. An APC germline mutation, 3184–3187del leading to Q1062fs was identified.
She was placed under oestroprogestative oral contraception use during 5 years from 1986 to 1991. Compliance to endoscopic follow-up of the rectum was poor and she was convinced to be reoperated in 2001 in order to perform proctectomy and ileo-anal anastomosis. Pre-operative abdominal ultrasound performed to explore intermittent slight right upper abdominal pain revealed a slightly hyper-echoïc homogeneous 7 cm mass on the right liver. Magnetic Resonance Imaging showed a T1 hypointense homogeneous mass of liver segment VI, slighty hyper intense in T2 sequence and moderately enhanced at arterial phase by intravenously injected contrast. The definite diagnosis of hepatic adenoma was confirmed by intra-operative biopsies of the tumor and of non-tumor tissues performed during proctectomy. Pathology of the resected rectum showed multiple adenomatous polyps with low grade dysplasia. Two weeks after proctectomy, sudden right hypocondrium pain and hypotension occurred, attributed by CT-scan to an intra tumorous hemorrhage of the adenoma. Conservative treatment was chosen and successful. A right hepatectomy was performed a few months later to remove this symptomatic adenoma, which size of 7 cm remained stable after resorption of the intra-tumorous hematoma. Tumor and non-tumor liver samples were frozen immediately after surgery and stored at −80°C. The liver specimen was then fixed in 10% buffered formalin. Liver samples were paraffin embedded. Post-operative course was uneventful. The patient was re-operated in 2005 to resect two recent desmoid tumors of 10 and 6 cm developed in the anterior abdominal wall.
Mutation screening
Exons 1 to 10 of TCF1, 2 to 4 of CTNNB1 and codons 1001 to 1111 of APC were screened for mutations in the hepatocellular adenoma, using direct sequencing of the exons after amplification of the genomic DNA. Protocols were previously described in Bluteau et al. and Laurent Puig et al. (3, 12) and are available upon request.
Quantitative RT-PCR
RNA was extracted using RNeasy kit (Qiagen, Valencia, CA) and quantitative RT-PCR was performed to quantify the mRNA expression level of GLUL and GPR49, two target genes of β-catenin in the liver (13, 14) as previously described (7). The results were normalized using the expression level of the ribosomal RNA R18S and expressed as the n-fold ratio of the gene expression in the tested sample compared with the mean of 11 non tumor liver tissues. We used as positive controls 10 samples identified as activated for β-catenin from our previous study (7).
Immunohistochemistry
Paraffin sections were stained with hematein-eosin. Immunostainings were performed with an anti-β-catenin antibody (SantaCruz) and an anti-Glutamine synthetase antibody (BD Transduction Laboratories).
Results and discussion
On gross examination the lesion measured 7 cm and was well demarcated from the surrounding liver, but not encapsulated. It was soft, yellow with no hemorraghe or necrosis (Figure 1A). Light microscopy observation revealed that it was composed of benign appearing hepatocytes arranged in two-cell thick plates separated by sinusoids and intermingled with numerous thin-walled vessels. Hepatocytes showed a marked steatosis (Figure 1B). There was no portal triad, ductules, fibrosis, hemorrhage, peliosis or necrosis within the lesion. The surrounding non-tumor liver was normal.
Figure 1.
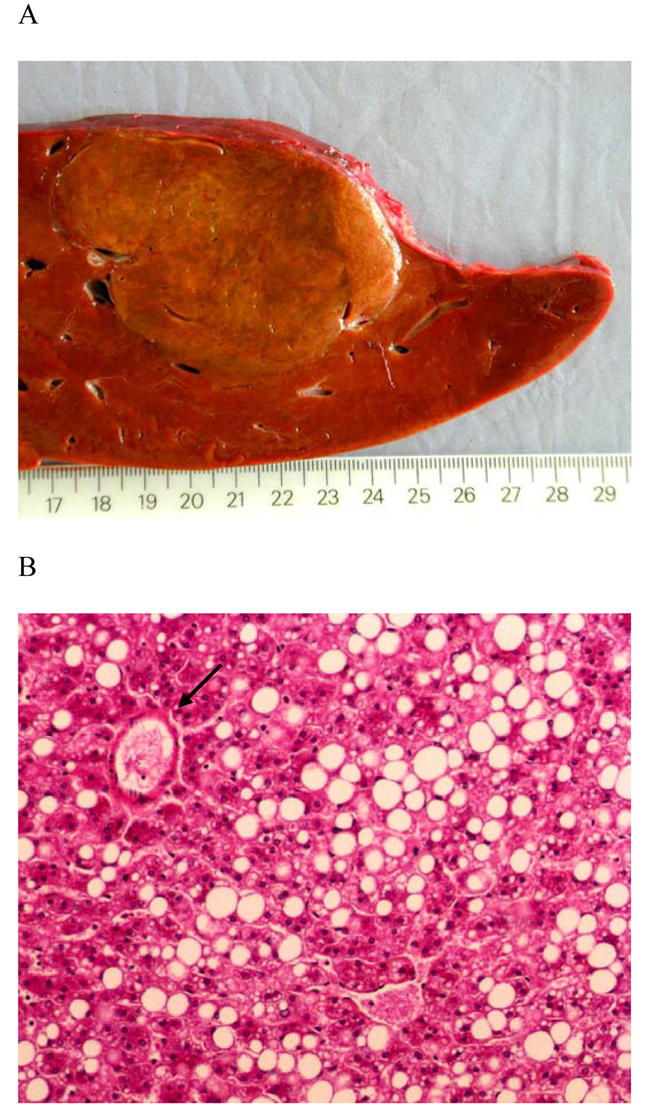
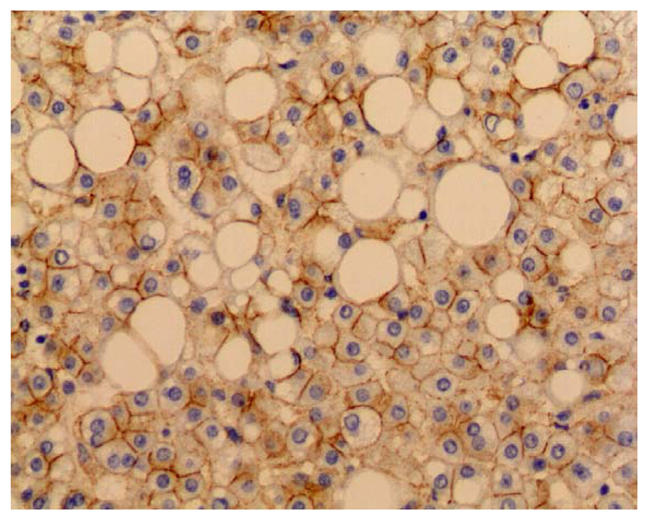
A: Macroscopy: the liver lesion measured 7 cm, was soft, yellow, well demarcated with no hemorraghe or necrosis, B: Microscopy: the lesion was composed of benign appearing hepatocytes with marked steatosis, intermingled with thin walled vessels (arrow) (HES staining, original magnification ×100) C: β-catenin immunostaining: hepatocytes plasma membrane staining with no nuclear reactivity in the hepatocellular adenoma (original magnification ×200)
A somatic mutation of HNF1α (787 C>T) leading to an amino acid substitution (R263S) was found in the adenoma sample. The mutation appeared homozygous at the RNA level. Moreover, genotyping of 3 polymorphisms in the HNF1α gene comparing the non-tumor and tumor samples indicated a loss of heterozygosity (LOH) in the tumor. Altogether, these results showed a somatic mutation of one HNF1α allele and a somatic deletion of the second HNF1α allele leading to a biallelic inactivation of the protein.
Sequencing of the APC gene revealed a heterozygous 4 nucleotides deletion at codon 1062, leading to a frameshift. Relative intensity of the mutated peak compared to the normal one was similar in both the tumor and the non-tumor sample, indicating the absence of a loss of heterozygosity at this locus. No additional somatic mutations of the APC gene were found in exon 14. No mutations were identified in CTNNB1, gene encoding the β-catenin. Consistently with the absence of a second event of mutation in APC and the absence of β-catenin mutation, no activation of the Wnt/β-catenin pathway was detected in the tumor. Indeed, β-catenin target genes were expressed at the same level in the adenoma and in the non-tumor liver samples of the FAP patient even when compared to control non-mutated normal liver tissues. The lack of Wnt/β-catenin activation was confirmed by immunohistochemistry: β-catenin immunostaining was observed at the hepatocyte plasma membranes, and no overexpression or nuclear reactivity in the adenoma was detected (Figure 1C). Moreover, there was no Glutamine Synthetase overexpression in the adenoma compared to the non tumor tissue.
Two hepatocellular adenoma cases, developed in FAP patients, previously described in the literature were described as displaying a biallelic APC inactivation (10, 11). The first patient was a 2-years-old girl presenting a non-sense APC germline mutation at codon 1451 and a deletion of the second allele in the tumor. In the same tumor, a TP53 mutation (R175H) was found. This last result was unusual since TP53 mutations are observed in 20 to 50% of the cases of hepatocellular carcinoma (15), whereas no TP53 mutation were found in a series of 13 adenomas in Taiwan (6) nor in a series of 30 adenomas in our lab (unpublished data). The second case described was a hepatocellular adenoma developed in a 27-years-old woman presenting a germline APC mutation at codon 1156 associated with a somatic APC mutation at codon 1464, leading to a cytoplasmic accumulation of β-catenin in the hepatocellular adenoma (11). In this last case the information about an oral contraceptive use was not available.
In this present work, morphological characteristics of the hepatocellular adenoma (i.e. steatosis in more than 30% of the hepatocytes with no cytological abnormalities, no inflammatory infiltrate and lobulated frontiers of the tumors) were characteristic of the HNF1α mutated cases described in our recent hepatocellular adenoma classification (7). It is very different from the β-catenin mutated adenomas that usually exhibit cytological abnormalities and pseudoglandular formations, without steatosis.
In conclusion, the present case shows that apart from the Wnt/β-catenin activation through the complete inactivation of APC, benign hepatocarcinogenesis can also occur by an inactivation of the tumor suppressor gene encoding HNF1α in a patient with FAP.
Acknowledgments
We thank Cristel Thomas for critical reading of this manuscript, Sylviane Olschwang for the genetic data, Benoît Terris for Glutamine Synthetase immunohistochemistry and Sandra Rebouissou for her help. We thank all the other members of the GENTHEP (Groupe d’étude Génétique des Tumeurs Hépatiques) network. This work was supported by the Association pour la Recherche sur le Cancer (ARC n°3108), the INSERM (Réseaux de recherche clinique et réseaux de recherche en santé des populations), the Fondation de France and the SNFGE. EJ is supported by an ARC doctoral fellowship.
References
- 1.Edmondson HA, Henderson B, Benton B. Liver-cell adenomas associated with use of oral contraceptives. N Engl J Med. 1976;294:470–472. doi: 10.1056/NEJM197602262940904. [DOI] [PubMed] [Google Scholar]
- 2.Terminology of nodular hepatocellular lesions. International Working Party. Hepatology. 1995;22:983–993. doi: 10.1016/0270-9139(95)90324-0. [DOI] [PubMed] [Google Scholar]
- 3.Bluteau O, Jeannot E, Bioulac-Sage P, Marques JM, Blanc JF, Bui H, Beaudoin JC, et al. Bi-allelic inactivation of TCF1 in hepatic adenomas. Nat Genet. 2002;32:312–315. doi: 10.1038/ng1001. [DOI] [PubMed] [Google Scholar]
- 4.Bacq Y, Jacquemin E, Balabaud C, Jeannot E, Scotto B, Branchereau S, Laurent C, et al. Familial liver adenomatosis associated with hepatocyte nuclear factor 1alpha inactivation. Gastroenterology. 2003;125:1470–1475. doi: 10.1016/j.gastro.2003.07.012. [DOI] [PubMed] [Google Scholar]
- 5.Reznik Y, Dao T, Coutant R, Chiche L, Jeannot E, Clauin S, Rousselot P, et al. Hepatocyte nuclear factor-1 alpha gene inactivation: cosegregation between liver adenomatosis and diabetes phenotypes in two maturity-onset diabetes of the young (MODY)3 families. J Clin Endocrinol Metab. 2004;89:1476–1480. doi: 10.1210/jc.2003-031552. [DOI] [PubMed] [Google Scholar]
- 6.Chen YW, Jeng YM, Yeh SH, Chen PJ. P53 gene and Wnt signaling in benign neoplasms: beta-catenin mutations in hepatic adenoma but not in focal nodular hyperplasia. Hepatology. 2002;36:927–935. doi: 10.1053/jhep.2002.36126. [DOI] [PubMed] [Google Scholar]
- 7.Zucman-Rossi J, Jeannot E, Nhieu JT, Scoazec JY, Guettier C, Rebouissou S, Bacq Y, et al. Genotype-phenotype correlation in hepatocellular adenoma: new classification and relationship with HCC. Hepatology. 2006;43:515–524. doi: 10.1002/hep.21068. [DOI] [PubMed] [Google Scholar]
- 8.Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 9.Joslyn G, Carlson M, Thliveris A, Albertsen H, Gelbert L, Samowitz W, Groden J, et al. Identification of deletion mutations and three new genes at the familial polyposis locus. Cell. 1991;66:601–613. doi: 10.1016/0092-8674(81)90022-2. [DOI] [PubMed] [Google Scholar]
- 10.Bala S, Wunsch PH, Ballhausen WG. Childhood hepatocellular adenoma in familial adenomatous polyposis: mutations in adenomatous polyposis coli gene and p53. Gastroenterology. 1997;112:919–922. doi: 10.1053/gast.1997.v112.pm9041254. [DOI] [PubMed] [Google Scholar]
- 11.Blaker H, Sutter C, Kadmon M, Otto HF, Von Knebel-Doeberitz M, Gebert J, Helmke BM. Analysis of somatic APC mutations in rare extracolonic tumors of patients with familial adenomatous polyposis coli. Genes Chromosomes Cancer. 2004;41:93–98. doi: 10.1002/gcc.20071. [DOI] [PubMed] [Google Scholar]
- 12.Laurent-Puig P, Legoix P, Bluteau O, Belghiti J, Franco D, Binot F, Monges G, et al. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology. 2001;120:1763–1773. doi: 10.1053/gast.2001.24798. [DOI] [PubMed] [Google Scholar]
- 13.Cadoret A, Ovejero C, Terris B, Souil E, Levy L, Lamers WH, Kitajewski J, et al. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene. 2002;21:8293–8301. doi: 10.1038/sj.onc.1206118. [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto Y, Sakamoto M, Fujii G, Tsuiji H, Kenetaka K, Asaka M, Hirohashi S. Overexpression of orphan G-protein-coupled receptor, Gpr49, in human hepatocellular carcinomas with beta-catenin mutations. Hepatology. 2003;37:528–533. doi: 10.1053/jhep.2003.50029. [DOI] [PubMed] [Google Scholar]
- 15.Laurent-Puig P, Plomteux O, Bluteau O, Zinzindohoue F, Jeannot E, Dahan K, Kartheuser A, et al. Frequent mutations of hepatocyte nuclear factor 1 in colorectal cancer with microsatellite instability. Gastroenterology. 2003;124:1311–1314. doi: 10.1016/s0016-5085(03)00268-3. [DOI] [PubMed] [Google Scholar]