

Abstract
Background
β-blocker treatment has emerged as an effective treatment modality for heart failure. Interestingly, β-blockers can activate both pro-apoptotic and anti-apoptotic pathways. Nevertheless, the mechanism for improved cardiac function seen with β-blocker treatment remains largely unknown. Carvedilol is a non-selective β-blocker with α-receptor blockade and antioxidant properties. We therefore studied the impact of the effects of carvedilol in an animal model of end-stage heart failure.
Results
To test whether chronic treatment with β-blockade decreases apoptosis, we treated myopathic turkeys with two dosages of carvedilol, 1 mg/kg (DCM1) and 20 mg/kg (DCM20), for four weeks and compared them to non-treated DCM animals (DCM0) and to control turkeys (CON). Echocardiographic measurements showed that non-treated DCM animals had a significantly lower fractional shortening (FS) when compared to CON (68.73 ± 1.37 vs. 18.76 ± 0.59%, p < 0.001). Both doses of carvedilol significantly improved FS (33.83 ± 10.11 and 27.73 ± 6.18% vs. 18.76 ± 0.59 % for untreated DCM, p < 0.001). DCM left ventricles were characterized by a higher percentage of apoptotic nuclei when compared to CON (5.64 ± 0.49 vs. 1.72 ± 0.12%, respectively p < 0.001). Both doses of carvedilol significantly reduced the number of apoptotic nuclei (2.32 ± 0.23% and 2.36 ± 0.26% 1 mg and 20 mg/kg respectively).
Conclusions
Carvedilol improves ventricular function. Furthermore, treatment with carvedilol decreased the incidence of apoptosis in cardiac myocytes from failing hearts at both doses. These data suggest that the inhibition of apoptosis with carvedilol may lead to improvement in ventricular function and may underlie a beneficial effect of β-blockade independent of heart rate lowering effects.
Keywords: Heart failure, carvedilol myocyte, β-blocker
Background
Loss of myocytes is thought to contribute to the progressive decline in left ventricular function and the development of congestive heart failure. Recent studies have proposed that myocyte loss in cardiomyopathy can occur by apoptosis without an inflammatory response [reviewed in [1-3]]. In heart failure, growth stimulation initially occurs as a compensatory effort to meet chronically altered hemodynamic demands and is mediated by systemic and/or local up-regulation of mediators of adrenergic pathways and by various cytokines [4,5]. However, cytokines can be directly toxic to cardiac myocytes and result in increased apoptotic cell death [3,6]. Local up-regulation of angiotensin II induces immediate-early genes, which may lead to increased protein synthesis and myocardial hypertrophy or, alternatively, may up-regulate expression of apoptotic proteins (such as p53) in cardiac myocytes. The initial increase in circulating norepinephrine is thought to maintain cardiac function through inotropic mechanisms. However, a direct cardiac myocyte toxicity from norepinephrine is well recognized [6,7]. Furthermore, pressure-overload and stretch associated release of angiotensin II has been shown to induce myocyte apoptosis [8-10]. In an elegant paper by Telger et al., it has clearly been demonstrated that cardiac hypertrophy is initially preceded by a wave of apoptosis of cardiac myocytes followed by cell growth and a decrease in programmed cell death [10]. Apoptosis may, therefore, be the consequence of prolonged growth stimulation (adrenergic and renin-angiotensin axes) of adult cardiac myocytes, which are terminally differentiated. Similarly, certain cytokines (such as tumor necrosis factor) can induce growth as well as apoptosis [11]. As proposed by Telger et al. it is becoming clear that cell growth and programmed cell death are in fact two linked processes [10].
Over the last five years, β-blocker therapy has become one of the main treatment modalities for heart failure. Of particular interest is the use of carvedilol [12-16]. Carvedilol is a novel multiple-action neurohormonal antagonist. Its primary activities are nonselective β-adrenoceptor blockade, vasodilatation (mediated through α1-adrenoceptor blockade), and antioxidant activity [12]. In many clinical studies, carvedilol has been shown to improve left ventricular function and symptoms in patients with ischemic heart disease or idiopathic dilated cardiomyopathy [13-16] and to dramatically reduce mortality [17]. However, it remains unclear how carvedilol improves ventricular function in heart failure.
In an animal model of cardiomyopathy that has many similarities to human heart failure, we tested the hypothesis that a reduction in apoptosis may be responsible for the beneficial effects of carvedilol. As such, attenuation of apoptosis in the myocardium may represent a novel and important mechanism that contributes beneficial effects on the myocardium.
Results
Baseline Characteristics of Dilated Cardiomyopathic Animals
We have previously shown that removal of furazolidone from the feed after three weeks results in progressive cardiac dilatation and heart failure [18]. At the time of randomization of the DCM group into three groups i.e., DCM0 (no treatment), DCM1 (lower dose carvedilol) or DCM20 (higher dose carvedilol), fractional shortening was not different between the groups (p > 0.1). Evidence for severe heart failure was marked by significantly reduced left ventricle fractional shortening (controls 63.67 ± 1.04% n = 34 vs. DCM 11.95 ± 0.59% n = 18, p < 0.001).
Effect of Carvedilol on Ventricular Function
We investigated the effect of carvedilol by administering two different dosages (1 mg and 20 mg/kg body weight per day) to control and DCM turkeys for four weeks. We studied two dosages (pharmacological and non-pharmacological) as a difference in dose-dependency for cardiac function improvement has previously been demonstrated by us with another non-selective beta-blocker [18]. Furthermore, we wanted to determine if carvedilol might exert beneficial effects independent of heart rate effects. The lower dose (1 mg/kg) did not reduce heart rate. However, the higher dose reduced HR by approximately 15% for 8 hours.
In vivo contractile performance of the heart was assessed by the determination of fractional shortening of the left ventricle. As shown in Table 1 and Figure 1, fractional shortening was increased by 32 and 45% in DCM1 and DCM20 animals, respectively, compared to non-treated DCM animals (DCM0). Compared with baseline values at the time of randomization, the improvement in fractional shortening was of 57% in DCM1 and 65% in DCM20. Associated with improved cardiac function was a reduction in the incidence of apoptotic nuclei (see below).
Table 1.
Inhibition of Apoptosis by Carvedilol Correlates with Improvement in LV Function.
| Cont n = 13 | Cont1 n = 11 | Cont20 n = 10 | DCM0 n = 18 | DCM1 n = 13 | DCM20 n = 12 | |
| FS % | 68.73* ± 1.37 | 66.09* ± 0.90 | 66.99* ± 1.37 | 18.76 ± 1.83 | 27.73* ± 6.18 | 33.83* ± 10.11 |
| Apoptosis % | 1.72* ± 0.12 | 2.10 * ± 0.25 | 2.20* ± 0.24 | 5.64 ± 0.49 | 2.32* ± 0.23 | 2.36* ± 0.26 |
Fractional shortening (FS%) was calculated from echocardiographic images as described in the methods. Apoptotic nuclei were counted as described in methods. *p < 0.001 compared to DCM0.
Figure 1.
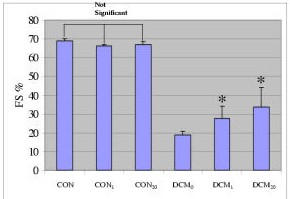
Effect of carvedilol on fractional shortening (FS) of the left ventricle. Carvedilol administration is associated with a improvement in left ventricular function DCM0 (n = 18), DCM1 (n = 13), DCM20 (n = 12). *p < 0.001 compared to DCM0. Healthy control turkeys (CON); control turkeys treated with two dosages of carvedilol, 1 mg/kg (CON1) and 20 mg/kg (CON20); dilated cardiomyopathic turkeys (DCM); non-treated DCM animals (DCM0); myopathic turkeys treated with two dosages of carvedilol, 1 mg/kg (DCM1) and 20 mg/kg (DCM20).
The body weight was significantly decreased in DCM animals compared to control animals (p < 0.001 compared to control) (Table 2). This animal model is associated with cachexia. Carvedilol treatment resulted in a slight gain of body weight, 15% in the DCM1 group and 20% in the DCM20 group compared with the non-treated DCM group (DCM0). Carvedilo also increased the body weight of control animals (p < 0.001). These data indicate a favorable effect on cachexia. In non-treated DCM animals the hearts weighed significantly less than control hearts (p = 0.01). DCM animals treated with carvedilol showed a significant increase in HW compared to hearts from non-treated DCM animals (p < 0.001). Therefore the HW/BW ratio was lower for hearts from carvedilol treated DCM animals compared to hearts from non-treated DCM animals. Interestingly, the HW/BW ratio was lower for carvedilol treated control animals compared to non-treated control animals. Left ventricle volume was significantly larger for non-treated DCM hearts. Carvedilol significantly decreased LV volume at both dosages.
Table 2.
Effect of Carvedilol on Gross Morphology
| HW (kg) | BW (kg) | HW/BW (%) | LV vol (ml) | |
| Control (n = 13) | 0.018 |
4.36# ± 0.16 | 0.4 |
0.24• ± 0.04 |
| Cont1 (n = 11) | 0.018 |
5.3# ± 0.14 | 0.35 |
0.37• ± 0.06 |
| Cont20 (n = 10) | 0.02 |
5.65# ± 0.09 | 0.35 |
0.37• ± 0.04 |
| DCM0 (n = 18) | 0.017 ± 0.001 | 2.61 ± 0.23 | 0.64 ± 0.049 | 9.36 ± 2.0 |
| DCM1 (n = 13) | 0.019 |
3.07# ± 0.29 | 0.60 |
6.28• ± 2.54 |
| DCM20 (n = 12) | 0.019 |
3.26# ± 0.29 | 0.58 |
5.38• ± 1.95 |
HW: heart weight ![]() p ≤ 0.01 compared to non-treated DCM group;
BW: body weight #p < 0.001 compared to non-treated DCM group;
HW/BW ratio
p ≤ 0.01 compared to non-treated DCM group;
BW: body weight #p < 0.001 compared to non-treated DCM group;
HW/BW ratio ![]() p < 0.04 compared to non-treated DCM
group; LV vol: atria were removed and the left ventricles were arrested
in diastole and filled with saline • p ≤ 0.008 compared to non-treated
DCM. Note the increase in body weight and heart weight with carvedilol
treatment. This resulted in a significant improvement in HW/BW ratio
despite having heavier hearts. The reduction in heart weight seen
in the non-treated DCM group was associated with a higher incidence
of apoptosis while the increased heart weight in hearts from DCM
animals treated with carvedilol was associated with a lower incidence
of apoptosis.
p < 0.04 compared to non-treated DCM
group; LV vol: atria were removed and the left ventricles were arrested
in diastole and filled with saline • p ≤ 0.008 compared to non-treated
DCM. Note the increase in body weight and heart weight with carvedilol
treatment. This resulted in a significant improvement in HW/BW ratio
despite having heavier hearts. The reduction in heart weight seen
in the non-treated DCM group was associated with a higher incidence
of apoptosis while the increased heart weight in hearts from DCM
animals treated with carvedilol was associated with a lower incidence
of apoptosis.
Effect of Carvedilol on Cardiac Myocyte Apoptosis
Evaluation of tissue sections by the TUNEL technique revealed only small numbers of TUNEL-positive cells in the control hearts (Table 1 and Figure 2A). Hearts from DCM animals had a significant increase in the number of TUNEL-positive cells compared to controls (5.64 ± 0.49% vs. 1.72 ± 0.12% DCM and control respectively, p < 0.001), indicating the occurrence of apoptosis (Figure 2B).
Figure 2.
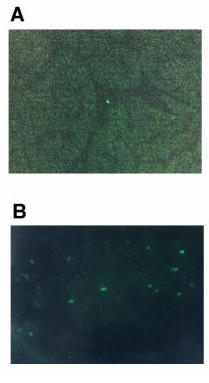
Detection of apoptotic cells by fluorescence microscopy. The TUNEL assay with fluorescein as the tag was used to stain the nuclei of apoptotic cells in frozen cardiac sections. (A) Control heart (containing essentially no TUNEL-positive cells in this field) and (B) a fluorescent micrograph of a representative field from a failing heart (DCM) containing a higher number of apoptotic nuclei. Micrographs were taken with 40X objective and reduced for reproduction.
In contrast to non-treated DCM hearts, myocardial tissue sections prepared from hearts from animals treated with carvedilol showed a significant reduction in the numbers of TUNEL-positive myocyte nuclei. An example of such an observation is depicted in Figure 3A and 3B (DCM1 and DCM20 respectively). Both the lower and the higher doses of carvedilol were effective in reducing apoptosis from 5.64 ± 0.49% in non-treated DCM animals to almost control levels 2.32 ± 0.23% (DCM1) and 2.36 ± 0.26% (DCM20), p < 0.001, compared to DCM0 respectively (Figure 3C).
Figure 3.
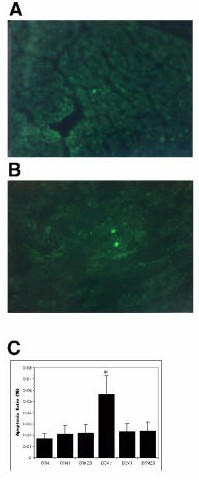
Effect of carvedilol on cardiac myocyte apoptosis. Failing hearts treated with carvedilol show reduced number of TUNEL-positive nuclei, (A) DCM1 and (B) DCM20. Both low and high doses of the drug inhibited myocyte apoptosis to almost control levels (C). No effect of carvedilol on apoptosis in control groups was observed (C). *p < 0.001 vs. control and carvedilol treated groups. Micrographs in (A) and (B) were taken with 40X objective and reduced for reproduction.
Discussion
Effect of Carvedilol on a Heart Failure Model
The furazolidone-induced turkey model is a well characterized experimental model of dilated cardiomyopathy with features very similar to those found in human heart failure [19-22]. Decreased contractile function (reduced fractional shortening of the left ventricle) and morphological changes (increased heart volumes, left ventricular dilatation, and myocyte hypertrophy) are characteristic features of this model [21,22].
β-blocker treatment has emerged as an effective treatment modality for heart failure. Both in animal and human studies of heart failure, β-adrenergic blockade intervention appears to have beneficial effects on cardiac myocyte function [1,23,24]. Of particular importance, studies using carvedilol have shown remarkable improvements in cardiac performance [12,13,15,16] and reduction in mortality in humans [17]. We have previously shown that propranolol, a nonselective β-blocker, is cardioprotective and prevents the development of heart failure in turkeys when given concurrently with furazolidone [21]. Similarly, carteolol, a nonselective β-blocker, resulted in improvement in ejection fraction, reduction in ventricular volumes, an increase in developed pressure as well as an increase in rate of survival in our turkey model [18]. Improvement in cardiac function was associated with cellular remodeling of cardiac myocytes. Cardiac myocytes from hearts treated with a non-selective β-blocker i.e., carteolol, had normal calcium cycling, normal myocardial energetics, normal β1 receptor density as well as regression of myocyte hypertrophy and reduced connective tissue content [18]. Furthermore, administration of carteolol has been reported to prevent the development of virally induced cardiomyopathy in a murine model [25]. In a canine model with left ventricular dysfunction produced by multiple sequential intracoronary microembolizations, long term treatment with metoprolol, a β1-selective blocker, has been reported to prevent the progression of LV systolic dysfunction and LV chamber dilatation [26]. Similarly, our present findings on the effect of carvedilol indicated improvement of left ventricular function and smaller LV volumes. Carvedilol prevented progression of LV systolic dysfunction and LV dilatation as previously reported in other models using β-blockers [18,26].
Animals with dilated cardiomyopathy which received carvedilol showed significant improvement in LV systolic function after 4 weeks. Notably, the improvement of LV systolic function in animals treated with carvedilol was associated with a decrease of LV volumes compared with non-treated DCM animals. Importantly, both doses of carvedilol improved LV contractile performance. Fractional shortening (%) was increased by an average of approximately 38% and impressively by 80% in some individual animals. These data seem to agree with the results of human clinical studies, and are suggestive that carvedilol not only slows deterioration of cardiac function, but also improves cardiac function.
The trend that the 20 mg/kg body weight dose might not have been well tolerated was demonstrated by some animals becoming moribund for a short period after dosing which might be due to a reduction in heart rate and cardiac output with resultant reflect tachycardia. We have previously shown a negative force-interval relationship (i.e., negative treppe) in failing hearts with this model [18]. This is in line with data from human studies, which showed that when first administered, β-blockers actually slow heart rate and diminish ejection fraction [27]. The higher dose of carvedilol significantly reduced heart rate and blood pressure for up to eight hours. Animals that tolerated the higher dose did nevertheless benefit from the treatment. This is similar to human clinical studies where greater benefit is derived from higher dosages, if tolerated [28].
Apoptosis in Animal Models of Heart Failure
Heart failure is characterized by progressive deterioration of global left ventricular function over time. The mechanisms responsible for the worsening of cardiac function are not clear. Loss of cardiac myocytes has been suspected to be a feature of the cardiomyopathic process that contributes to progressive decline in left ventricular function and the development of congestive heart failure [29]. Evidence supporting the concept of myocyte apoptosis occurrence and contribution in the progression of heart failure has been obtained from a variety of observations, including in vitro studies, experimental animal models of cardiac dysfunction, and studies on cardiac tissue obtained from patients with end-stage heart failure [5,7-9,29].
We sought to evaluate the incidence and the extent of apoptosis in our animal model of heart failure. Using the TUNEL technique as a means of detection, we were able to document the occurrence of apoptosis in ventricles from failing hearts, which was characterized by a higher percentage of apoptotic nuclei when compared to controls (5.64% vs. 1.72%). These apoptotic indexes are within the range of values in heart failure reported in the literature (0.2% to 35%) [3,30-33]. The high degree of variability reported in the literature in the magnitude of apoptosis may be due to the diversity and specificity of methods used to quantify apoptotic nuclei.
When reporting apoptotic nuclei using the TUNEL assay, two observations should be considered; 1) although apoptosis occurred in our model of heart failure, we might have missed, in random histological sections, evidence of apoptosis thereby under estimating the severity of programmed cell death due to the fact that apoptotic cells undergo rapid phagocytosis with the entire process lasting less than two hours in some cell systems [34], and 2) apoptosis in the present study was evaluated at a single time point when the heart was still undergoing LV remodeling. Hence, the documentation of apoptosis at this stage may not reflect the true magnitude of its occurrence during the initiation and transition to heart failure. In a rat model of pressure-overload hypertrophy produced by aortic banding, apoptosis appeared to peak at four days and gradually subsided after one month of aortic banding [9]. Therefore, a longitudinal study of apoptosis in failing hearts from initiation to transition to failure is worth pursuing. In this way, the incidence of apoptosis can be documented as the heart moves progressively from normal to compensated hypertrophy and finally to overt decompensated heart failure. These animals were in compensated heart failure and, often in the non-treated group, went into decompensated heart failure when stressed during echocardiographic examination.
With improved cardiac function, there should be a reduction in activation of the adrenergic-renin-angiotensin axes as well as a decrease in activated cytokines such as tumor-necrosis factor-α. In our model of heart failure, cardiac function correlated with the incidence of apoptosis. As described in Table 1 and Figure 3, there were a higher number of apoptotic cells in hearts with a reduction in fractional shortening. In contrast, control hearts with normal ventricular function showed low rates of apoptosis. The extent and occurrence of apoptosis may partially explain the lack of an overall increase in heart weight in hearts from non-treated DCM animals, despite a pronounced increase in heart size. These observations, and the results from several animal models, demonstrate that apoptotic cells are present in failing hearts, suggesting that apoptosis might be a mechanism involved, at least in part, in the progression of heart failure and the reduction of myocyte mass.
Carvedilol Inhibition of Apoptosis and Improvement of Ventricular Function
The extent of apoptosis in cardiomyopathic animals was significantly reduced by treatment with carvedilol. Both the lower and higher doses of carvedilol were effective in decreasing the rate of apoptosis (approximately a 58% reduction). This observation represents an important finding in demonstrating the effectiveness of a cardiovascular drug to protect against continual cardiac myocyte apoptosis in failing hearts in vivo. Furthermore, the inhibition of apoptosis by carvedilol was accompanied by signs of improvement in left ventricular function and heart size (i.e. reduced LV volume) in the myopathic animals. Whether the improved function, small heart size and decreased wall stress were responsible for a reduction in apoptosis is not known. However, the derived cardiac benefit was independent of heart rate effects because benefit was obtained at both dosages.
Our data support the conclusions of Rossig et al. in endothelial cells. They reported levels of apoptotic cells for control and NYHA III-IV hearts that were similar to our observations [35]. We similarly believe that the suppression of apoptosis by carvedilol is likely due to its antioxidative properties rather than the β-blocking effects [35]. We base this on our observation that a similar reduction in the incidence of apoptosis was seen at both a non-pharmacological (no effect on heart rate or blood pressure) as well as a pharmacological dose of carvedilol. In a canine model of chronic heart failure produced by multiple sequential intracoronary embolizations, metprolol, a β1 selective blocker, reduced the incidence of apoptosis supporting our experimental observations with carvedilol [36]. The incidence of apoptosis was 5.32 ± 0.77 in heart failure animals that were not treated which is similar to our findings of 5.64 ± 0.49 in hearts from untreated animals with heart failure. Recently, Li et al. have demonstrated that spontaneously hypertensive rats (SHR) with symptoms of heart failure had significantly higher levels of apoptotic myocytes than control myocytes [37]. When treated with an angiotensin-converting enzyme inhibitor, the number of apoptotic cells in the SHR with symptoms of heart failure was dramatically reduced to control levels; controls were SHR without symptoms of heart failure [37]. Kajstura et al. have also reported that angiotensin II increased the percentage of apoptotic cells in isolated adult rat ventricular myocytes, and this effect was mediated by AT-1 receptors [38]. Although ventricular function was not assessed in these studies, the observations raise the possibility that, like our findings with carvedilol, the beneficial effect of ACE-inhibitors or AT-1 receptor blockers in heart failure may in part be attributed to an inhibition of myocyte apoptosis with a resultant improvement in in vivo cardiac function, a concept that needs further study.
The data presented suggest that attenuation of apoptosis may underlie the beneficial effect of β-blockade on ventricular function, and that the inhibition of apoptosis can possibly lead to improvement in ventricular function. This represents a novel mechanism to slow the progressive deterioration of myocardial function that can occur in patients with cardiac failure. Because reactive oxygen radicals play a role in inducing cell apoptosis [39] and the potent antioxidant property of carvedilol [12], we speculate that the beneficial effects attributed to carvedilol (i.e. inhibition of apoptosis and improvement of LV function) are mediated, in part, by its prevention of oxygen-derived free radical damage [35,40]. Our data supports this idea.
Therapeutic Significance
With the clear demonstration that cardiac myocyte apoptosis is present in our heart failure model strategies for prevention of apoptosis can be tested. The clinical consensus has been that higher doses of β-blockade should incur more benefits on ventricular remodeling. However, our studies demonstrated similar benefits at a therapeutic and sub-therapeutic dose. In light of the beneficial effect of carvedilol on the progression of heart failure, and its unique multi-actions that are not shared by any other β-blocking drug or by any other agent currently used in the treatment of heart failure, our findings represent an important new mechanism of improved cardiac function.
The development of animal models of heart failure in which apoptosis is an important feature will allow the modulation of cell death pathways through targeted interventions. Understanding the role of specific signaling pathways in cardiac myocyte apoptosis and developing strategies to manipulate these intracellular pathways may provide in the near future novel therapeutic approaches for the management of heart failure.
Conclusions
In conclusion, our model of heart failure, which shows a high level of congruence with the human condition, has been shown to have a high level of apoptosis that corresponds with cardiac failure. Carvedilol, a non-selective β-blocker with α-blocking properties as well as antioxidant properties, significantly reduced the incidence of programmed cell death while improving cardiac function. This effect was independent of heart rate lowering effects, i.e. β-blockade. The beneficial effects of carvedilol can therefore be obtained with subtherapeutic doses. This is a novel observation and important in treating patients who may not tolerate higher doses (heart rate and blood pressure lowering) of carvedilol. The apoptotic process is linked to the development and progression of heart failure as well as the improvement in cardiac function seen with β-blockade.
Methods
Experimental Design
One day-old broad-breasted white turkey poults, obtained from a commercial breeder, were wing-banded for easy identification. At seven days of age, they were weighed and randomly divided into two groups using a random number generator. The control group (n = 34) was maintained on a normal ration, free from any additives, and the experimental group (n = 91) was fed 700 parts per million furazolidone (Fz) for three weeks. Furazolidone was stopped after day twenty-eight and the animals were randomized into three groups: one group did not receive carvedilol treatment, DCM0 (n = 52), whereas the other two groups, DCM1 (n = 19) and DCM20 (n = 20), were treated with carvedilol in the same dosages as respective aged-matched control animals (see following). Similarly, age matched control animals were randomized into three groups: one group received no pharmacological agent (Con0. n = 13), whereas the other two groups received different dosages of carvedilol, 1 mg/kg (Con1 n = 11) or 20 mg/kg body weight (Con20 n = 10). The lower dose of carvedilol did not lower heart rate or blood pressure. However, the higher dose of carvedilol resulted in a significant reduction in heart rate (15%) and blood pressure (9%) for up to eight hours. We performed dose range studies using nine concentrations of carvedilol (data not shown). At the time of euthanasia, body weights, heart weights (after removal of the atria), and LV volumes were obtained on all experimental animals.
Echocardiographic Measurements
Echocardiographic views were obtained using a 7.5 MHz transducer on unsedated and quietly resting animals. Several cardiac cycles were recorded on a videotape and the two dimensional images subsequently played back for analysis. Diastolic left ventricular internal dimension (LVIDd) and systolic left ventricular internal dimension (LVIDs) were also measured and used to determine the fractional shortening. Fractional shortening (%) of the left ventricle was calculated as (LVIDd - LVIDs/ LVIDd) × 100.
TUNEL
Randomly selected hearts from control and cardiomyopathic turkey poults were rapidly removed, flushed with PBS and infused with a tissue embedding medium (Tissue-Tek, OCT Compound, Miles Inc., Elkhart, IN) and frozen in liquid nitrogen. Serial 7 μm cross-sections from heart blocks were cut and fixed to coated slides. Frozen heart sections were fixed in 10% neutral formalin (4% formaldehyde) for 10 minutes at room temperature and post-fixed in Methanol/Acetone (1:1) for 10 minutes at -20°C. Detection of apoptotic cardiac myocytes was achieved by direct immunofluorescence detection of digoxigenin-labeled genomic DNA using the ApopTag Plus in Situ apoptosis detection kit – Fluorescein (Intergen, Purchase, NY). This method used the TUNEL technique to stain DNA fragments in nuclei of apoptotic cells. Tissue sections were then counter-stained with Hoechst 33258 stain (1 μg/ml) (Sigma), and viewed with an epifluorescence microscope (Zeiss Axiophot) equipped with filter sets for fluorescein and Hoechst staining. To quantify apoptosis, four to five randomly selected microscopic fields per section were examined. The percentage of apoptotic cells was determined by counting the total number of nuclei and TUNEL positive nuclei (apoptotic myocytes). Samples were numbered to conceal the identity of different groups during counting. Sections of interest were photographed using a microscope-integrated 35-mm camera.
Statistical Analysis
Data given in the text are means ± SD. The difference between the means was evaluated using student's t test. P < 0.05 was considered significant.
Authors' contributions
CCO drafted and edited the manuscript. AAD carried out creation of the animal model. ECHOs and the echocardiographic measurements were analyzed by NL and JKG. DL, KS and GJ carried out the TUNEL assays. CCO, CPM, RJH and JKG participated in the design of the study. CCO and JKG performed the statistical analysis, and data interpretation. MXL performed figure generation and animal dosing. RJH and JKG conceived of the study, and participated in its design and coordination. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
This work is supported by a research contract from SmithKline Beecham Pharmaceuticals to Gwathmey, Inc. and in part by grants from NIH: HL-49574 & HL-52249 (JKG), NIH HL49574 Minority Supplement (CCO) and T32 HL07374-23 (MXL).
Contributor Information
Chukwuka C Okafor, Email: okaforc1@mail.nih.gov.
Cynthia Perreault-Micale, Email: cperreault@gwathmey.com.
Roger J Hajjar, Email: rhajjar@partners.org.
Djamel Lebeche, Email: lebeche@cvrc.mgh.harvard.edu.
Klara Skiroman, Email: kskiromam@gwathmey.com.
George Jabbour, Email: gjabbour@cvrc.mgh.harvard.edu.
Angelia A Doye, Email: adoye@gwathmey.com.
Michael X Lee, Email: mlee@gwathmey.com.
Nancy Laste, Email: nlaste@gwathmey.com.
Judith K Gwathmey, Email: gwathmey@earthlink.com.
References
- Eichhorn EJ, Bristow MR. Medical therapy can improve the biological properties of the chronically failing heart. A new era in the treatment of heart failure. Circulation. 1996;94:2285–2296. doi: 10.1161/01.cir.94.9.2285. [DOI] [PubMed] [Google Scholar]
- Yeh ETH. Life and death in the cardiovascular system. Circulation. 1997;95:782–786. doi: 10.1161/01.cir.95.4.782. [DOI] [PubMed] [Google Scholar]
- Haunstetter A, Izumo S. Apoptosis: basic mechanisms and implications for cardiovascular disease. Circ Res. 1998;82:1111–1129. doi: 10.1161/01.res.82.11.1111. [DOI] [PubMed] [Google Scholar]
- Chien KR, Zhu H, Knowlton KU, Miller-Hance W, van-Bilsen M, O'Brien TX, Evans SM. Transcriptional regulation during cardiac growth and development. Annu Rev Physiol. 1993;55:77–95. doi: 10.1146/annurev.ph.55.030193.000453. [DOI] [PubMed] [Google Scholar]
- Katz AM. Cell death in the failing heart: role of an unnatural growth response to overload. Clinical Cardiology. 1995;18:IV36–44. doi: 10.1002/clc.4960181607. [DOI] [PubMed] [Google Scholar]
- Singh K, Xiao L, Remondino A, Sawyer DB, Colucci WS. Adrenergic regulation of cardiac myocyte apoptosis. J Cell Physiol. 2002;201: 189:257–265. doi: 10.1002/jcp.10024. [DOI] [PubMed] [Google Scholar]
- Mann DL, Kent RL, Parsons B, Cooper G. Adrenergic effects on the biology of the adult mammalian cardiocyte. Circulation. 1992;85:790–804. doi: 10.1161/01.cir.85.2.790. [DOI] [PubMed] [Google Scholar]
- Leri A, Claudio PP, Li Q, Wang X, Reiss K, Wang S, Malhotra A, Kajstura J, Anversa P. Stretch-mediated release of angiotensin II induces myocyte apoptosis by activating p53 that enhances the local renin-angiotensin system and decreases the Bcl-2-to-Bax protein ratio in the cell. J Clin Invest. 1998;101:1326–1342. doi: 10.1172/JCI316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez J, Fortuno MA, Ravassa S. Apoptosis in hypertensive heart disease. Curr Opin Cardiol. 1998;13:317–325. doi: 10.1097/00001573-199809000-00005. [DOI] [PubMed] [Google Scholar]
- Teiger E, Than VD, Richard L, Wisnewsky C, Tea BS, Gaboury L, Tremblay J, Schwartz K, Hamet P. Apoptosis in pressure overload-induced heart hypertrophy in the rat. J Clin Invest. 1996;97:2891–2897. doi: 10.1172/JCI118747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krown KA, Page MT, Nguyen C, Zechner D, Gutierrez V, Comstock KL, Glembotski CC, Quintana PJ, Sabbadini RA. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J Clin Invest. 1996;98:2854–2865. doi: 10.1172/JCI119114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerstein GZ, Poste G, Ruffolo RR. Carvedilol update III: rationale for use in congestive heart failure. Drugs of Today. 1995;31:307–326. [Google Scholar]
- Olsen SL, Gilbert EM, Renlund DG, Taylor DO, Yanowitz FD, Bristow MR. Carvedilol improves left ventricular function and symptoms in chronic heart failure: a double-blind randomized study. J Am Coll Cardiol. 1995;25:1225–1231. doi: 10.1016/0735-1097(95)00012-S. [DOI] [PubMed] [Google Scholar]
- Di Lenarda A, Sabbadini G, Salvatore L, Sinagra G, Mestroni L, Pinamonti B, Gregori D, Ciani F, Muzzi A, Klugmann S, Camerini F. Long-term effects of carvedilol in idiopathic dilated cardiomyopathy with persistent left ventricular dysfunction despite chronic metoprolol. The Heart-Muscle Disease Study Group. J Am Coll Cardiol. 1999;33:1926–1934. doi: 10.1016/S0735-1097(99)00134-5. [DOI] [PubMed] [Google Scholar]
- Quaife RA, Gilbert EM, Christian PE, Datz FL, Mealey PC, Volkman K, Olsen SL, Bristow MR. Effects of carvedilol on systolic and diastolic left ventricular performance in idiopathic dilated cardiomyopathy or ischemic cardiomyopathy. Am J Cardiol. 1996;78:779–784. doi: 10.1016/S0002-9149(96)00420-1. [DOI] [PubMed] [Google Scholar]
- Frishman WH. Drug therapy: Carvedilol. N Engl J Med. 1998;339:1759–1765. doi: 10.1056/NEJM199812103392407. [DOI] [PubMed] [Google Scholar]
- Packer M, Bristow MR, Cohn JN, Colucci WS, Fowler MB, Gilbert EM, Shusterman NH. The Effect of Carvedilol on Morbidity and Mortality in Patients with Chronic Heart Failure. N Engl J Med. 1996;334:1349–1355. doi: 10.1056/NEJM199605233342101. [DOI] [PubMed] [Google Scholar]
- Gwathmey JK, Kim CS, Hajjar RJ, Khan F, DiSalvo TG, Matsumori A, Bristow MR. Cellular and molecular remodeling in a heart failure model treated with the beta-blocker carteolol. Am J Physiol. 1999;276:H1678–1690. doi: 10.1152/ajpheart.1999.276.5.H1678. [DOI] [PubMed] [Google Scholar]
- Jankus EF, Noren GR, Staley NA. Furazolidone-induced cardiac dilatation in turkeys. Avian Dis. 1972;16:958–961. [PubMed] [Google Scholar]
- Czarnecki CM, Jankus EF, Hultgren BD. Effects of furazolidone on the development of cardiomyopathies in turkey poults. Avian Dis. 1974;18:125–133. [PubMed] [Google Scholar]
- Glass MG, Fuleihan F, Liao R, Lincoff AM, Chapados R, Hamlin R, Apstein CS, Allen PD, Ingwall JS, Hajjar RJ. Differences in cardioprotective efficacy of adrenergic receptor antagonists and Ca2+ channel antagonists in an animal model of dilated cardiomyopathy. Effects on gross morphology, global cardiac function, and twitch force. Circ Re. 1993;73:1077–1089. doi: 10.1161/01.res.73.6.1077. [DOI] [PubMed] [Google Scholar]
- Hajjar RJ, Liao R, Young JB, Fuleihan F, Glass MG, Gwathmey JK. Pathophysiological and biochemical characterisation of an avian model of dilated cardiomyopathy: comparison to findings in human dilated cardiomyopathy. Cardiovasc Res. 1993;27:2212–2221. doi: 10.1093/cvr/27.12.2212. [DOI] [PubMed] [Google Scholar]
- Mann DL, Kent RL, Parsons B, Cooper G., 4th Adrenergic effects on the biology of the adult mammalian cardiocyte. Circulation. 1992;85:790–804. doi: 10.1161/01.cir.85.2.790. [DOI] [PubMed] [Google Scholar]
- Doughty RN, MacMahon S, Sharpe N. Beta-blockers in heart failure: promising or proved? J Am Coll Cardiol. 1994;23:814–821. doi: 10.1016/0735-1097(94)90773-0. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Matsumori A, Okada I, Yamada T, Kawai C. Beta-blocker treatment of dilated cardiomyopathy. Beneficial effect of carteolol in mice. Circulation. 1991;83:2021–2028. doi: 10.1161/01.cir.83.6.2021. [DOI] [PubMed] [Google Scholar]
- Sabbah HN, Shimoyama H, Kono T, Gupta RC, Sharov VG, Scicli G, Levine TB, Goldstein S. Effects of long-term monotherapy with enalapril, metoprolol, and digoxin on the progression of left ventricular dysfunction and dilation in dogs with reduced ejection fraction. Circulation. 1994;89:2852–2859. doi: 10.1161/01.cir.89.6.2852. [DOI] [PubMed] [Google Scholar]
- Hall SA, Cigarroa CG, Marcoux L, Risser RC, Grayburn PA, Eichhorn EJ. Time course of improvement in left ventricular function, mass and geometry in patients with congestive heart failure treated with beta-adrenergic blockade. J Am Coll Cardiol. 1995;25:1154–1161. doi: 10.1016/0735-1097(94)00543-Y. [DOI] [PubMed] [Google Scholar]
- Bristow MR, Gilbert EM, Abraham WT, Adams KF, Fowler MB, Hershberger RE, Kubo SH, Narahara KA, Ingersoll H, Krueger S, Young S, Shusterman N. Carvedilol produces dose-related improvements in left ventricular function and survival in subjects with chronic heart failure. MOCHA Investigators. Circulation. 1996;94:2807–2816. doi: 10.1161/01.cir.94.11.2807. [DOI] [PubMed] [Google Scholar]
- MacLellan WR, Schneider MD. Death by design: Programmed cell death in cardiovascular biology and disease. Circ Res. 1997;81:137–144. doi: 10.1161/01.res.81.2.137. [DOI] [PubMed] [Google Scholar]
- Sharov VG, Sabbah HN, Shimoyama H, Goussev AV, Lesch M, Goldstein S. Evidence of cardiocyte apoptosis in myocardium of dogs with chronic heart failure. Am J Pathol. 1996;148:141–149. [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Cigola E, Cheng W, et al. Myocyte nuclear mitotic division and programmed myocyte cell death characterize the cardiac myopathy induced by rapid ventricular pacing in dogs. Laboratory Investigation. 1995;73:771–787. [PubMed] [Google Scholar]
- Olivetti G, Abbi R, Quaini F, et al. Apoptosis in the failing human heart. N Engl J Med. 1997;336:1131–1141. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- Narula J, Haider N, Virmani R, et al. Apoptosis in myocytes in end-stage heart failure. N Engl J Med. 1996;335:1182–1189. doi: 10.1056/NEJM199610173351603. [DOI] [PubMed] [Google Scholar]
- McCarthy NJ, Evan GI. Methods for detecting and quantifying apoptosis. Curr Top Dev Bio. 1998;36:259–278. doi: 10.1016/s0070-2153(08)60507-4. [DOI] [PubMed] [Google Scholar]
- Rossig L, Haendeler J, Mallat Z, Hugel B, Freyssinet J, Tedgui A, Dimmeler S, Zeiher AM. Congestive Heart Failure Induces Endothelial Cell Apoptosis: Protective Role of Carvedilol. J Am Coll Cardiol. 2000;36:2081–2089. doi: 10.1016/S0735-1097(00)01002-0. [DOI] [PubMed] [Google Scholar]
- Sabbah HN, Sharov VG, Gupta RC, Todor A, Singh V, Goldstein S. Chronic Therapy With Metoprolol Attenuates Cardiomyocyte Apoptosis in Dogs With Heart Failure. J Am Coll Cardiol. 2000;36:1698–1705. doi: 10.1016/S0735-1097(00)00913-X. [DOI] [PubMed] [Google Scholar]
- Li Z, Bing OH, Long X, Robinson KG, Lakatta EG. Increased cardiomyocyte apoptosis during the transition to heart failure in the spontaneously hypertensive rat. Am J Physiol. 1997;272:H2313–H2319. doi: 10.1152/ajpheart.1997.272.5.H2313. [DOI] [PubMed] [Google Scholar]
- Kajstura J, Cigola E, Malhotra A, Li P, Cheng W, Meggs LG, Anversa P. Angiotensin II induces apoptosis of adult ventricular myocytes in vitro. J Mol Cell Cardiol. 1997;29:859–870. doi: 10.1006/jmcc.1996.0333. [DOI] [PubMed] [Google Scholar]
- Buttle TM, Sandstrom PA. Oxidative stress as a mediator of apoptosis. Immunol Today. 1994;15:7–10. doi: 10.1016/0167-5699(94)90018-3. [DOI] [PubMed] [Google Scholar]
- Flesch M, Maack C, Cremers B, Baumer AT, Sudkamp M, Bohm M. Effect of beta-blockers on free radical-induced cardiac contractile dysfunction. Circulation. 1999;100:346–353. doi: 10.1161/01.cir.100.4.346. [DOI] [PubMed] [Google Scholar]