

Abstract
Background
Adipocyte-derived leucine aminopeptidase (ALAP) is a recently identified member of the M1 family of zinc-metallopeptidases and is thought to play a role in blood pressure control through inactivation of angiotensin II and/or generation of bradykinin. The enzyme seems to be particularly abundant in the heart. Recently, the Arg528-encoding allele of the ALAP gene was shown to be associated with essential hypertension.
Methods
We evaluated the influence of this polymorphism on the change in left ventricular mass index in 90 patients with essential hypertension and echocardiographically diagnosed left ventricular hypertrophy, randomised in a double-blind study to receive treatment with either the angiotensin II type I receptor antagonist irbesartan or the beta1-adrenoceptor blocker atenolol for 48 weeks. Genyotyping was performed using minisequencing.
Results
After adjustment for potential covariates (blood pressure and left ventricular mass index at baseline, blood pressure change, age, sex, dose and added antihypertensive treatment), there was a marked difference between the Arg/Arg and Lys/Arg genotypes in patients treated with irbesartan; those with the Arg/Arg genotype responded on average with an almost two-fold greater regression of left ventricular mass index than patients with the Lys/Arg genotype (-30.1 g/m2 [3.6] vs -16.7 [4.5], p = 0.03).
Conclusions
The ALAP genotype seems to determine the degree of regression of left ventricular hypertrophy during antihypertensive treatment with the angiotensin II type I receptor antagonist irbesartan in patients with essential hypertension and left ventricular hypertrophy. This is the first report of a role for ALAP/aminopeptidases in left ventricular mass regulation, and suggests a new potential target for antihypertensive drugs.
Keywords: Aminopeptidase, irbesartan, hypertension, polymorphism, left ventricular hypertrophy, angiotensin, pharmacogenomics, bradykinin.
Background
Hypertension is associated with a number of adverse morphologic and functional changes in the cardiovascular system such as left ventricular (LV) hypertrophy, a condition associated with an increased risk of both cardiovascular morbidity and mortality and also all-cause mortality [1-4]. The effects of angiotensin II on blood pressure and on structural cardiac and vascular remodelling indicate that the renin-angiotensin system plays an important part in the development of LV hypertrophy. Most angiotensin II is generated in two sequential steps: renin catalyzes the conversion of angiotensinogen to angiotensin I, which is subsequently hydrolyzed by angiotensin-converting enzyme (ACE) to form angiotensin II [5].
ACE, however, not only generates angiotensin II but also degrades bradykinin. Kinetic studies have shown that bradykinin is the preferred ACE substrate [6]. Bradykinin mediates important cardiovascular effects that may protect against LV hypertrophy. All components of a functional bradykinin system are expressed in the heart, and bradykinin clearly mediates important cardiovascular effects, such as negative inotropism and inhibition of myocardial growth [6]. For instance, a B2 bradykinin-receptor antagonist significantly attenuated the antihypertensive effect of ACE inhibitors in humans, and the cardioprotective effect of ACE inhibitors was lost in B2 bradykinin-receptor knock-out mice [6]. It thus seems probable that bradykinin is an important counteractive substance of the cardiac effects of the renin-angiotensin system.
Aminopeptidases play a role in the metabolism of several peptides [7] that may be involved in blood pressure regulation and the pathogenesis of hypertension [8,9]. Adipocyte-derived leucine aminopeptidase (ALAP) has recently been identified as a member of the M1 family of zinc-metallopeptidases [10,11]. ALAP was shown to hydrolyze a variety of bioactive peptides in vitro, including angiotensin II and kallidin [10]. ALAP is widely expressed in human tissues. Its presence in the cortex of the kidney may be especially interesting due to the presence of kallikrein. It also seems to be particularly abundant in the heart [12]. The enzyme is thought to play a role in the regulation of blood pressure through inactivation of angiotensin II and/or generation of bradykinin [10].
Recently, the Arg528 variant of the Lys528Arg (A1583G) polymorphism in the ALAP gene was shown to be associated with essential hypertension [13]. Since ALAP seems to be particularly abundant in the heart, it would seem reasonable to believe that the enzyme could be involved in the modulation of LV mass. No study has yet investigated the effect of this polymorphism on the change in LV mass during antihypertensive treatment. Therefore we chose to study the relationship between this specific polymorphism and the change in LV mass index (LVMI) in hypertensive patients with LV hypertrophy during antihypertensive treatment with either the AT1-receptor antagonist irbesartan or the beta1-adrenoceptor blocker atenolol.
Methods
The subjects participated in the SILVHIA trial, which has been described in detail earlier [14]. Briefly, Caucasian men and women with mild-to-moderate essential hypertension, and echocardiographically verified LV hypertrophy were enrolled, with the primary goal of evaluating the efficacy of irbesartan compared to atenolol on blood pressure reduction and regression of LV hypertrophy. LV hypertrophy was considered present if LVMI was >131 g/m2 for men and >100 g/m2 for women. The Penn convention was used for calculation of LV mass, which was corrected for body mass index (LVMI). The inclusion criteria constituted a diastolic blood pressure (DBP) of 90–115 mmHg. Secondary hypertension was excluded by means of a physical examination and routine laboratory tests. All antihypertensive agents were withdrawn before the start of a 4–6 week, single-blind, placebo lead-in period, after which the patients received either irbesartan 150 mg or atenolol 50 mg once daily as monotherapy in a double-blind fashion. The doses were doubled after six weeks if DBP was ≥ 90 mmHg. If DBP remained ≥ 90 mmHg at week 12, hydrochlorothiazide (12.5–25 mg once daily) was added. At week 24, felodipine (5–10 mg once daily) was added if required. In all, 101 patients completed the whole 48 week study, for whom DNA and echocardiographic data were available in 90. The appropriate ethics committees approved the study, the participating patients gave their informed consent, and the study was completed in accordance with institutional guidelines.
Genomic DNA was extracted from EDTA-blood using spin columns (QIAamp® DNA Blood Mini Kit, QIAGEN, Germany). PCR was conducted using AmpliTaq™ Gold DNA polymerase (Applied Biosystems, USA) with primers described earlier[13] manufactured by Interactiva (The Virtual Laboratory, Germany) with the exception of biotinylation at the 5'-end of the forward primer. 50 μl reaction volumes containing PCR Buffer II, 3.5 mmol/L MgCl2, 0.25 mmol/L of each dNTP, 0.4 μmol/L of each primer, 1 U of Taq DNA polymerase and approximately 100 ng of genomic DNA were used. PCR conditions consisted of an initial activation step at 95°C for 10 min, followed by 40 cycles of 95°C for 30 s, 61°C for 30 s and 72°C for 60 s, after which a final elongation step at 72°C for 10 min concluded the reaction (GeneAmp PCR system 9700, Applied Biosystems, USA). Genotyping for the ALAP polymorphism was conducted with solid phase minisequencing follwing a protocol described earlier [15-17]. The detection primer used was 5'-GAA CAC TTG GAC ACT GCA GA-3'.
Data are presented as mean values ± SE. The estimated adjusted mean difference in LVMI change, as well as echocardiographic measurements, at 48 weeks between the genotypes (three categories) was calculated with the GLM (general linear models) procedure of the SAS software (SAS Institute, USA) for each treatment group. Two different models were used: one univariate and one multivariate model including the potential covariates age, systolic and diastolic blood pressure (SBP and DBP), LVMI or the specific echocardiographic parameter at study entry, dose of atenolol or irbesartan, change in SBP and DBP (all continuous), sex, as well as the addition of hydrochlorthiazide or felodipine (yes/no). BMI was also included in the multivariate model for changes in echocardiographic measurements. A p value of <0.05 was considered significant.
Results
Genotype distribution (46 Arg/Arg [51%], 35 Lys/Arg [39%], and 9 Lys/Lys [10%]) was consistent with Hardy-Weinberg equilibrium. Fourty seven patients (22 Arg/Arg, 19 Lys/Arg and 6 Lys/Lys) were given atenolol and 43 (24 Arg/Arg, 16 Lys/Arg and 3 Lys/Lys) irbesartan. Among patients given atenolol, 57% received 50 mg, 39% 100 mg and the remaining either 75 mg or 25 mg. Among patients given irbesartan, 66% received 300 mg, 29% 150 mg, and the remaining either 75 or 225 mg. A total of 57% of the patients required additional treatment with felodipine and/or hydrochlorothiazide. Baseline characteristics of the patients stratified by treatment and genotype are shown in table 1. There were no significant differences between the genotypes in any group, except for baseline DBP in the atenolol group, where patients with the Lys/Lys genotype had a significantly higher level than those with the Lys/Arg genotype (p = 0.01),
Table 1.
Baseline patient characteristics stratified by treatment and genotype.
| Irbesartan | Lys/Lys (n = 3) | Lys/Arg (n = 16) | Arg/Arg (n = 24) |
| Gender (male/female) | 1/2 | 11/5 | 13/11 |
| Age (years) | 57.3 [4.1] | 52.6 [1.6] | 53.7 [1.8] |
| BMI (kg/m2) | 26.8 [1.8] | 27.2 [0.7] | 27.0 [0.7] |
| SBP (mmHg) | 159.6 [10.6] | 161.7 [4.8] | 164.0 [4.0] |
| DBP (mmHg) | 105.6 [2.9] | 104.1 [2.0] | 104.2 [1.2] |
| IVS (mm) | 1.2 [0.1] | 1.4 [0.05] | 1.4 [0.05] |
| PWT (mm) | 1.0 [0.04] | 1.1 [0.03] | 1.1 [0.04] |
| LVEDD (mm) | 4.9 [0.2] | 5.1 [0.1] | 5.1 [0.1] |
| LVMI (g/m2) | 126.2 [7.6] | 143.5 [5.0] | 154.8 [8.5] |
| Atenolol | Lys/Lys (n = 6) | Lys/Arg (n = 19) | Arg/Arg (n = 22) |
| Gender (male/female) | 6/0 | 11/8 | 14/8 |
| Age (years) | 51.8 [3.5] | 55.5 [1.9] | 52.7 [1.8] |
| BMI (kg/m2) | 26.9 [0.9] | 26.9 [0.9] | 27.8 [0.9] |
| SBP (mmHg) | 170.1 [8.6] | 159.8 [5.4] | 154.0 [3.2] |
| DBP (mmHg) | 109.0 [2.2] | 99.8 [2.0] | 102.6 [1.6] |
| IVS (mm) | 1.3 [0.09] | 1.3 [0.05] | 1.3 [0.04] |
| PWT (mm) | 1.0 [0.03] | 1.0 [0.03] | 1.1 [0.03] |
| LVEDD (mm) | 5.6 [0.3] | 5.1 [0.1] | 5.1 [0.1] |
| LVMI (g/m2) | 159.7 [12.5] | 140.5 [5.5] | 146.3 [5.6] |
BMI = body mass index; SBP = systolic blood pressure; DBP = diastolic blood pressure; IVS = intraventricular septum; PWT = posterior wall thickness; LVEDD = left ventricular end-diastolic diameter; LVMI = left ventricular mass index. Mean values ± SE.
As was previously shown in the SILVHIA trial [14], both irbesartan and atenolol progressively reduced LVMI by 26 (p < 0.001) and 14 g/m2 (p < 0.001) (16 and 9%), respectively, at week 48, with a greater reduction in the irbesartan group (p = 0.02). After stratification by ALAP genotype, there was a statistically significant regression of LVMI only among patients with the Arg/Arg genotype in both treatment groups (-33.6 g/m2 [6.1] in the irbesartan group and -15.5 g/m2 [3.0] in the atenolol group, p < 0.0001 for both), and among patients with the Lys/Arg genotype in the irbesartan group (-14.9 g/m2 [5.6], p = 0.02). There were no statistically significant differences between genotypes and change in intraventricular septum and posterior wall thickness or left ventricular end diastolic diameter in the two treatment groups (table 2). Overall uni- and multivariate changes in LVMI and in blood pressure change at 48 weeks, stratified by treatment and genotype, are shown in table 3. According to the multivariate model there was a significant difference in LVMI change between the Arg/Arg and Lys/Arg genotype among patients given irbesartan (figure 1). Patients with the Arg/Arg genotype responded with a greater regression of LVMI than patients with the Lys/Arg genotype (-30.1 g/m2 [3.6] vs -16.7 [4.5], p = 0.03). Patients with the Lys/Lys genotype were too few (n = 3) to allow statistical comparison in the irbesartan group. No significant differences were found between genotypes among patients given atenolol. Nor were there any significant differences in blood pressure change between genotypes in any treatment group at 48 weeks (table 3).
Table 2.
Change in echocardiographic measurements at 48 weeks stratified by treatment and genotype
| Irbesartan | Atenolol | |||||
| Lys/Lys (n = 3) | Lys/Arg (n = 16) | Arg/Arg (n = 24) | Lys/Lys (n = 6) | Lys/Arg (n = 19) | Arg/Arg (n = 22) | |
| IVS (mm) | -0.12 [0.1] | -0.21 [0.04] | -0.19 [0.04] | -0.15 [0.06] | -0.11 [0.04] | -0.14 [0.03] |
| IVS (mm) – multivariate | -0.11 [0.1] | -0.22 [0.04] | -0.18 [0.03] | -0.20 [0.07] | -0.14 [0.04] | -0.099 [0.04] |
| PWT (mm) | -0.043 [0.07] | -0.055 [0.03] | -0.13 [0.03] | -0.11 [0.05] | -0.041 [0.03] | -0.13 [0.03] |
| PWT (mm) – multivariate | -0.032 [0.07] | -0.073 [0.03] | -0.12 [0.02] | -0.070 [0.06] | -0.051 [0.03] | -0.13 [0.03] |
| LVEDD (mm) | 0.077 [0.2] | 0.17 [0.09] | -0.063 [0.07] | -0.037 [0.1] | 0.11 [0.07] | 0.17 [0.07] |
| LVEDD (mm) – multivariate | -0.13 [0.2] | 0.15 [0.07] | -0.029 [0.06] | 0.016 [0.2] | 0.11 [0.08] | 0.17 [0.07] |
IVS = intraventricular septum; PWT = posterior wall thickness; LVEDD = left ventricular end-diastolic diameter. Mean values ± SE.
Table 3.
LVMI and blood pressure change at 48 weeks stratified by treatment and genotype
| Irbesartan | Atenolol | |||||
| Lys/Lys (n = 3) | Lys/Arg (n = 16) | Arg/Arg (n = 24) | Lys/Lys (n = 6) | Lys/Arg (n = 19) | Arg/Arg (n = 22) | |
| DBP change (mmHg) | -14.8 [8.1] | -18.2 [2.3] | -19.5 [1.5] | -19.6 [3.3] | -12.8 [1.8] | -17.5 [1.4] |
| DBP change (mmHg) | -14.8 [8.1] | -18.2 [2.3] | -19.5 [1.5] | -19.6 [3.3] | -12.8 [1.8] | -17.5 [1.4] |
| SBP change (mmHg) | -9.1 [15.3] | -26.3 [5.1] | -31.5 [4.1] | -27.1 [5.9] | -19.3 [3.7] | -21.2 [3.3] |
| LVMI change (g/m2) | -12.2 [15.3] | -14.9 [6.6] | -33.6 [5.4] | -25.2 [8.6] | -9.0 [4.8] | -15.5 [4.7] |
| LVMI change (g/m2) – multivariate | -31.6 [11.6] | -16.7 [4.5] | -30.1 [3.6] * | -22.2 [9.6] | -13.3 [5.0] | -12.6 [4.5] |
SBP = systolic blood pressure; DBP = diastolic blood pressure; LVMI = left ventricular mass index. Mean values ± SE. * p = 0.03 between Arg/Arg and Lys/Arg.
Figure 1.
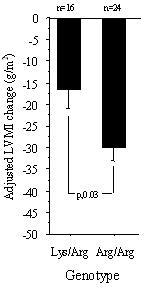
Adjusted mean LVMI change in the irbesartan group. LVMI = left ventricular mass index. Adjustment was made for age, dose, baseline systolic and diastolic blood pressure (SBP and DBP) and LVMI, changes in SBP and DBP, as well as additional antihypertensive medication. Mean values ± SE.
Discussion
To our knowledge, this is the first study investigating the effect of the Lys528Arg polymorphism in the ALAP gene on the change in LV mass during antihypertensive treatment. Our results suggest that patients with hypertensive LV hypertrophy that are homozygous for the Arg-encoding allele respond with a greater reduction of LV mass to the AT1 receptor antagonist irbesartan than patients with the Lys/Arg phenotype. At present, there is no evidence that this polymorphism causes functional alterations in the ALAP protein. However, the Lys at amino acid 528 is conserved in ALAP, leucyl/cystinyl aminopeptidase (LNPEP), rat puromycin-insensitive leucyl-specific aminopeptidase and mouse vascular endothelial growth factor (VEGF)-induced aminopeptidase [13]. Albeit, conservation of an amino acid does not necessarily indicate functional importance of the site. Recombinant expression experiments comparing activities of the two variants of the enzyme would elucidate functional alterations caused by this polymorphism.
In one of the subgroups, the Arg/Arg genotype in the irbesartan treatment group, baseline LVMI was not completely normally distributed when normality was analyzed by the Shapiro-Wilks test. This, however, did not affect our conclusions. There was no significant baseline difference in LVMI between the Arg/Arg and Arg/Lys group given irbesartan. The p value for a difference between the groups with a t-test was 0.32 for the original data and 0.43 for the log-transformed data. The p values for the univariate and the multivariate adjusted difference in mean LVMI difference after 48 weeks between the groups remained nearly identical after log transformation of the LVMI data, and therefore skewness can not explain our finding. The LVMI data in the other subgroups displayed a normal distribution by the original data.
The hypothesis raised by Yamamoto et al [13] was that a reduced ALAP activity in the Arg528 form compared with the Lys528 form resulted in high blood pressure and hypertension due to insufficient inactivation of angiotensin II or decreased generation of bradykinin, or both. This may imply that patients homozygous for the Arg-encoding allele benefit more from treatment with an AT1 receptor antagonist than a beta1-adrenoceptor blocker, which was the case in our study. The results also suggest a role for ALAP/aminopeptidases in the regulation of LV mass, which is in accordance with Hattori et al that ALAP is particularly abundant in the heart. If so, aminopeptidases might be a new potential target for antihypertensive drugs.
The major limitation of the present study is the small number of subjects not allowing us to evaluate the regression of LVMI among those with the more uncommon Lys/Lys genotype in the irbesartan group. Also, the possibility that the studied polymorphism could be in linkage disequilibrium with another polymorphism cannot be ruled out. The strength of the study, on the other hand, is that the subjects represent a clinically well-characterised group, randomised to treatment in a prospective, double-blind trial. It would be of great interest to study the impact of this polymorphism on the incidence of cardiovascular end-points during AT1-receptor antagonist treatment in larger trials.
Conclusions
Our results suggest that patients with essential hypertension and LV hypertrophy and who carry the Arg528Arg ALAP phenotype respond with a greater regression of LVMI during treatment with the AT1 receptor antagonist irbesartan than those with the Lys528Arg genotype. To our knowledge, our results are the first to suggest that ALAP/aminopeptidases may play a role in the control of LV mass. This might be a potential target for new antihypertensive drugs.
Competing interests
Drs Lars Lind and Karl Peter Öhman became affiliated with AstraZeneca AB after completion of the SILVHIA trial.
Authors' contributions
PH participated in the current raising of the hypothesis, the DNA extraction, setting up and performance of the PCR and genotyping and in the writing of the manuscript. LL participated in the initiation of the study and the writing of the manuscript. KM participated in the statistical analyses and the writing of the manuscript. LK took part in the collection of the study material, scientific discussions and the writing of the manuscript. TK participated in the design of the study protocol, and acted as chairman of the steering committee of the SILVHIA-trial, was responsible for the core laboratory and took part in the writing of the manuscript. KM included and followed the main part of the patients in the study, prepared the database used and took part in the writing of the manuscript. KPÖ participated in the design, patient examination, laboratory analyses, data evaluation and manuscript preparation of the underlying SILVHIA-trial, and contributed in the current manuscript preparation. FN took part in the recruitment and treatment of the patients, participated in the analyses of the study results and in the writing of the manuscript. UL and ACS took part in the setting up of the genotyping equipment and in the writing of the manuscript. HM took part in the design of the present genetic study, the genetic analyses and in the writing of the manuscript. All authors have read and approved the final version of the manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
This work was supported by grants from the Swedish Foundation for Strategic Research and the Swedish Research Commitee. The SILVHIA trial was supported by grants from Karolinska Institutet, Stockholm, Sweden, Bristol-Meyers Squibb Pharmaceutical Research Institute, Princeton, NJ, USA, and Sanofi-Synthélabo, Paris, France.
Contributor Information
Par Hallberg, Email: par.hallberg@medsci.uu.se.
Lars Lind, Email: lars.lind@medsci.uu.se.
Karl Michaëlsson, Email: karl.michaelsson@ortopedi.uu.se.
Lisa Kurland, Email: lisa.kurland@medsci.uu.se.
Thomas Kahan, Email: th_kahan@algonet.se.
Karin Malmqvist, Email: per.malmqvist@mbox2.swipnet.se.
Karl Peter Öhman, Email: Peter.Ohman@astrazeneca.com.
Fredrik Nyström, Email: fredrik.h.nystrom@telia.com.
Ulrika Liljedahl, Email: ulrika.liljedahl@medsci.uu.se.
Ann-Christine Syvänen, Email: ann-christine.syvanen@medsci.uu.se.
Hakan Melhus, Email: hakan.melhus@medsci.uu.se.
References
- Koren MJ, Devereux RB, Casale PN, Savage DD, Laragh JH. Relation of left ventricular mass and geometry to morbidity and mortality in uncomplicated essential hypertension. Ann Intern Med. 1991;114:345–352. doi: 10.7326/0003-4819-114-5-345. [DOI] [PubMed] [Google Scholar]
- Schillaci G, Verdecchia P, Porcellati C, Cuccurullo O, Cosco C, Perticone F. Continuous relation between left ventricular mass and cardiovascular risk in essential hypertension. Hypertension. 2000;35:580–586. doi: 10.1161/01.hyp.35.2.580. [DOI] [PubMed] [Google Scholar]
- Verdecchia P, Carini G, Circo A, Dovellini E, Giovannini E, Lombardo M, Solinas P, Gorini M, Maggioni AP. Left ventricular mass and cardiovascular morbidity in essential hypertension: the MAVI study. J Am Coll Cardiol. 2001;38:1829–1835. doi: 10.1016/S0735-1097(01)01663-1. [DOI] [PubMed] [Google Scholar]
- Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–1566. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- Unger T. The role of the renin-angiotensin system in the development of cardiovascular disease. Am J Cardiol. 2002;89:3A–9A; discussion 10A. doi: 10.1016/S0002-9149(01)02321-9. [DOI] [PubMed] [Google Scholar]
- Zuraw B. Bradykinin in protection against left-ventricular hypertrophy. Lancet. 2001;358:1116–1118. doi: 10.1016/S0140-6736(01)06300-0. [DOI] [PubMed] [Google Scholar]
- Taylor A. Aminopeptidases: structure and function. Faseb J. 1993;7:290–298. doi: 10.1096/fasebj.7.2.8440407. [DOI] [PubMed] [Google Scholar]
- Healy DP, Song L. Kidney aminopeptidase A and hypertension, part I: spontaneously hypertensive rats. Hypertension. 1999;33:740–745. doi: 10.1161/01.hyp.33.2.740. [DOI] [PubMed] [Google Scholar]
- Song L, Healy DP. Kidney aminopeptidase A and hypertension, part II: effects of angiotensin II. Hypertension. 1999;33:746–752. doi: 10.1161/01.hyp.33.2.746. [DOI] [PubMed] [Google Scholar]
- Hattori A, Kitatani K, Matsumoto H, Miyazawa S, Rogi T, Tsuruoka N, Mizutani S, Natori Y, Tsujimoto M. Characterization of recombinant human adipocyte-derived leucine aminopeptidase expressed in Chinese hamster ovary cells. J Biochem (Tokyo) 2000;128:755–762. doi: 10.1093/oxfordjournals.jbchem.a022812. [DOI] [PubMed] [Google Scholar]
- Matsumoto H, Rogi T, Yamashiro K, Kodama S, Tsuruoka N, Hattori A, Takio K, Mizutani S, Tsujimoto M. Characterization of a recombinant soluble form of human placental leucine aminopeptidase/oxytocinase expressed in Chinese hamster ovary cells. Eur J Biochem. 2000;267:46–52. doi: 10.1046/j.1432-1327.2000.00949.x. [DOI] [PubMed] [Google Scholar]
- Hattori A, Matsumoto H, Mizutani S, Tsujimoto M. Molecular cloning of adipocyte-derived leucine aminopeptidase highly related to placental leucine aminopeptidase/oxytocinase. J Biochem (Tokyo) 1999;125:931–938. doi: 10.1093/oxfordjournals.jbchem.a022371. [DOI] [PubMed] [Google Scholar]
- Yamamoto N, Nakayama J, Yamakawa-Kobayashi K, Hamaguchi H, Miyazaki R, Arinami T. Identification of 33 polymorphisms in the adipocyte-derived leucine aminopeptidase (ALAP) gene and possible association with hypertension. Hum Mutat. 2002;19:251–257. doi: 10.1002/humu.10047. [DOI] [PubMed] [Google Scholar]
- Malmqvist K, Kahan T, Edner M, Held C, Hagg A, Lind L, Muller-Brunotte R, Nystrom F, Ohman KP, Osbakken MD, Ostergern J. Regression of left ventricular hypertrophy in human hypertension with irbesartan. J Hypertens. 2001;19:1167–1176. doi: 10.1097/00004872-200106000-00023. [DOI] [PubMed] [Google Scholar]
- Syvanen AC. Detection of point mutations in human genes by the solid-phase minisequencing method. Clin Chim Acta. 1994;226:225–236. doi: 10.1016/0009-8981(94)90217-8. [DOI] [PubMed] [Google Scholar]
- Suomalainen A, Syvanen AC. Analysis of nucleotide sequence variations by solid-phase minisequencing. Methods Mol Biol. 1996;65:73–79. doi: 10.1385/0-89603-344-9:73. [DOI] [PubMed] [Google Scholar]
- Syvanen AC. Solid-phase minisequencing as a tool to detect DNA polymorphism. Methods Mol Biol. 1998;98:291–298. doi: 10.1385/0-89603-443-7:291. [DOI] [PubMed] [Google Scholar]