

Abstract
Heteroduplex DNA lacking d(GATC) methylation is subject to mismatch-provoked double-strand cleavage at d(GATC) sites in a reaction dependent on MutH, MutL, MutS, and ATP. We have exploited this reaction to develop a method for removal of polymerase-produced mutant sequences that arise during sequence amplification by PCR. After denaturation and reannealing, the PCR product pool is subjected to MutH, MutL, and MutS mismatch repair proteins under double-strand cleavage conditions, followed by isolation of uncleaved product by size selection. Use of an Escherichia coli lac forward mutation assay has shown that this procedure reduces the incidence of polymerase-induced mutant sequences by an order of magnitude. Twenty mutants that originated from three independent PCR amplification reactions and survived MutHLS treatment all were found to contain an infrequently occurring A⋅T → T⋅A transversion mutation at a unique position within the product. By contrast, the majority of mutations in untreated PCR products were transitions occurring throughout the amplified region, although frameshifts and transversions also were observed. The MutHLS method thus can be used to effectively remove the majority of mutant sequences produced by polymerase errors during PCR amplification.
Keywords: mutation, mismatch repair, MutH, MutL, MutS
PCR amplification of DNA sequences with thermostable DNA polymerases (1, 2) has had a profound impact on molecular biology and genetics. One shortcoming of the PCR method is the production of mutant sequences that result from misincorporation by the DNA polymerase used for amplification (3–6). The frequency of polymerase errors during PCR can be estimated from equations 1 and 6 of Luria and Delbrück (7) as
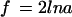 |
where f is the expected fraction of product molecules that contain a mutation somewhere in their sequence, l is the length of the amplified sequence in base pairs, n is the number of cycles, and a is the error rate for the polymerase expressed per nucleotide incorporated. Because the error rate for Taq DNA polymerase is about 10−5 (5, 6), the expected mutant frequency after 20 template doublings of a 1,000-bp sequence is about 40%. This problem is obviated to some extent with Vent and Pfu polymerases, which in contrast to Taq possess editing exonuclease activity and have error rates of about 10−6 (5, 6). However, inasmuch as the mutant frequency is a linear function of the size of the DNA segment, mutations produced by the latter polymerases become a problem when larger sequences are amplified. The significance of polymerase errors for PCR applications in genetic diagnostics and cDNA cloning has been documented in the literature (8–10).
Because increased genetic integrity of amplification products should enhance the utility of PCR, we have developed a method, based on use of bacterial mismatch repair proteins, for removal of polymerase-produced mutant sequences from amplified product pools. Initiation of Escherichia coli methyl-directed mismatch repair occurs via the mismatch-provoked, MutS-, MutL-, and ATP-dependent activation of a MutH-associated endonuclease that incises the helix at unmethylated d(GATC) sequences (11). Although the natural substrate for activated MutH is a hemimethylated d(GATC) site, unmethylated d(GATC) sequences are subject to cleavage on both strands when reactions are allowed to progress for extended periods of time (11). We show here that the mismatch-provoked, double-strand cleavage reaction coupled with a size selection step can be used to effectively remove most mutant sequences from PCR products.
MATERIALS AND METHODS
PCR Amplification.
A 1,611-bp sequence spanning M13 mp18 lacZα sequences of interest (12) was amplified in reactions (100 μl) containing 10 mM Tris⋅HCl at pH 8.3, 50 mM KCl, 2 mM MgCl2, 200 μM each dNTP (Pharmacia), 100 pmol of each primer, 5 μg of T4 gene 32 protein (Boehringer Mannheim), 50 ng of template DNA (M13 mp18), and 2.5 units of AmpliTaq DNA polymerase (Perkin–Elmer). PCR primers (Oligos Etc., Guilford, CT) were d(TTATACGTGCTCGTCAAAGCA) and d(AATGCCTGAGTAATGTGTAGG) corresponding to M13 mp18 nucleotides 5458–5478 and 7048–7069, respectively (Fig. 1). Thirty-cycle amplification used a Perkin–Elmer Gene Amp 9600 thermocycler with incubations at 94°C for 15 sec, 60°C for 15 sec, and 72°C for 1 min. Products were denatured and reannealed immediately after amplification by heating to 95°C for 1 min and incubation at 65°C for 60 min followed by incubation at 37°C for 30 min. EDTA was added to 20 mM, reactions extracted with phenol, and the PCR product purified by binding to a silica matrix spin column (Pierce Xtreme DNA purification columns) and elution with distilled H2O. Product DNA was quantitated by an ethidium bromide fluorescence dot method as follows: samples (2.0 μl of an appropriate dilution) were added to 8 μl of 1 μg/ml ethidium bromide and spotted onto plastic wrap. UV-induced fluorescence was measured using a photometric grade cooled charge-coupled device imager (Photometrics, Tucson, AZ), and concentrations of PCR products determined by comparison to the fluorescence of DNA samples of known concentration. PCR product yield ranged from 4.3 to 7.3 μg (n = 12), corresponding to 8.6 to 9.4 template doublings.
Figure 1.

Forward mutation assay with M13 mp18. The location of PCR primers is indicated by hatched lines. After amplification, denatured and reannealed PCR products were hydrolyzed with MutHLS proteins and/or DraIII and BglII, and the DNA fragment corresponding to the full-length DraIII/BglII product was isolated by size (see Materials and Methods). This product was cloned into an M13 mp18 derivative from which the DraIII/BglII fragment had been removed. In the orientation shown, the DraIII site is 256 bp clockwise from the nearest end of the PCR product while the BglII site is 134 bp counterclockwise from the other end.
MutHLS Reactions.
Reactions (50 μl total) were assembled as follows: 20 μl of 125 mM Hepes, pH 8.0/50 mM KCl/2.5 mM DTT/125 μg/ml BSA/5 mM ATP/10 mM MgCl2, and 1 μg of denatured and reannealed PCR DNA were preincubated at 37°C for 8 min. Reactions were initiated by adding 30 μl of a premixed solution of 5 μg of MutS (13), 12 μg of MutL (14), and 18 ng of MutH (15) in 20 mM potassium phosphate, pH 7.4/50 mM KCl/0.1 mM EDTA/1 mM DTT/1 mg/ml BSA. After incubation for 45 min at 37°C, reactions were supplemented with an additional 30 μl of a premixed solution of MutS, MutL, and MutH as described above, as well as 3 μl of a solution containing 500 mM Hepes at pH 8.0, 200 mM KCl, 10 mM DTT, 20 mM ATP, and 40 mM MgCl2, and incubation continued at 37°C for 45 min. A second MutHLS/buffer supplementation was performed in an identical manner, and after a third 45-min incubation, reactions were terminated by addition of EDTA to 10 mM and extraction with phenol, followed by extraction with ether. DNA was concentrated and purified by binding to a QIAquick spin column (Qiagen) and elution with distilled H2O. Concentration was determined as described above.
Experiments with model heteroduplexes, as well as PCR-amplified samples, demonstrated that a single MutHLS treatment was not sufficient to cleave all the mismatch-containing molecules. The multiple-treatment protocol summarized above is necessary to drive the reaction to completion.
Forward Mutation Assay.
Untreated or MutHLS-treated PCR products were digested for 2 hr at 37°C with 10 units of DraIII (Amersham) and 4 units of BglII (New England Biolabs) in 20-μl reactions containing 1 μg of DNA, 100 mM NaCl, 50 mM Tris⋅HCl (pH 7.5), 10 mM MgCl2, 1 mM DTT, and 100 μg/ml BSA. After termination of hydrolysis by addition of EDTA to 10 mM, DNA products were collected by ethanol precipitation and subjected to electrophoresis through 1% agarose in 40 mM Tris-acetate/1 mM EDTA (final pH 7.5). The DNA species corresponding to the full-length DraIII/BglII fragment was recovered using a Gene Clean Kit (Bio 101) according to recommendations of the manufacturer, and quantitated using the ethidium bromide dot method described above. Recovery of full-length fragment after MutHLS and DraIII/BglII cleavage ranged from about 20% to 50%. The gel-purified fragment was ligated to the larger DraIII/BglII fragment of M13 mp18 (Fig. 1), which was prepared by endonucleolytic digestion and removal of the smaller fragment by agarose gel electrophoresis. Reactions (20 μl) contained 50 ng of gel-purified PCR DNA, 50 ng of M13 mp18 derivative, 66 mM Tris⋅HCl (pH 7.6), 6.6 mM MgCl2, 10 mM DTT, 66 μM ATP, and 0.5 Weiss unit of T4 DNA ligase (Amersham). Incubation was at 16°C for 12–16 hr.
Ligation products were transfected into XL2-Blue Ultracompetent Cells (Stratagene) according to recommendations of the manufacturer. A 10-μl sample of the ligation reaction was added to 100 μl of competent cells, and the mixture was incubated on ice for 30 min. After a 45-sec heat pulse at 42°C, the mixture was supplemented with 0.9 ml of prewarmed (42°C) SOB broth (20 g of Bacto-tryptone, 5 g of Bacto-yeast extract, and 0.5 g of NaCl). A 50-μl sample of the transfection reaction was added to a tube at 49°C containing 4 ml of Luria–Bertani medium soft agar (10 g of Bacto-tryptone, 5 g of Bacto-yeast extract, 10 g of NaCl, and 7.5 g of agar per liter), 4 mg of X-Gal (5-bromo-4-chloro-3-indolyl β-d-galactoside, Amersham), and 800 μg of IPTG (isopropyl thiogalactoside, Amersham). Two-hundred microliters of a log-phase culture of XL1-Blue (Stratagene) was added, and the soft agar mixture poured onto a Luria–Bertani medium plate. After incubation at 37°C for 12–16 hr, 3,000 to 12,000 plaques were scored according to color for wild-type (blue) or mutant (colorless or pale blue) β-galactosidase phenotype (16). The frequency of plaques obtained when ligation was performed in the absence of PCR product was 0.3–0.6% of that observed in the presence of amplified DNA. Because all of these plaques were lacZα +, they presumably reflect trace presence of the smaller M13 mp18 DraIII/BglII fragment (above and Fig. 1).
Sequence Analysis of Mutants.
Phage stocks prepared from colorless and light-blue mutant plaques were used to make replicative-form DNA. After linearization with BglII, the sequence of the lac region of interest (16) was determined on both strands by dye terminator cycle sequencing using AmpliTaq DNA Polymerase (Perkin–Elmer). The primer used to determine the sequence of the viral strand was d(TAGATGGGCGCATCGTAACCG), while that for the complementary strand was d(AGGGCCAGGCGGTGAAGGGCA). Sequence determination used a Perkin–Elmer/Applied Biosystems Model 377 DNA Sequencer.
RESULTS
Removal of Sequences Containing Mutations from PCR Product Pools by Treatment with MutHLS Proteins.
Random mutations resulting from polymerase errors during amplification will be converted to mismatched base pairs when a PCR product is denatured and reannealed. Because helices lacking d(GATC) modification are subject to MutH-, MutL-, and MutS-dependent, mismatch-provoked double-strand cleavage at d(GATC) sites (11), we reasoned that treatment of a denatured and reannealed PCR product with these proteins followed by size fractionation might permit removal of mutant sequences from an amplified product.
To test the efficiency of this approach, we have used a variation of the E. coli lac forward mutation assay developed to measure the fidelity of DNA polymerases (16). This method is capable of scoring 241 substitution mutations at 125 positions and 199 frameshift mutations within the lacZα regulatory and coding regions (positions 6109–6417 of M13 mp18). A segment of the M13 mp18 sequence (nucleotides 5458–7069) spanning the region of interest (Fig. 1) was PCR-amplified with Taq polymerase for 30 cycles, and the product pool denatured and reannealed. Half of each resultant hybrid preparation was treated with MutH, MutL, and MutS under conditions sufficient to elicit double-strand cleavage at d(GATC) sites in those molecules containing mismatches (see Materials and Methods). After digestion of untreated and MutHLS-treated samples with DraIII and BglII, products corresponding to the full-length DraIII/BglII fragment (Fig. 1) were isolated by gel electrophoresis and cloned into an M13 mp18 derivative from which the lac region had been removed. These molecules were tested for their activity in the lacZα complementation assay (17).
As summarized in Table 1, the frequency lacZα− mutations ranged from 3.1–3.7% for three independently amplified samples that were not treated with MutH, MutL, and MutS. Inasmuch as the genetic target in these experiments was 308 bp, including the multiple cloning site of the vector (12, 16), and amplification was about nine template doublings (see Materials and Methods), these values correspond to a Taq polymerase error rate of 6 × 10−6 per nucleotide incorporated per cycle, ignoring the incidence of silent mutations. This value is in excellent agreement with previous determinations of the fidelity of the Taq enzyme (4–6). By contrast, the mutation frequency in MutHLS-treated samples derived from the same amplification reactions ranged from 0.3% to 0.4%, 10-fold lower than that observed with the untreated DNAs.
Table 1.
Reduction of mutant frequency in PCR products by MutHLS treatment.
| Experiment | MutHLS | Mutant plaques | Wild-type plaques | Mutant frequency,* % |
|---|---|---|---|---|
| 1 | No | 127 | 3,546 | 3.58 |
| 2 | No | 204 | 5,591 | 3.65 |
| 3 | No | 213 | 6,947 | 3.07 |
| 1 | Yes | 23 | 5,191 | 0.44 |
| 2 | Yes | 31 | 11,339 | 0.27 |
| 3 | Yes | 45 | 11,941 | 0.38 |
Three independently amplified samples were split, with one-half of each receiving MutH, MutL, and MutS treatment as described in Materials and Methods. After cleavage of untreated and MutHLS-treated DNAs with DraIII and BglII, the full-length DraIII/BglII product was isolated by gel electrophoresis and cloned into DraIII/BglII-cleaved M13mp18. The lacZα complementation phenotype was scored by X-Gal (5-bromo-4-chloro-3-indolyl β-d-galactoside) color assay (17). Values shown are not corrected for background lac mutations present in nonamplified DNA (0.02%).
Mutant frequency = 100 × [mutant plaques/total plaques].
lacZα Mutation Spectra for Untreated and MutHLS-Treated DNAs.
To clarify the reduction in mutant frequency resulting from MutHLS treatment, representative samples of lacZα mutant phage were sequenced. This analysis (Table 2) showed the majority of mutations in phage prepared from untreated DNA samples to be transitions (13 of 18), although three frameshifts and two transversions were also identified, with one of the latter being in a phage that harbored a double mutation. These mutations were distributed throughout the lacZα region. A dramatic difference was observed with mutant phage derived from MutHLS-treated DNA samples. Twenty of 20 mutants were found to contain the same A⋅T → T⋅A transversion mutation at position 6340. This mutation was also identified in one phage prepared from a DNA sample that was not treated with the Mut proteins (mutant 5 in Table 1). Because the background frequency of lacZα mutant sequences in the M13 mp18 DNA preparation used as PCR substrate was about 5% of that observed in MutHLS-treated samples (Table 1), we infer that this A⋅T → T⋅A transversion arose during amplification.
Table 2.
The lacZα mutation spectra of untreated and MutHLS-treated amplified DNA samples.
| PCR | Mutant number | Nucleotide change | Position |
|---|---|---|---|
| Untreated | |||
| 1 | 1 | C → T | 6121 |
| 3 | T → C | 6199 | |
| 2 | G → A | 6400 | |
| 5 | A → T | 6341 | |
| 4 | TAATA → TATA | 6384–6385 | |
| 2 | 6, 8, 9 | C → T | 6289, 6146, 6360 |
| 12 | T → C | 6173 | |
| 11, 13 | A → G | 6340, 6340 | |
| 7 | T → C and T → A | 6144 and 6166 | |
| 3 | 14, 15 | C → T | 6146, 6360 |
| 17 | T → C | 6352 | |
| 10 | CCCCC → CCCC | 6363–6367 | |
| 16 | AAAA → AAA | 6322–6325 | |
| MutHLS-treated | |||
| 1 | 1–6 | A → T | 6340 only |
| 2 | 7–13 | A → T | 6340 only |
| 3 | 14–20 | A → T | 6340 only |
All mutations were confirmed by sequencing both DNA strands.
DISCUSSION
These experiments show that the mismatch-provoked MutHLS-double-strand cleavage reaction can be exploited to reduce the incidence of mutations in PCR products by an order of magnitude. Our results are consistent with previous studies of Taq DNA polymerase fidelity, which have shown that errors producing transition mutations are much more frequent than those that yield insertion/deletions or transversions (2, 3, 10, 18–20). The sequence results summarized in Table 2 indicate that the MutHLS reaction can be used to effectively remove transition mutations, and although the sample size is small, probably frameshifts and some transversions as well. This conclusion is consistent with the known mismatch specificity of the methyl-directed reaction deduced from in vivo and in vitro studies (reviewed in ref. 21). Thus, G-T, A-C, G-G, A-A, and small insertion/deletion mispairs are usually well repaired. Individual T-T, T-C, and A-G mismatches are corrected with intermediate to low efficiency depending on sequence context, and C-C is not processed. Perhaps surprisingly, we did not observe any G⋅C → C⋅G transversions in the mutant DNAs sequenced. On the other hand, three independent samples of MutHLS-treated PCR products were dramatically enriched for a unique A⋅T → T⋅A transversion. We attribute this result to a sequence context effect.
Because they are more or less random, polymerase errors are without significant effect on DNA sequence results obtained with amplified samples. However, such errors can compromise genetic diagnostic methods based on mismatch detection with amplified DNA (8, 9) and render results obtained with cloned PCR products problematic (10), with the severity of this problem increasing with DNA chain length and the number of doublings. By permitting removal of most mutant sequences generated during amplification, the method described here should prove useful in circumventing these difficulties.
Acknowledgments
P.M. is a consultant to Amersham, which has a license from Duke University to commercialize mismatch repair activities. This work was supported in part by Grant GM27319 from the National Institute of General Medical Sciences.
References
- 1.Saiki R, Scharf S, Faloona F, Mullis K B, Horn G T, Erlich H A, Arnheim N. Science. 1985;230:1350–1354. doi: 10.1126/science.2999980. [DOI] [PubMed] [Google Scholar]
- 2.Saiki R K, Gelfand D H, Stoffel S, Scharf S J, Higuchi R, Horn G T, Mullis K B, Erlich H A. Science. 1988;239:487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- 3.Keohavong P, Thilly W G. Proc Natl Acad Sci USA. 1989;86:9253–9257. doi: 10.1073/pnas.86.23.9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eckert K A, Kunkel T A. Nucleic Acids Res. 1990;18:3739–3744. doi: 10.1093/nar/18.13.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lundberg K S, Shoemaker D D, Adams M W W, Short J M, Sorge J A, Mathur E J. Gene. 1991;108:1–6. doi: 10.1016/0378-1119(91)90480-y. [DOI] [PubMed] [Google Scholar]
- 6.Cline J, Braman J C, Hogrefe H H. Nucleic Acids Res. 1996;24:3546–3551. doi: 10.1093/nar/24.18.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luria S E, Delbrück M. Genetics. 1943;28:491–511. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reiss J, Krawczak M, Schloesser M, Wagner M, Cooper D N. Nucleic Acids Res. 1990;18:973–978. doi: 10.1093/nar/18.4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith J, Modrich P. Proc Natl Acad Sci USA. 1996;93:4374–4379. doi: 10.1073/pnas.93.9.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ennis P D, Zemmour J, Salter R D, Parham P. Proc Natl Acad Sci USA. 1990;87:2833–2837. doi: 10.1073/pnas.87.7.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Au K G, Welsh K, Modrich P. J Biol Chem. 1992;267:12142–12148. [PubMed] [Google Scholar]
- 12.Yanisch-Perron C, Vieira J, Messing J. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 13.Su S-S, Modrich P. Proc Natl Acad Sci USA. 1986;83:5057–5061. doi: 10.1073/pnas.83.14.5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grilley M, Welsh K M, Su S-S, Modrich P. J Biol Chem. 1989;264:1000–1004. [PubMed] [Google Scholar]
- 15.Welsh K M, Lu A-L, Clark S, Modrich P. J Biol Chem. 1987;262:15624–15629. [PubMed] [Google Scholar]
- 16.Bebenek K, Kunkel T A. Methods Enzymol. 1995;262:217–232. doi: 10.1016/0076-6879(95)62020-6. [DOI] [PubMed] [Google Scholar]
- 17.Messing J. Methods Enzymol. 1983;101:20–78. doi: 10.1016/0076-6879(83)01005-8. [DOI] [PubMed] [Google Scholar]
- 18.Tindall K R, Kunkel T A. Biochemistry. 1988;27:6008–6013. doi: 10.1021/bi00416a027. [DOI] [PubMed] [Google Scholar]
- 19.Dunning A M, Talmud P, Humphries S E. Nucleic Acids Res. 1988;16:10393. doi: 10.1093/nar/16.21.10393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen J, Sahota A, Stambrook P J, Tischfield J A. Mutat Res. 1991;249:169–176. doi: 10.1016/0027-5107(91)90143-c. [DOI] [PubMed] [Google Scholar]
- 21.Modrich P. Annu Rev Genet. 1991;25:229–253. doi: 10.1146/annurev.ge.25.120191.001305. [DOI] [PubMed] [Google Scholar]