

Abstract
It has long been suspected that maize is the product of an historical tetraploid event. Several observations support this possibility, including the fact that the maize genome contains duplicated chromosomal segments with colinear gene arrangements. Some of the genes from these duplicated segments have been sequenced. In this study, we examine the pattern of sequence divergence among 14 pairs of duplicated genes. We compare the pattern of divergence to patterns predicted by four models of the evolution of the maize genome—autotetraploidy, genomic allotetraploidy, segmental allotetraploidy, and multiple segmental duplications. Our analyses indicate that coalescent times for duplicated sequences fall into two distinct groups, corresponding to roughly 20.5 and 11.4 million years. This observation strongly discounts the possibility that the maize genome is the product of a genomic allotetraploid event, and it is also difficult to reconcile with either autotetraploidy or multiple independent segmental duplications. However, the presence of two (and only two) coalescent times is predicted by the segmental allotetraploid model. If the maize genome is the product of a segmental allotetraploid event, as these data suggest, then its two diploid progenitors diverged roughly 20.5 million years ago (Mya), and the allotetraploid event probably occurred approximately 11.4 Mya. Comparison of maize and sorghum sequences suggests that one of the two ancestral diploids shares a more recent common ancestor with sorghum than it does with the other ancestral diploid.
Keywords: duplicated loci, chromosomal evolution
Because of its economic importance, maize (Zea mays ssp. mays) has long been a focus of genetic and evolutionary analyses. Yet, many aspects of the evolutionary genetics of maize remain a mystery. One such mystery surrounds the origin of the maize genome. Maize belongs to the grass tribe Andropogoneae, which has a haploid or base chromosome number of 5 (1). However, maize and some other members of the Andropogoneae, including sorghum (Sorghum bicolor), have 10 haploid chromosomes (2–4).
The presence of twice the Andropogoneae base chromosome number in maize has prompted several researchers to speculate that maize is of tetraploid origin (2). Some empirical evidence is consistent with this view. For example, it has long been known that the maize genome contains duplicated genes (4), and both isozyme (5) and restriction fragment length polymorphism (RFLP) (6) analyses have demonstrated that the maize genome contains duplicated chromosomal segments with colinear gene arrangements. Although these observations are consistent with the hypothesis of a tetraploid origin, maize is clearly not a tetraploid. Both cytological (7) and RFLP (6) data indicate that the genome does not contain five homeologous chromosome pairs. If maize is the product of an ancient tetraploid event, then the maize genome has since undergone extensive repatterning (6).
As an alternative to ancient tetraploidy, it has been postulated that duplications in the maize genome were produced by multiple independent segmental duplications, whereby chromosomal regions duplicate without changes in ploidy (4–6, 8). This model has been considered less likely because it does not readily account for the doubling of haploid chromosome number in maize (n = 10) relative to the base number (n = 5) for the Andropogoneae.
Sequence data are now available from many of the loci that have been duplicated in maize. The pattern of sequence divergence among these duplicated loci may provide insights into the mode of maize sequence duplication. In this study, we examine four potential models of the evolution of the maize genome, use these models to generate predictions about patterns of sequence coalescence at duplicated loci, and test the predictions with sequence data from duplicated loci.
MODELS AND PREDICTIONS
Duplication in the maize genome could be the result of either segmental duplication or one of several possible modes of tetraploid formation. Each mode of tetraploid formation leads to a different pattern of coalescence between sequences from duplicated loci. Here we consider three known forms (models) of tetraploid formation—autopolyploidy, genomic allopolyploidy, and segmental allopolyploidy—together with segmental duplication.
Autotetraploids, which can result from somatic doubling or the fusion of unreduced gametes within a diploid species, possess four sets of homologous chromosomes. Before the time of chromosome doubling, there is relatively little divergence among alleles at most loci because genetic drift and directional selection limit divergence among alleles. After autotetraploid formation, individual loci exhibit tetrasomic inheritance. During the period of tetrasomic inheritance, each locus contains four alleles, and genetic drift and selection continue to limit diversification among alleles. With the shift from tetrasomic inheritance to disomic inheritance, the single locus becomes two separate but duplicated loci, and sequences from these duplicated loci begin to diverge. Extant sequences sampled from these duplicated loci will have a coalescent time that reflects roughly the time of onset of disomic inheritance. The switch from tetrasomic to disomic inheritance is the key variable in this model. If the switch is coordinated among chromosomes, then all pairs of duplicated sequences are expected to coalesce at roughly the same time (Fig. 1, column A). If the switch from tetrasomic to disomic inheritance is not well coordinated among chromosomes, then coalescent times could be scattered over a broad range.
Figure 1.

Diagrammatic representation of the expected distribution of coalescent times under the four models of chromosomal duplication. Columns: A, The autotetraploid model under the assumption that the switch to disomy is coordinated among chromosomes; B, the genomic allotetraploid model; C, the segmental allotetraploid model; D, the model of segmental duplication.
Allotetraploids typically arise from interspecific hybridization, so that the four chromosome sets of a tetraploid are of two distinct types. “Genomic” allotetraploids exhibit bivalent formation and disomic inheritance (9). With exclusive disomic inheritance, homologous loci from the two ancestral species remain distinct. Sequences from “duplicated” loci have a coalescent time that corresponds to the date of the divergence of the two ancestral diploid species (Fig. 1, column B). The species’ divergence time predates tetraploid formation.
“Segmental” allotetraploids arise from the hybridization of species with only partially differentiated chromosome sets (9). They thus exhibit a mixture of bivalent and tetravalent formation during meiosis. Chromosomal pairs that undergo only bivalent formation contain sequences whose coalescence corresponds to the time of the divergence of the ancestral species (e.g., Fig. 1, column B). Genes located on chromosomes that exhibit tetrasomic inheritance have two possibilities. At tetraploid formation, each tetrasomic locus has four alleles: two from parental species A and two from parental species B, i.e., its genotype is aabb. During the period of tetrasomic inheritance, genetic drift (or selection) could bring the allele(s) of one parent to fixation (aaaa or bbbb), or the locus could remain heterozygous for alleles from both parents (aaab, aabb, or abbb). With the shift to disomic inheritance, the tetrasomic locus becomes two distinct (duplicated) loci. Sequences sampled from duplicated loci that have experienced a period of tetrasomic inheritance can exhibit two distinct coalescent times. If the duplicated loci become fixed for alleles from the same parental species, then the coalescent time of the sequences reflects the time of onset of disomic inheritance (e.g., Fig. 1, column A). Alternatively, if the duplicated loci become fixed for alleles from different parents, then the coalescent time reflects the time of divergence between species A and species B (e.g., Fig. 1, column B). Taken over numerous pairs of duplicated loci, this model predicts a bimodal distribution of coalescent times, with the older coalescent times reflecting the time of species divergence and the more recent coalescent times reflecting the switch from tetrasomic to disomic inheritance (Fig. 1, column C).
An important corollary prediction of the segmental allotetraploid model is that two pairs of duplicated loci can exhibit bimodality of coalescent times, even if they are located on the same chromosome. In the absence of selection, fixation of ancestral parental alleles at a tetrasomic locus is a function of genetic drift. This random process can result in the fixation of alleles from one parent at one tetrasomic locus, whereas a linked tetrasomic locus can remain heterozygous for alleles from both parents. When disomy ensues, the two linked duplicates will exhibit different coalescent times, despite their location on the same chromosome.
Finally, the model of segmental duplication posits that numerous independent events led to the duplication of loci in the maize genome. This model predicts that all loci within a single duplicated chromosomal region diverged at the same time, and this model also predicts that loci on different duplicated chromosomal segments did not necessarily duplicate at the same time (Fig. 1, column D). In other words, the coalescent time of sequences should be related to chromosomal position, such that sequences from duplicated loci on the same chromosomal pairs have similar coalescent times, but sequences from duplicated loci on different chromosome pairs have different coalescent times.
MATERIALS AND METHODS
Data.
Because we wished to examine loci that have been duplicated by chromosomal rearrangement, we tried to exclude loci duplicated by other means. For example, we did not include data from multigene families because plant multigene families undergo dynamic fluctuations in copy number (10–12) that appear to be largely independent of large chromosomal rearrangements. We also did not include highly diverged duplicate loci (e.g., Adh1 and Adh2, which duplicated before the divergence of the grass family; unpublished data) because this study focuses on relatively recent duplication events.
The duplicated sequences and outgroup sequences used in this study are presented in Table 1. Some of the maize loci have been mapped to a chromosomal location (Table 1). All of the duplicate sequences that have been mapped are found on chromosomal pairs that are known to contain duplicated segments (6). We used sequences from maize (GenBank accession nos. X16084, X03935, and X02913), sorghum (GenBank accession nos. M31965 and U23945) and Pennisetum (GenBank accession no. X16547) for interspecific comparisons.
Table 1.
Sequences analyzed in this study
| No. | Duplicated loci* (GenBank accession nos./other sources) | Chromosomal location† | Outgroup taxon (GenBank accession no.) |
|---|---|---|---|
| 1 | orp1, orp2 (M76684, M76685) | 4S, 10L | — |
| 2 | ant1, ant2 (X57556, X59086) | — | Rice (D12637) |
| 3 | ohp1, ohp2 (L00623, L06478) | 1L, 5S | Rice (D78609) |
| 4 | r, b (X60706, X57276) | 2S, 10L | Rice (U39860) |
| 5 | cpna, cpnb (Z12114, Z12115) | — | — |
| 6 | cdc2a, cdc2b (M60526, ref. 13) | — | Rice (X60374) |
| 7 | whp1, c2 (X60204, X60205) | 2L, 4L | Rice (X89859) |
| 8 | fer1, fer2 (X61392, X61391) | — | — |
| 9 | c1, pl1 (X52201, L13454) | 9S, 6L | Rice (X96749) |
| 10 | ibp1, ipb2 (X79085, X79086) | 9L, 1S | — |
| 11 | tbp1, tbp2 (L13301, L13302) | 1L, 5S | Wheat (Z18804) |
| 12 | vpl4a, vp14b (D. McCarty, personal communication) | 1L, 5S | — |
| 13 | obf1, obf2 (X69152, X69153) | 1S, 9L | Wheat (D12921) |
| 14 | pgpa1, pgpa2 (X15406, X15407) | — | — |
The names or functions of the loci are as follows: orp, orange pericarp (tryptophan synthase β subunit); ant, mitochondrial adenine nucleotide translocator; ohp, opaque2 heterodimerizing protein; r/b, basic helix–loop–helix transcriptional regulator; cpn, mitochondrial chaperonin-60; cdc2, cell division control protein 2; whp1, white pollen; c2, colorless (chalcone synthase); fer, ferritin; cl, colored aleurone; pl, purple plant (Myb-like DNA binding protein); ibp, initiator binding protein; tbp, TATA box-binding protein; bp, viviparous; obf, octopine synthase binding factor; pgpa, pseudo-glyceraldehyde-3-phosphate dehydrogenase subunit A.
L, the long arm of the chromosome; S, the short chromosome arm.
Only exon sequences were aligned. For alignments, amino acid sequences were inferred from nucleotide sequences, and amino acid sequences were aligned manually. Some alignments include outgroup sequences, which were identified by blast searches of GenBank. The length of each alignment is given (Tables 2 and 3). In some cases, the length of an alignment changed with the inclusion of an outgroup, either because the outgroup sequence was incomplete or because the outgroup was difficult to align.
Table 2.
Distances between duplicated sequences
| No. | Duplicated loci | Length, bp | Syn. dist.* | Nonsyn. dist.† |
|---|---|---|---|---|
| 1 | orp1, orp2 | 1,170 | 0.298 (1.44) | 0.0114 (1.31) |
| 2 | ant1, ant2 | 1,173 | 0.277 (1.32) | 0.0114 (1.32) |
| 3 | ohp1, ohp2 | 1,200 | 0.254 (1.19) | 0.0593 (7.25) |
| 4 | r, b | 1,677 | 0.241 (0.83) | 0.0841 (7.84) |
| 5 | cpna, cpnb | 1,734 | 0.186 (0.55) | 0.0126 (0.97) |
| 6 | cdc2a, cdc2b | 882 | 0.177 (1.04) | 0.0097 (1.46) |
| 7 | whp1, c2 | 1,206 | 0.169 (0.66) | 0.0286 (3.29) |
| 8 | fer1, fer2 | 627 | 0.168 (1.44) | 0.0189 (4.01) |
| 9 | c1, pl1 | 777 | 0.159 (1.05) | 0.0462 (9.25) |
| 10 | ibp1, ipb2 | 2,061 | 0.150 (0.36) | 0.0482 (3.62) |
| 11 | tbp1, tbp2 | 606 | 0.147 (1.20) | 0.0066 (1.45) |
| 12 | vpl4a, vpl4b | 1,821 | 0.121 (0.29) | 0.0346 (2.69) |
| 13 | obf1, obf2 | 1,026 | 0.104 (0.48) | 0.0196 (2.95) |
| 14 | pgpa1, pgpa2 | 1,170 | 0.102 (0.39) | 0.0494 (5.97) |
| χhomo2 | P < 0.0001 | P < 0.0001 | ||
Syn. dist., synonymous distance estimates between sequences from duplicated loci. Variance (×103) is given in parentheses.
Nonsyn. dist., nonsynonymous distance estimates between sequences from duplicated loci. Variance (×105) is given in parentheses.
Table 3.
Relative rate tests
| No. | Duplicated pair† | Length, bp | Syn.‡ | Nonsyn.§ |
|---|---|---|---|---|
| 2 | ant1, ant2 | 1,173 | — | — |
| 3 | ohp1, ohp2 | 1,200 | — | — |
| 4 | r, b | 1,035 | — | — |
| 6 | cdc2a, cdc2b | 882 | — | — |
| 7 | whp1, c2 | 1,206 | — | ∗∗ (1) |
| 9 | c1, pl1 | 330 | — | — |
| 11 | tbp1, tbp2 | 606 | — | — |
| 13 | obf1, obf2 | 1,026 | — | ∗ (2) |
, P < 0.05; ∗∗, P < 0.01; —, nonsignificant result.
Outgroup sequences for relative rate tests are given in Table 1.
Syn., results of tests comparing synonymous substitution rates between sequences.
Nonsyn., results of tests comparing nonsynonymous substitution rates between sequences. Numbers in parentheses indicate whether the first or second sequence listed is inferred to evolve more rapidly.
Analyses.
Pairwise distances were based on the method of Nei and Gojobori (14). This method estimates synonymous and nonsynonymous substitutions separately and reasonably accurately (15). For relative rate tests, we employed the method of Muse and Gaut (16), which tests for both synonymous and nonsynonymous rate heterogeneity between evolutionary lineages.
Homogeneity of distance estimates was tested by a χ2 test that incorporates variance information. The χ2 statistic takes the form
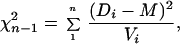 |
where there are n pairs of sequences with distance estimate D and variance V. The weighted mean M is calculated by
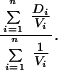 |
RESULTS
Pairwise Distance Estimates Among Pairs of Duplicated Loci.
Synonymous and nonsynonymous distances between sequences from duplicated loci are reported in Table 2. Synonymous distance estimates vary among pairs of duplicated loci. For example, the pair with the highest distance between sequences (orp1:orp2) is nearly 3-fold more diverged than the pair with the smallest distance between them (pgpa1:pgpa2). The χ2 homogeneity test over all synonymous distance estimates is highly significant, indicating that these distances are not estimates of a single underlying distance parameter (Table 2). Pairwise nonsynonymous distances vary even more dramatically. The largest nonsynonymous distance estimate (r:b) is 10-fold greater than the smallest nonsynonymous distance estimate (tbp1:tbp2). Heterogeneity in nonsynonymous distance estimates is also highly significant (Table 2).
Estimates of synonymous distance between duplicated loci are represented graphically in Fig. 2. Two groups of synonymous distance estimates do not have overlapping standard deviations. One group, which we will denote group A, consists of four relatively highly diverged duplicated sequence pairs: r:b, orp1:orp2, ant1:ant2, and ohp1:ohp2. Group B consists of the remaining 10 pairs of sequences, which are less highly diverged. We applied the χ2 homogeneity test to each group to determine whether distance estimates are homogeneous within each of these two groups. The test was not significant for either group (group A, P = 0.65; group B, P = 0.08).
Figure 2.

A graph of synonymous distances between duplicated sequences and between interspecific sequences. Shaded boxes represent the standard deviation of distance estimates. The time scale is based on an assumed rate of synonymous nucleotide substitution (see text).
The mean distance of duplicated pairs in group A is 0.267 synonymous substitutions per site (sampling variance among distance estimates = 6.21 × 10−4). The mean distance of group B pairs is 0.148 substitutions per synonymous site (sampling variance = 9.02 × 10−4). A Student’s t test indicates that the difference in the mean distance between group A and group B is highly significant (t = 7.62, df = 12, p = 3.1 × 10−6). However, this is an a posteriori test, and the significance level of the t test must be corrected for the fact that our grouping of distance estimates is one combination out of 9,907 possible two-grouped combinations of 14 distance estimates. Accordingly, we adjusted the critical value of the t test with the Dunn–Sidak correction, which is conservative for nonindependent comparisons (17). The Dunn–Sidak correction adjusts the a posteriori t test significance level to 5.2 × 10−6 (based on an experiment-wide significance level of 0.05), which is higher than the P level of the t test. Thus, the t test remains significant, indicating that the group A sequence pairs are significantly more diverged than the group B sequence pairs.
Alternatively, the distance measures in Fig. 2 could be organized to represent three groups. The three groups are group A′, which is defined to be the same as group A; group B′, which includes duplicates tbp1:tbp2, whp1:c2, cpna:cpnb, fer1:fer2, ibp1:ibp2, cdc2a:cdc2b, and c1:pl1; and group C′, which contains the three least diverged duplicates obf1:obf2, pgpa1:pgpa2, and vpl4a:vpl4b. Distances within these three groups are homogeneous by the χ2 test criterion (data not shown). However, t test comparisons among these groups are not significant (A vs. B′, P = 1.6 × 10−5; A vs. C′, P = 4.4 × 10−5; B′ vs. C′, P = 5.8 × 10−5; the a posteriori significance level, based on an experiment-wide significance level of 0.05 and adjusted for all possible three-grouped combinations of 14 distance estimates, is 2.0 × 10−8). Thus, there is no statistical support for defining three groups, and there is evidence for two and only two homogeneous groups of synonymous distance estimates with these data.
Rates of Nucleotide Substitution.
The predictions of the tetraploid and the segmental duplication models are formulated in terms of coalescent times (see above). We have shown that there are two distinct groups of sequence pairs—a more highly diverged group and a less highly diverged group—but we have yet to investigate the relationship between distance estimates and coalescent times. Comparing distances across loci is equivalent to comparing time across loci when the following two conditions hold: condition 1, duplicated sequences evolve with equal nucleotide substitution rates after duplication; condition 2, synonymous substitution rates are equal across pairs of duplicated sequences. In this section, we examine whether these two conditions hold.
To fully examine the two conditions, it is necessary to have at least one outgroup sequence for each pair of duplicated sequences. Unfortunately, the outgroup data are limited. We have found outgroup sequences for 8 of the 14 pairs of duplicated loci (Table 1). Only 6 of these pairs share an outgroup sequence from the same taxon (rice).
Equality of synonymous substitution rates between duplicated sequences (condition 1) was examined by relative rate tests. We applied relative rate tests (16) to the 8 pairs of duplicated loci for which an outgroup is available (Table 3). Some duplicated sequences have evolved at significantly different nonsynonymous substitution rates since the time of their duplication. For example, relative rate tests indicate that whp1 has evolved more rapidly than c2 at nonsynonymous sites. Similarly, obf2 has evolved more rapidly than obf1 at nonsynonymous sites. However, there is no evidence for synonymous rate variation between duplicated sequences, indicating that pairs of duplicated sequences have evolved at roughly similar synonymous rates since the time of their duplication.
One method to examine whether synonymous substitution rates vary across duplicate pairs (condition 2) is to compare k, the average number of synonymous substitutions from duplicated sequences to an outgroup sequence, among pairs. The parameter k is estimated by d10 + d20/2, where d10 is the synonymous distance from one of a pair of duplicated maize sequences to the outgroup sequence, and d20 is the synonymous distance from the second maize sequence in the duplicated pair to the outgroup.
We estimated k for those duplicate pairs for which a rice outgroup sequence is available (Fig. 3). Two observations about k estimates should be noted. First, the k value for the r:b duplicate pair is quite high relative to other duplicate pairs. This high value could reflect rapid synonymous substitution rates at r:b loci, or it could indicate that the rice sequence is not strictly orthologous to the maize sequences. [The latter is distinctly possible, given that r:b genes exist as a multigene family in both rice (18) and Pennisetum (19).] Second, k values are homogeneous for the remaining five loci (χ2 homogeneity, P = 0.054). In this case, acceptance of the null hypothesis of homogeneity may merely reflect low statistical power with a sample size of five, but k is nearly identical for three duplicate pairs (ant1:ant2, c1:pl1, and cdc2a:cdc2b).
Figure 3.

Graphical representation of k, the average synonymous distance between duplicated maize sequences and a rice outgroup sequence. Shaded boxes represent standard deviations.
Nearly identical k values suggest that synonymous substitution rates are very similar among these three sequence pairs. Note that one of these three duplicate pairs (ant1:ant2) is in the more diverged set (group A) and that the other two duplicate pairs (c1:pl1 and cdc2a:cdc2b) are in the less diverged set (group B) (Table 2 and Fig. 2). Thus, divergence among duplicated sequences varies despite similar synonymous substitution rates. These results lead to two conclusions. First, there is no common time of duplication for these three pairs of duplicated sequences. Second, although the relationship between distance and divergence time may not be perfect (e.g., the r:b pair may have evolved with a more rapid synonymous substitution rate), many of the duplicate sequence pairs appear to be evolving at roughly similar synonymous substitution rates.
Dates and Systematic Placement of Duplication Events.
There are two groups of duplicated sequence pairs—a more highly diverged group (group A) and a less highly diverged group (group B). If the two groups are the result of two distinct duplication events, it is of interest to know when these duplication events occurred, both in terms of time and in terms of the systematic history of the grass family.
One cannot estimate duplication times without assuming a rate of nucleotide substitution. The average synonymous substitution rate at the Adh1 and Adh2 loci of grasses has recently been estimated to be 6.5 × 10−9 substitutions per synonymous site per year (20). We make the assumption that Adh substitution rates are representative of synonymous substitution rates for duplicated sequence pairs. Given this rate, sequences in group A duplicated approximately 20.5 Mya [i.e., 0.267/(2)6.5 × 10−9], and pairs of duplicated sequences in group B diverged approximately 11.4 Mya (Fig. 2).
It is of interest to explore how duplication events within maize are related to the time of divergence of maize and sorghum sequences. Fig. 2 shows the average distance between maize and sorghum sequences based on two genes, mdh and waxy. Remarkably, the maize and sorghum pairwise distances fall between the group A distances and the group B distances. This result suggests that the coalescent time of group A sequences is greater than the divergence time between the sorghum and maize sequences and, therefore, implies that the sorghum genome is more closely related to one of the two 20.5-million-year-old maize subgenomes than it is to the other subgenome. Fig. 2 also provides a distance estimate between a maize and a Pennisetum Adh1 sequence. [Pennisetum and maize are members of the same grass subfamily, but Pennisetum is not in the tribe Andropogoneae (21).] The distance between maize and Pennisetum sequences is greater than the distance between any two maize duplicated sequences, suggesting that the coalescence of maize duplicated sequences is more recent than the divergence between maize and Pennisetum.
DISCUSSION
Assumptions of Distance Analyses.
Before the significance of our results can be explored fully, it is important to discuss the methods and assumptions of analyses. The data were chosen with the assumption that they reflect adequately the process of genomic duplication in maize since the origin of the Andropogoneae. Maize contains a great many other duplicated sequences, but they do not fall within the framework of our study. For example, some maize duplicate pairs like Adh1:Adh2, Zag1:Zmm2, and Pyrde1:Pyrde2 appear to have diverged before the divergence of rice and maize, ≈50 Mya (22; data not shown). Multigene families like Sod and Cab contain elements that diverged recently and other elements that diverged early in the history of the grass family (data not shown). Clearly, gene duplication is an active process in maize; this study was designed to illuminate only the pattern of relatively recent chromosomal duplications.
We have focused on synonymous distances rather than nonsynonymous distances, for three reasons. First, nonsynonymous rates have long been known to show more variation among genes than synonymous rates (23, 24); this is seen in the duplicate data (Table 2). For this study, it is important that variation in rates among genes is minimized, and we accordingly focused on synonymous substitutions. Second, nonsynonymous rates are expected to deviate from clock-like behavior after gene duplication (25, 26), whereas synonymous rates are not necessarily expected to vary between the two duplicates (20). Some of the relative rate tests in this study reveal differences in nonsynonymous rates between duplicated sequences, making nonsynonymous rates unreliable for estimating coalescent times. Third, there are no easily discernible groupings of nonsynonymous distances among pairs of duplicated sequences (Table 2), suggesting that nonsynonymous rates are not informative for investigating the timing of duplication events.
The analysis of sequence data suggests that duplicated loci belong to two different groups that diverged roughly 20.5 and 11.4 Mya. The reliability of these time estimates depends both upon the substitution rate, which we have assumed to be 6.5 × 10−9 substitutions per synonymous site per year (20), and upon the two conditions discussed above, i.e., the amount of substitution rate variation between duplicated sequences and the amount of substitution rate variation among pairs of duplicated sequences. Our analyses suggest that the former factor is unlikely to contribute much uncertainty to our coalescent time estimates, but the latter may be a source of error.
We also made simplifying assumptions regarding the process of random genetic drift in the formulation of ploidy models. Random genetic drift contributes to variance in genetic diversity among loci. The amount of genetic diversity at a locus at the time of a chromosomal duplication will ultimately affect the inference of coalescent times. For example, some Adh1 allelic lineages in maize have existed for ≈2.0 Mya (27). If these allelic lineages were involved in the process of gene duplication at time x Mya, we would overestimate the divergence time between loci as x + 2 Mya. Although it is not strictly correct to ignore drift in our models, the scale of variation in coalescent times contributed by drift is expected to be much smaller than the scale of coalescent time variation seen between group A and group B duplicate pairs (Fig. 2). However, the scale of genetic drift is probably such that it contributes to stochastic variation among within-group distance estimates.
Support for the Segmental Allotetraploid Model.
Statistical analyses indicate that there are two different groups of distance estimates. This result is inconsistent with a single divergence time for all duplicate sequence pairs. To see this crucial point, imagine either an autopolyploid or a genomic allotetraploid event in which all loci were duplicated at roughly the same time. To get two groups of distance estimates, one must postulate that the loci have evolved with two (and only two) distinct synonymous substitution rates since the ploidy event. Although several studies have found evidence of synonymous rate variation among loci (28, 29), there is no precedence demonstrating two (and only two) different substitution rates. Our results strongly discount models of genome evolution that predict a single coalescent time among pairs of duplicated loci.
Of the models we have considered, only one—segmental allotetraploidy—is consistent with two distinct coalescent times, and the data provide compelling evidence for two distinct coalescent times. We see no plausible way to modify the genomic allotetraploid model to accommodate the data. Under the autotetraploid model, one could argue that after tetraploid formation there was an initial switch to disomy for one set of loci, then an extended time period during which inheritance patterns remained unchanged, and finally a period in which all remaining tetrasomic loci switched to disomy. However, there are no known genetic mechanisms that predict this pattern, and this scenario is less parsimonious than the segmental allotetraploid model. Similarly, one could construe the segmental duplication model such that there was an initial period during which one set of chromosome segments was duplicated, then an extended period without further duplication, followed by second round of segmental duplications. However, this model is also unparsimonious, lacks a known underlying genetic mechanism, and does not adequately explain a doubling of chromosome number in maize relative to the base number of the Andropogoneae.
The relationship of coalescent time with the physical location of duplicated loci also supports the segmental allotetraploid hypothesis. A corollary prediction of the segmental allotetraploid model is that pairs of duplicate loci on the same chromosomes can have different coalescent times. This is the case with these data. For example, three pairs of duplicated loci (ohp1:ohp2, tbp1:tbp2, and vpl4a:vpl4b) are located on the long arm of chromosome 1 and the short arm of chromosome 5, and these pairs differ in their coalescent times (ohp1:ohp2 sequences are in the more highly diverged group and sequences from tbp1:tbp2 and vpl4a:vpl4b belong to the less diverged group). Conversely, the data indicate that sequences from duplicated loci on different chromosomal pairs have similar coalescent times. For example, group A contains sequence pairs from chromosomes 2 and 10, 4 and 10, and 1 and 5 (Table 1), and group B also contains sequences from a wide array of chromosomal pairs, i.e., chromosomes 1 and 5, 6 and 9, and 2 and 4. Both group A and group B likely contain sequences from other chromosomes as well because some of the duplicate pairs in the study have not been mapped to a chromosomal location (Table 1). These patterns are difficult to reconcile with the segmental duplication and the autotetraploid models.
Evolution of the Maize Genome.
The pattern of divergence times among duplicated loci provides key insights into the timing and mode of evolution of the maize genome. First, under the segmental allotetraploid model (Fig. 1, column C), the coalescent time for group A sequences represents the time of divergence of the two ancestral diploid (n = 5) species. We estimate that the two ancestral species diverged roughly 20.5 Mya, a time more recent than the divergence of the maize and Pennisetum lineages (Fig. 2). Second, the divergence time for maize–sorghum mdh and waxy sequences is approximately 16.5 Mya, i.e., more recent than the divergence of the two diploid progenitors of maize. This observation suggests that at least some elements of the sorghum genome share a more recent common ancestor with one of the two maize subgenomes than the two maize subgenomes share with each other. This suggestion can be tested once sequence data for sorghum genes corresponding to group A duplicated pairs are available. Third, under the segmental allotetraploid model, the coalescent time of 11.4 Mya for group B sequences represents both a minimal estimate of the time of interspecific hybridization and an estimate of the time of the onset of disomy. It should be noted that the group B sequences form a relatively tight group with no statistical evidence for heterogeneity (Fig. 2). This may indicate that the maize genome switched from tetrasomic to disomic inheritance in a concerted fashion; rapid genomic evolution has been found with experimentally created polyploids (30). Finally, of the 14 duplicated pairs analyzed, only 4 belong to the older of the two coalescent groups. This may indicate that either selection or drift acted to homogenize allelic diversity at tetrasomic loci such that there was preferential retention of the alleles of only one of the diploid parental species. Preferential elimination of one parental genome has been inferred for other polyploid species (30, 31).
Polyploidy has been a potent force in plant evolution and the subject of intense interest over the past 60 years (32). Many new insights into polyploid formation and subsequent genomic evolution are now being garnered from the application of modern molecular methods to this classical issue (30, 31). With the advent of large scale genome projects, it is likely that extensive DNA sequence data will soon be available for maize and other cereal crops. In this environment, molecular sequence analyses can help resolve modes of polyploid formation and subsequent genome evolution.
Acknowledgments
We thank Drs. S. Muse, A. Eyre-Walker, H. Hilton, J. Kermicle, J. Birchler, and J. Wendel and two anonymous reviewers for comments and ideas, and Drs. D. McCarty and R. Schmidt for sharing unpublished sequence data. This research supported by U.S. Department of Agriculture Grants 95-00566 (to B.S.G.) and 93-37300-8773 (to J.F.D), and National Science Foundation Grant MCB-9513573 (to J.F.D).
ABBREVIATION
- Mya
million years ago
References
- 1.Celarier R P. Rhodora. 1956;58:135–143. [Google Scholar]
- 2.Anderson E. Chron Bot. 1945;9:88–92. [Google Scholar]
- 3.Garber E D. Univ Calif Pub Bot. 1950;23:283–362. [Google Scholar]
- 4.Rhoades M M. Am Nat. 1951;85:105–110. [Google Scholar]
- 5.Wendel J F, Stuber C W, Goodman M M, Beckett J B. J Hered. 1989;80:218–228. doi: 10.1093/oxfordjournals.jhered.a110839. [DOI] [PubMed] [Google Scholar]
- 6.Helentjaris T, Weber D, Wright S. Genetics. 1988;118:353–363. doi: 10.1093/genetics/118.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rhoades M M. In: Corn and Corn Improvement. Sprague G F, editor. New York: Academic; 1955. pp. 123–219. [Google Scholar]
- 8.Whitkus R, Doebley J, Lee M. Genetics. 1992;132:1119–1130. doi: 10.1093/genetics/132.4.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stebbins G L. Chromosomal Evolution in Higher Plants. London: Arnold; 1971. [Google Scholar]
- 10.Waters E R. Genetics. 1995;141:785–795. doi: 10.1093/genetics/141.2.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morton B R, Gaut B S, Clegg M T. Proc Natl Acad Sci USA. 1996;93:11735–11739. doi: 10.1073/pnas.93.21.11735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Durbin M L, Learn G H, Huttley G A, Clegg M T. Proc Natl Acad Sci USA. 1995;92:3338–3342. doi: 10.1073/pnas.92.8.3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Colasanti J, Tyers M, Sundaresan V. Proc Natl Acad Sci USA. 1991;88:3377–3381. doi: 10.1073/pnas.88.8.3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nei M, Gojobori T. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 15.Muse S V. Mol Biol Evol. 1996;13:105–114. doi: 10.1093/oxfordjournals.molbev.a025549. [DOI] [PubMed] [Google Scholar]
- 16.Muse S V, Gaut B S. Mol Biol Evol. 1994;11:715–724. doi: 10.1093/oxfordjournals.molbev.a040152. [DOI] [PubMed] [Google Scholar]
- 17.Sokal R R, Rholf F J. Biometry. New York: Freeman; 1995. [Google Scholar]
- 18.Hu J P, Anderson B, Wessler S R. Genetics. 1996;142:1021–1031. doi: 10.1093/genetics/142.3.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Purugganan M D, Wessler S R. Genetics. 1994;138:849–854. doi: 10.1093/genetics/138.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaut B S, Morton B R, McCaig B M, Clegg M T. Proc Natl Acad Sci USA. 1996;93:10274–10279. doi: 10.1073/pnas.93.19.10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kellogg E A, Watson L. Bot Rev. 1993;59:273–343. [Google Scholar]
- 22.Wolfe K H, Gouy M, Yang Y-W, Sharp P M, Li W-H. Proc Natl Acad Sci USA. 1989;86:6201–6205. doi: 10.1073/pnas.86.16.6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li W-H, Graur D. Fundamentals of Molecular Evolution. Sunderland, MA: Sinauer; 1991. p. 69. [Google Scholar]
- 24.Muse S V, Gaut B S. Genetics. 1997;146:393–399. doi: 10.1093/genetics/146.1.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kimura M, Ohta T. Proc Natl Acad Sci USA. 1974;71:2848–2852. doi: 10.1073/pnas.71.7.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li W-H. In: Population Genetics and Molecular Evolution. Ohta T, Aoki K, editors. Tokyo: Japan Scientific Societies Press; 1985. pp. 333–352. [Google Scholar]
- 27.Gaut B S, Clegg M T. Proc Natl Acad Sci USA. 1993;90:5095–5099. doi: 10.1073/pnas.90.11.5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bulmer M, Wolfe K H, Sharp P M. Proc Natl Acad Sci USA. 1991;88:5974–5978. doi: 10.1073/pnas.88.14.5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolfe K H, Sharp P M, Li W-H. J Mol Evol. 1989;29:208–211. [Google Scholar]
- 30.Song K, Lu P, Tang K, Osborn T C. Proc Natl Acad Sci USA. 1995;92:7719–7723. doi: 10.1073/pnas.92.17.7719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soltis D E, Soltis P S. Crit Rev Plant Sci. 1993;12:243–273. [Google Scholar]
- 32.Stebbins G L. Variation and Evolution in Plants. New York: Columbia Univ. Press; 1950. [Google Scholar]