

Abstract
Infection by an endogenous pararetrovirus using forms of both episomal and chromosomal origin has been demonstrated and characterized, together with evidence that petunia vein clearing virus (PVCV) is a constituent of the Petunia hybrida genome. Our findings allow comparative and direct analysis of horizontally and vertically transmitted virus forms and demonstrate their infectivity using biolistic transformation of a provirus-free petunia species. Some integrants within the genome of P.hybrida are arranged in tandem, allowing direct release of virus by transcription. In addition to known inducers of endogenous pararetroviruses, such as genome hybridization, tissue culture and abiotic stresses, we observed activation of PVCV after wounding. Our data also support the hypothesis that the host plant uses DNA methylation to control the endogenous pararetrovirus.
Keywords: Caulimoviridae/integration/petunia/petunia vein clearing virus/tandem array
Introduction
The genotypes and phenotypes of eukaryotes are greatly influenced by retroelements. As retroviruses and pararetroviruses, they are responsible for several diseases and cause distinct symptoms in infected tissue. As retrotransposons, they have a fundamental impact on genome shape, evolution and adaptation to different environments (Kumar and Bennetzen, 1999; Pardue and DeBaryshe, 1999; Kalendar et al., 2000). The defining feature of a retroelement is the use of reverse transcription in the replication cycle. The gag-pol core (encoding proteins necessary for genome encapsidation, and DNA synthesis from RNA) is common to most retroelements, regardless of their classification. However, this classification is not always straightforward, and recent findings indicate the existence of a number of transitional elements that combine features of different retroelement subgroups (Song et al., 1994; Danilevskaya et al., 1997; Vicient et al., 2001).
Insertion into the host chromatin is an essential step in the replication of retroviruses and retrotransposons. The inserted retroviral DNA is termed ‘provirus’, since it represents an intermediate DNA stage in the life cycle of these RNA viruses (Varmus, 1982). From the inserted copy, terminally redundant genomic RNA can be produced under the control of promoter- and polyadenylation-sequences present in the long terminal repeats (LTRs). Pararetroviruses, comprising the families of vertebrate Hepadnaviridae and the six different genera of plant Caulimoviridae (Pringle, 1999), lack both a functional integrase and LTRs. Pararetroviral DNA usually accumulates as circular minichromosomes in the nucleus, with terminally redundant RNA being produced in this case by transcribing part of the circular DNA sequence twice. Like DNA virus sequences (e.g. Orend et al. 1994), animal (zur Hausen, 1991) and plant pararetrovirus sequences (Harper et al., 2002) can also become incorporated into the host chromatin by accident. However, such sequences are usually fragmented and rearranged and, if full viral genomes are incorporated, their lack of LTRs precludes production of a (pre)genomic RNA. Although integrated hepatitis B virus (HBV) sequences are found in >80% of HBV-related hepatocellular carcinomas, their role in oncogenesis remains obscure (Tsuei et al., 2002) and no activation of HBV proviral sequences has been reported to date.
Sequence analysis has shown that endogenous pararetrovirus-like sequences can be major constituents of plant genomes [e.g. tobacco endogenous pararetrovirus (TEPRV) in Nicotiana tabacum; Jakowitch et al., 1999], and some authors have discussed their potential benefits to the host (Mette et al., 2002). Endogenous viral sequences are thought to be able, under certain conditions, to initiate viral infection after induction, a situation that has been observed in two genera of Caulimoviridae (Ndowora et al., 1999; Lockhart et al., 2000). Activation of banana streak (badna) virus (BSV) and tobacco vein clearing (cassava vein mosaic-like) virus (TVCV) requires a hybrid genome (Lockhart et al., 2000; Lheureux et al., 2003). In addition, changes in day length in the case of TVCV (Lockhart et al., 2000) and tissue culture in the case of BSV (Dallot et al., 2001) have been identified as inducing agents.
Induction of petunia vein clearing virus (PVCV) infection has been observed in healthy Petunia hybrida after grafting or heat stress (D.-E.Lesemann, personal communication; Zeidan et al., 2001), as well as after water stress (Lockhart and Lesemann, 1997).
Until now, no integrated cluster of pararetroviruses has been observed that would allow induction of virus infection via transcription rather than by sequence recombination. Here, we describe one such case in the genome of P.hybrida, in which PVCV DNA exists arranged in tandem arrays. This provides for sequence redundancy allowing escape by transcription, much like the escape of retroviruses due to their LTRs. Tandemly arranged pararetrovirus DNA might represent an intermediate (with respect to replication strategy) between retro- and pararetro-viruses. Here, for the first time, infection by an endogenous pararetrovirus using forms of both episomal and chromosomal origin has been demonstrated and characterized.
Results
Hosts containing integrated PVCV DNA
To investigate the host range of plants harbouring PVCV proviral DNA, we analysed genomic DNA preparations of several wild-type petunia species for the presence of integrated PVCV sequences. Southern hybridization (Figure 1A) revealed PVCV-specific sequences of various size and concentration in the genomic DNA of healthy Petunia axillaris and P.hybrida (cv. Himmelsröschen). Virus-specific PCR products were identified in P.axillaris ssp. axillaris and Petunia integrifolia ssp. integrifolia (Figure 1B). However, no endogenous PVCV sequences were detected in healthy Petunia parodii or Nicotiana glutinosa (Figure 1A and B); therefore, these plants were used for further infection experiments.

Fig. 1. Detection of PVCV DNA in different Petunia ssp. and N.glutinosa. (A) Southern hybridization of genomic DNA preparations digested with BstEII. Ten micrograms of DNA was loaded for samples from P.hybrida and 20 µg for each of the other plants. The probe covered 100 bp from the 3′-end and 600 bp from the 5′-end of the genome. The BstEII restriction site is at position 6885 within the PVCV sequence. The arrow marks the position of linearized PVCV-DNA. (B) PCR analysis of genomic DNA preparations of different Petunia ssp. and N.glutinosa with PVCV primers as illustrated in Figuire 3A. The generated PCR product was 500 bp in size. Asterisks indicate putative parental crossing partners of P.hybrida. (C) Southern hybridization of genomic DNA extracted from infected N.glutinosa (20 µg) and P.parodii (10 µg) using the complete PVCV genome as a probe. Typical topological forms of linear and circular nuclear Caulimoviridae DNA are indicated. Marker DNA (M, only bands of relevant length are shown) consists of a HindIII digest of digoxigenin-labelled λ DNA (Roche).
Induction of chromosomal PVCV copies
Wounding, which promotes cell proliferation, was tested for its ability to induce PVCV infection from integrated copies. For this purpose, 20–30 plants were grown in prolonged cultivation with regular trimming and also in tissue culture with regular transfer of the upper plant part. Both procedures led to virus induction in 30% of the plants after 2–3 months. Control plants (cv. Himmelsröschen) were not trimmed during greenhouse culture, or were transferred as whole plants during tissue culture, and showed symptoms in only 1–5% of the plants. The rare appearance of symptom expression in control plants may be explained by exposure to additional stresses due to the extended culture period.
Graft transmission of PVCV
We set up a number of grafts to demonstrate viral transmission in our experimental system. Petunia parodii plants were easily infected from grafted scions of PVCV-infected plants, as visualized by vein clearing after 6–8 weeks (Table I, columns 2 and 3; Figure 2); episomal forms of virus DNA could be detected by Southern hybridization (Figure 1C). Although N.glutinosa plants could also be infected, the hybridization signals were much lower. Indeed, the host plant range of PVCV seems to be limited even within the Solanaceae, since no symptoms were induced in N.tabacum (Table I). Scions from the original host plant (P.hybrida cv. Himmelsröschen), as well as scions from the newly introduced host plants (N.glutinosa and P.parodii), induced virus infection of provirus-free stock tissue (Table I, compare columns 2 and 3 with 4 and 5), indicating that passage through N.glutinosa or P.parodii does not affect viral infectivity. Both viral DNA and virions can be detected in chlorotic leaf tissue of these host plants.
Table I. Horizontal transmission of PVCV using grafting.
| Infected scion |
P.hybrida (cv. Himmelsröschen) |
N.glutinosa | P.parodiia | ||
|---|---|---|---|---|---|
| Stock | N.glutinosa | P.parodii | N.tabacum (SR1) | P.parodii | N.glutinosa |
| Vein clearing | + | + | – | + | + |
| Virionsb | + | + | Not determined | + | + |
| Infected/grafted plants | 6/9 | 15/23 | 0/5 | 3/6 | 3/5 |
| Infection rate (%) | 67 | 65 | 0 | 50 | 60 |
aPetunia parodii was infected with p72-2 using biolistic inoculation.
bVirions were detected using immunosorbent electron microscopy.
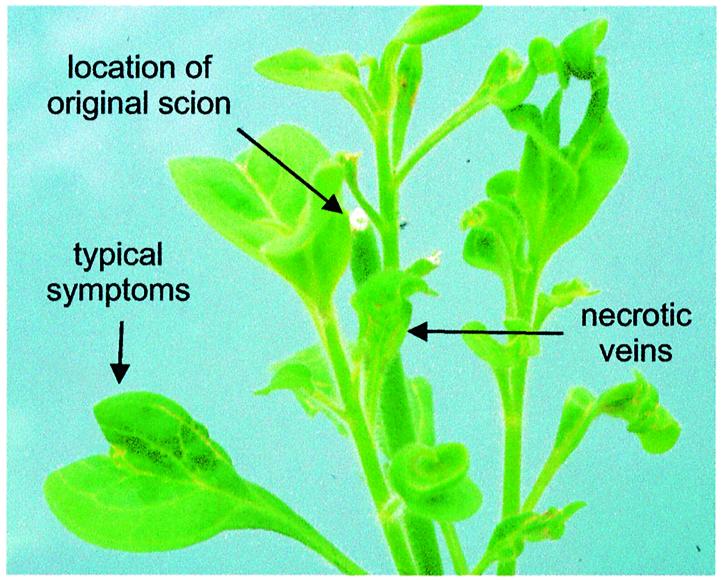
Fig. 2. PVCV transmission by grafting infected scions of P.hybrida to P.parodii. The original scion (arrow) had perished. Symptoms are epinasty of leaves, and vein clearing. In some cases chlorotic vein areas became necrotic during growth.
Vein clearing symptoms appear 2 months after grafting in newly developed leaves of the stock plant (Figure 2). Furthermore, virions can be detected in symptomatic leaves using immunosorbent electron microscopy. These are truly derived from the scion, since, unlike in P.hybrida, no endogenous PVCV sequences are detected in N.glutinosa or P.parodii (Figure 1A and B). In general, the stock plant tissue outgrew the scion. Therefore, scions were always taken from diseased plants and grafted onto healthy ones. The route of viral infection was similar to that observed in the source plant and occurred mainly in young (side) shoots.
Cloning of full-length episomal PVCV
Because of the low and uneven distribution of virus in infected plants, we used a PCR-based cloning strategy to obtain a full-length clone of episomal PVCV from infected N.glutinosa. Primers 32463 and 32462 (Figure 3B), which have overlapping complementary sequences at the single SacI restriction site, were used to generate a full-length PVCV genome from a circularized DNA template (see Materials and methods). The cloned viral population showed a heterologous pattern in restriction analysis, indicating several distinct variants (data not shown). One of these, p72-2 (Figure 3A), which was infectious in P.parodii (see below), was used for further experiments.

Fig. 3. Genome organization of episomal (A) and chromosomal (B and C) forms of infectious PVCV sequences. (A) Full-length PVCV clone (p72-2) derived from infected N.glutinosa. Conserved domains for movement protein (MP), RNA binding domain (RB), serine and aspartic protease (PRser and PRasp), reverse transcriptase (RT) and ribonuclease H (RH) are indicated. Note that the PRser domain overlaps the MP domain. RB and PRasp domains are not drawn to scale. By convention, the putative tRNA binding site for minus strand synthesis (tRNAMet) is used as the origin for nucleotide numbering. The position of the unique SacI site, the PCR-reporter product obtained with primers 16a and 16b, and the viral DNA probe used for screening the genomic library are indicated. The 3′-untranslated region contains regulatory elements (TATA box, poly A signal) for transcription. The region corresponding to the ‘quasi-LTR’ (QTR) is indicated. (B) Chromosomal organization of PVCV sequences in clone λ5 (17 kb) derived from a genomic library of P.hybrida cv. W138. The coding regions are boxed. Flat lines represent non-coding regions within the viral genome. PVCV sequences in reverse orientation (mirrored spelling) are found at the 5′-end of the λ insert. QTRs are indicated below, and SacI sites and the position of the infectious subclone λ5-7 are shown above the sequence. The interrupted lines below the QTRs indicate position and length of putative transcripts. The 3′-end of the transcript starting with QTR2 is not known, because it was not contained within the clone. An arrow marks the length and orientation of QTR within the untranslated region. Positions of primers used to analyse QTRs are indicated below the transcripts. Longer arrows represent reverse primers. The positions of the primers used to amplify fragments from subclone λ5-7 for in situ hybridization are indicated above this sequence: 32463 and 36828 to cover the left part of the genome (PVCV-L), 36191 and 34961 to cover the middle part (PVCV-M) and 34926 and 32462 to cover the intergenic region and surrounding sequences (PVCV-R). (C) Detection of complete PVCV DNA from tandem arrays within P.hybrida chromatin. DNA preparations of P.hybrida (cv. Himmelsröschen, lanes 2 and 3) served for identification of endogenous PVCV copies. For size comparisons of full-length viral genomes, an isolated SacI-fragment of PVCV full-length clone p72-2 was loaded (lane 4). Southern hybridization of an undigested genomic DNA preparation (lane 2) and of a DNA sample after incubation with SacI that has a single site within the viral genome (lane 3). DNA (5 µg) was loaded for samples from P.hybrida (cv. Himmelsröschen). The arrow marks the position of linearized PVCV DNA representing the full-length genome. As probe, ‘PVCV-M’ as described in Figure 3B was used. Marker DNA (lane 1) consists of a HindIII digest of digoxigenin-labelled λ DNA (Roche).
Cloning of integrated PVCV sequences
A genomic DNA library of P.hybrida was screened with a viral probe comprising consensus sequences for reverse transcriptase and RNaseH (Figure 3A). The haploid petunia genome comprises 1.3–1.5 × 109 bp (Mishiba et al., 2000), i.e. ∼10 times larger than the Arabidopsis thaliana genome. Accordingly, the 300 000 λ phage clones plated were considered sufficient to detect a single copy of an integrated PVCV genome. In fact, 72 positive clones were detected, suggesting that PVCV is a multicopy element. Of three clones analysed in more detail, two included junction sequences covering host and viral DNA. The viral borders of these originated from different positions close to the primer binding site. In clone λ3 (DDBJ/EMBL/GenBank accession No. AY334361) PVCV sequences comprised 420 nucleotides (nt) from the C-terminal end of the coding region adjacent to sequences upstream of the primer binding site, representing a quasi-LTR (QTR), as we define the corresponding region of caulimoviruses (the caulimovirus QTR domain comprises promoter and polyadenylation sequences but is present only as a single copy on the circular DNA, in contrast to the pair of LTRs of retrotransposons and retroviruses). These sequences showed 94% similarity to the QTR2 of clone λ5 (see below). The PVCV sequences in clone λ3 were flanked by metaviridae-like sequences (see Supplementary figure 1 available at The EMBO Journal Online).
Clone λ4 (DDBJ/EMBL/GenBank accession No. AY333912) contained a continuous stretch of viral DNA (nt 665–6153 within the PVCV genome) representing most of the coding region that showed a 664 nt deletion downstream of the primer binding site. The viral sequences were flanked at the 5′-end by plant DNA that contained two open reading frames (ORFs, cut-off >150 amino acids). One of them contained stretches of homology to the N-terminal part of the gag-pol polyprotein from metaviridae-like sequences in plants, whereas the other did not show any homology to known DNA sequences in BLAST searches (see Supplementary figure 1).
The third clone (λ5) contained mostly PVCV sequences and includes two slightly different QTRs flanking a full-length coding region (Figures 3B and 4). After subcloning with SacI, one full-length PVCV fragment (λ5-7) that led to symptom development when excised from the λ vector and used for bombardment of P.parodii plants (see below) was identified. Clone λ5-7 comprised 7250 nt, compared with the 7206 nt of clone p72-2, due to additional sequences within the QTR region (Figure 4).

Fig. 4. Arrangement and structure of QTRs in clone λ5 (DDBJ/EMBL/GenBank accession No. AY228106) and its progeny. The elements found within the U5 regions are indicated by letters, and the corresponding sequences are shown below. The nucleotide changes in the sequences labelled with (′) or (′′) are underlined.
Both episomal and chromosomal copies of PVCV are infectious in provirus-free P.parodii
To test our recombinant PVCV constructs for infectivity, we used biolistic delivery. Provirus-free N.glutinosa, with its thick leaves, proved to be unsuitable for biolistic inoculation. Instead, P.parodii plants were used successfully in the established infection assay.
Characteristic vein clearing symptoms occurred in P.parodii 6–8 weeks after inoculation. PVCV DNA was infectious, whether derived from episomal (p72-2; Figure 3A) or chromosomal (λ5, λ5-7; Figure 3B) viral copies. When linearized full-length copies (p72-2, λ5-7) of the viral genome were used for bombardment, infection rates of 11–16% were obtained (Table II). A marked increase in infectivity was obtained when the redundant form of the virus (λ5) was used as inoculum (infection rate up to 55%; Table II). In the latter case, viral RNA can probably be produced immediately via transcription from the tandem array, in contrast to linearized viral molecules, which first have to be circularized. As illustrated with p72-2, we were able to show that cloned PVCV introduced into P.parodii via bombardment can be transmitted to N.glutinosa using grafting, confirming its infectivity in both hosts and using both transmission modes (Table I, column 5).
Table II. Biolistic inoculations of P.parodii with recombinant PVCV DNA originating from episomal (p72-2) and chromosomal (λ5, λ5-7) forms.
| Origin of DNA | Construct | No. of plants | Infected plants | Infection rate (%) | Virionsa |
|---|---|---|---|---|---|
| Control | Blank vector | 16 | 0 | 0 | – |
| Infected N.glutinosa | P72 | 18 | 2 | 11 | + |
| Infected N.glutinosa | P72 | 15 | 2 | 13 | Not determined |
| Provirus P.hybrida W138 | λ5-7 | 32 | 5 | 16 | + |
| Provirus P.hybrida W138 | λ5-7 | 8 | 1 | 13 | + |
| Provirus P.hybrida W138 | λ5 | 11 | 6 | 55 | + |
| Provirus P.hybrida W138 | λ5 | 17 | 4 | 24 | + |
aVirions were detected using immunosorbent electron microscopy.
Sequence analysis and comparison of infectious episomal and chromosomal PVCV
The nucleotide sequence of the infectious full-length clone p72-2 originating from PVCV-infected N.glutinosa showed 98% homology to the nucleotide sequence assembled from subcloned viral DNA extracted from P.hybrida (cv. Himmelsröschen) with vein clearing symptoms (Richert-Pöggeler and Shepherd, 1997). However, the deduced amino acid sequence revealed that the entire coding region of p72-2 is assembled in a single ORF (DDBJ/EMBL/GenBank accession No. U95208, now updated with p72-2 sequence), in contrast to its distribution in two ORFs in the non-infectious sequence isolated from P.hybrida earlier (Figure 3A). A single nucleotide missing in the latter sequence (T3048) causes a frameshift and recognition of a stop codon at nt 3363. With the exception of the 38 amino acids following the frameshift, the amino acid sequence of the single ORF showed 99% homology when compared with the previously described ORFs (I, II), and the described consensus sequences (Richert-Pöggeler and Shepherd, 1997) are identical (Figure 3A). In addition to the known consensus sequences of caulimoviruses and retroelements, homology to the N-terminal half of serine proteases from capilloviruses (Yoshikawa et al., 1992) was detected in BLAST searches in a sequence overlapping the conserved domain of a putative PVCV movement protein (Figure 3A).
Sequence analysis of clone λ5 (DDBJ/EMBL/GenBank accession No. AY228106) revealed continuous stretches of PVCV DNA of various length and orientation (Figure 3B). The chromosomal forms of PVCV in P.hybrida cv. W138 showed high homology to p72-2. Within coding regions, the homology reached 98%, but it was reduced to 95% in the intergenic region. PVCV fragments within the λ clone showed even higher homology (99%) to each other. Within the λ insert, one unit-length PVCV genome, shown to be infectious after SacI restriction (Table II), was identified. This unit is followed by another continuous, almost full-length, PVCV sequence (Figure 3B), perhaps derived from another complete copy of PVCV, the 3′-end of which was not captured by cloning. The coding regions were flanked by QTRs that displayed sequence variation within the region upstream of the primer-binding site (Figure 4). Comparison with the QTR of the episomal virus p72-2 identified additional sequences with repeats and rearrangements of known viral (A, C and D) as well as non-viral (B, E and F) sequences (Figure 4). Viral progeny of plants bombarded with the genomic clone of PVCV (λ5) or its subclone λ5-7, lost these repetitive sequences. When total genomic DNA preparations from healthy P.hybrida cv. W138 were used as template for PCR, QTR1-specific primers synthesized a fragment of the expected size (primers 38956 and 38955; Figure 3B). Sequence analysis of these PCR products confirmed that the sequence arrangement observed in clone λ5 occurs in the plant genome and is therefore not a cloning artifact. At the 5′-end of clone λ5, pieces of PVCV coding regions in reverse orientation are interrupted by a non-coding region of PVCV, indicating that some rearrangements had occurred and that head-to-head arrangements of PVCV sequences are possible.
To analyse whether tandem arrangement of integrated PVCV is a more common phenomenon, total P.hybrida (cv. Himmelsröschen) DNA was separated on a 0.8% agarose gel after restriction with SacI, an enzyme that cleaves PVCV DNA just once. Southern hybridization revealed a prominent PVCV band migrating at 7.2 kbp (Figure 3C). This band comigrated with SacI-linearized PVCV DNA and amounts to about a quarter of the total PVCV DNA detected on the gel, a number that would correspond to the fraction of PVCV DNA present in tandem arrangement.
Methylation of PVCV sequences
Cytosine methylation of endogenous PVCV sequences in uninduced and induced P.hybrida was examined using the isoschizomer pair ScrFI (does not cleave if internal C of CCWGG is methylated) and BstNI (cleaves CCWGG regardless of methylation status) and a probe (PVCV-R) covering viral positions 5444–5671 (Figure 5), thus allowing detection of a variety of virus-specific fragments, including full-length copies, restriction fragments from both chromosomal and episomal forms, and integrated PVCV sequences flanked by plant DNA.
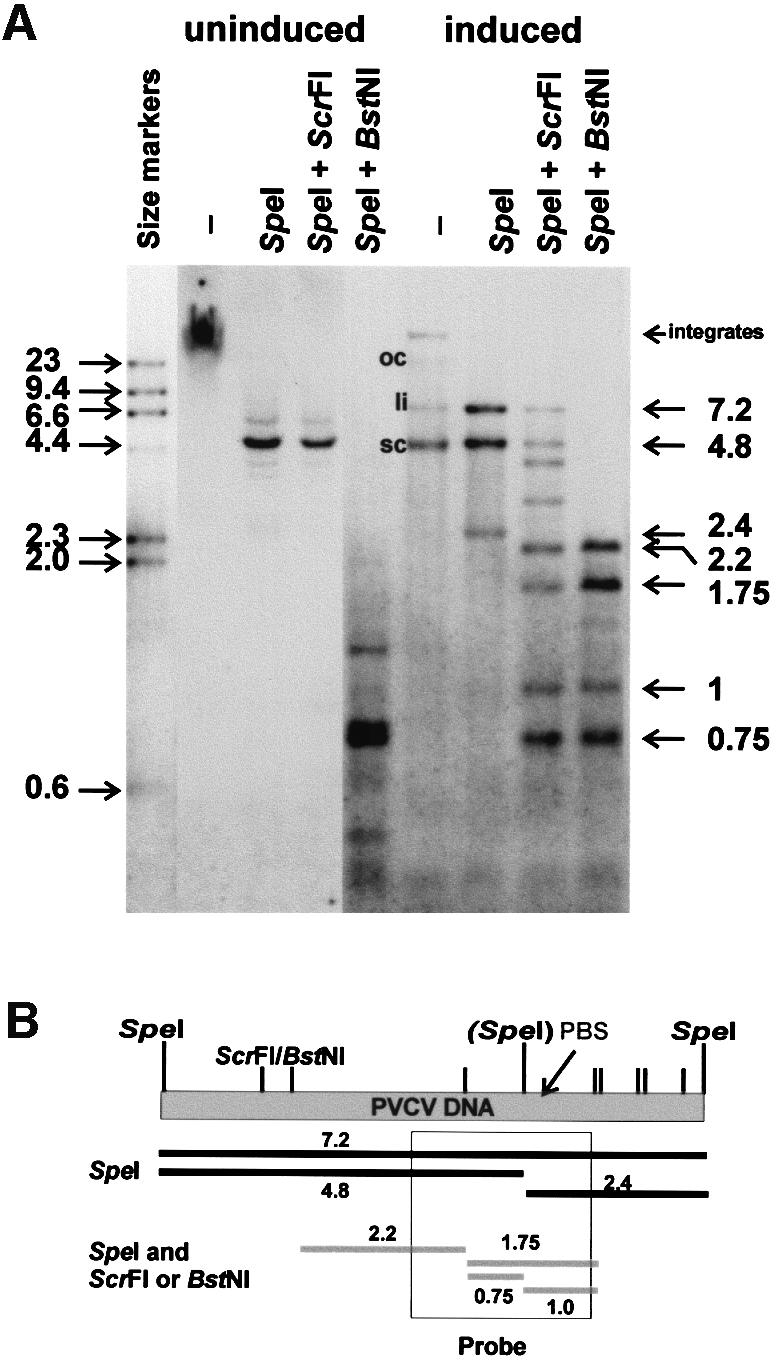
Fig. 5. Analysis of cytosine methylation. (A) Genomic DNA extracted from P.hybrida cv. Himmelsröschen was digested with SpeI alone or in combination with ScrFI (methylation sensitive) or BstNI (methylation insensitive). DNA from healthy plants (uninduced) was compared with that from symptom-expressing plants (induced). In undigested DNA from the induced sample, the positions of open circular (oc), linear (li) and supercoiled (sc) viral forms are indicated. Marker DNA consisted of a HindIII digest of digoxigenin-labelled λ DNA (Roche). The sizes of major SpeI/ScrFI(BstNI) fragments are shown on the right. (B) Linear representation of the PVCV genome, showing the positions of SpeI sites (in brackets, polymorphic site) and ScrFI/BstNI sites (smaller tags). PBS, primer binding site. The region of the genome covered by probe PVCV-R is indicated, along with the sizes (kb) of the major restriction fragments expected.
Interpretation of the hybridization pattern and identification of some fragments is complicated by the high numbers of integration events (estimated at ∼100–200), as well as polymorphism surrounding integration sites between PVCV copies. Nevertheless, a distinct methylation pattern was observed in plants with vein clearing symptoms compared with that in symptomless plants. In uninduced plants, undigested samples show a smear of high-molecular-weight DNA. SpeI digestion resolves this smear into a band of 4.8 kb, corresponding to the larger SpeI fragment of PVCV DNA. This band is not further digested by ScrFI; thus, this fragment contains no unmethylated C*CWGG sequences. In contrast, upon digestion with BstNI, the 4.8 kb band disappears in favour of a collection of smaller bands. This experiment clearly shows that the bulk of the integrated PVCV DNA is methylated. [Interestingly, the second (2.4 kb) SpeI fragment was not detected, indicating that integration and rearrangement events preferentially occur within this fragment. The two virus/host junction sequences analysed support this interpretation.]
Upon induction, the appearance of episomal virus is clearly evident. In the undigested samples, supercoiled, linear and (small amounts) of open circular PVCV DNA, as well as a weak band of PVCV integrates, can be discerned. Digestion with SpeI results in fragments of 7.2 kb, representing full-length linearized viral DNA that originated from circular forms, and 4.8/2.4 kb fragments, indicative of the polymorphism of the SpeI site at position 6984. The restriction pattern after SpeI–BstNI double digestion mainly follows the cloned sequence and is different from that of the uninduced sample, indicating that only a subpopulation of integrated sequences (corresponding to the cloned sequence) was induced and/or selected. A similar pattern is obtained using the methylation-sensitive ScrFI isoschizomer. However, this digestion was incomplete, indicating some methylation of the episomal (or packaged) viral DNA.
A restriction enzyme analysis using the HpaII–MspI isoschizomer pair provided similar results (see Supplementary data).
Identification of PVCV sequence integration sites
We used double target fluorescent in situ hybridization (FISH) to identify the chromosomal location of PVCV sequences integrated into the petunia genome. Chromosome nomenclature and identification criteria were according to Dietrich et al. (1981) and Fransz et al. (1996). Using chromosome lengths, arm ratios and location of nucleolus organizer regions, together with the sites of the 5S rDNA loci, chromosomes I, II, III and VII could be identified with certainty in all metaphases, and in some cases the very similar sized chromosomes IV, V and VI could also be distinguished. Three virus-specific probes covering the complete genome, representing the 5′ half (PVCV-L), middle part (PVCV-M) and 3′ half (PVCV-R), were used separately and in combination. Virus-specific hybridization signals were found with equal strength in the pericentromeric regions in five out of the seven chromosome pairs (Figure 6). The fluorescence of the viral probes was similar for all chromosomes except chromosome 3, which showed a much more intense signal. Separate use of the three virus-specific probes reduced the signal but generated a similar hybridization pattern (data not shown). With all probes tested, the strongest hybridization signal was identified on chromosome 3, suggesting a large cluster of PVCV copies at that location.

Fig. 6. In situ hybridization of PVCV probes L, M and R (as described in Figures 3B and 5) used on P.hybrida metaphase chromosomes. Chromosomes are stained in blue with the DNA stain DAPI. Virus sequence-specific signals are in red. In most cases, signals from both chromatids on homologous chromosomes are visible. The localization of the 5S RNA marker DNA is illustrated by the green signal on chromosome II. Unspecific signals are marked with ‘x’. Chromosomes are identified by numbers positioned to the right or above the chromosome.
Discussion
The universality of retroelements is a consequence of their ability to adapt specifically to their environment. In the case of PVCV, this adaptation has led to successful invasion of the petunia chromatin, making vertical transmission of the virus possible, and perhaps even more likely than horizontal transmission (Richert-Pöggeler and Shepherd, 1997). As the current study shows, PVCV uses specific mechanisms of genome expression to guarantee its survival in the host that have not so far been described in Caulimoviridae and Hepadnaviridae. Specifically, this plant virus can occasionally form head-to-tail concatamers within the host chromosomal DNA that give rise to structures known from retroviral proviruses. Thereby, the once singular QTR domain of the circular viral genome becomes duplicated and embraces a complete and functional viral genome. Our findings suggest that, in such an arrangement, the caulimoviral QTR actually functions as an LTR. The cryptic nature of viral infection and the limited transmissibility may imply that selection during evolution concentrated on chromosomal rather than episomal virus forms. Nevertheless, we were able to induce viral infection and link the viral progeny to its putative chromosomal origin. We also observed selective pressure against repetitive sequences within the QTR that were generated by the host DNA break repair machinery (see below).
The very high level of sequence conservation of endogenous PVCV would suggest that PVCV integration events happened rather recently in evolutionary history. Our PCR analysis indicated that PVCV sequences are present in several wild-type petunias. It remains to be determined whether, as in P.hybrida, complete and inducible viral genomes also exist in these species. The role of integrated viral sequences during the creation of hybrid petunia remains to be investigated in detail. For other endogenous pararetroviruses (BSV, TVCV and TEPRV), it has been proposed that the interaction of genetic factors after inter-specific genome hybridization supported their activation in the hybrid (Harper et al., 2002). Petunia is endemic to South America: the various species are distinct in their geographical distribution with partially overlapping areas (Wijsman, 1982). The original parental crossing partners of P.hybrida (P.axillaris ssp. axillaris and P.integrifolia ssp.) are found in areas to the east of the P.parodii territory. Thus, allopatric speciation of petunia might explain why PVCV-like sequences are lost in P.parodii, a subspecies of P.axillaris.
Interestingly, integrated PVCV sequences were found in the pericentromeric regions of chromosomes (Figure 6). These consist of heterochromatin (Dietrich et al., 1981), a region of condensed chromatin. In petunia, the inactivity of transgenes has been correlated with their integration into repetitive sequences of heterochromatin (Pröls and Meyer, 1992). This might also explain why no phenotypic changes, as would occur if gene-coding sequences were invaded, have been observed. On the other hand, other integration sites may have been occupied but led to lethality or infertility and therefore have not been maintained in the progeny.
Integrated PVCV sequences with true LTRs, typical of retroviruses and retrotransposons, have not been found. Isolated junction sequences between host and viral DNA occurred at terminal sites within the viral genome. These findings indicate that PVCV is a true pararetrovirus and integration is not obligatory for the replication cycle in the host plant, as shown in P.parodii and N.glutinosa. In the progeny of infected P.parodii and N.glutinosa, no vein clearing symptoms were detected and no PVCV-specific DNA could be amplified from leaf tissue using PCR (data not shown). Therefore, we assume that the observed vertical transmission of PVCV depends on viral sequences in the chromosome and not on invasion of generative tissues or seeds by episomal virus. Although short stretches of homology to amino acid sequence motifs of the catalytic domain of integrases can be identified in PVCV (Richert-Pöggeler and Shepherd, 1997), there are no further sequence homologies to recently identified putative integrase domains of retroelements (McClure et al., 2002). In pararetroviruses, the lack of a functional integrase suggests that host functions, such as the recombination and double-strand DNA break repair machinery, effect their integration. Infected nuclei contain both circular and linearized PVCV DNA (Figure 1C). Linearized DNA and other replication intermediates could act as filler DNA in the repair of double-strand breaks and thus lead to rearrangements during integration of PVCV. Such illegitimate recombination events have also been discussed as being responsible for transgene rearrangements close to the integration sites (Gorbanova and Levy, 1997; Svitashev et al., 2002) and to lead frequently to multiple integrations, including tandem arrays (Salomon and Puchta, 1998). Clone λ5 is a typical example, since it contains tandemly arranged viral sequences and slight rearrangements at their QTRs, which might have served as integration substrates, when containing the typical single-strand overhangs marking the start and endpoint of reverse transcription (Pfeiffer and Hohn, 1983). This direct head-to-tail array of integrated viral sequences provides a straightforward mechanism for viral induction. However, such an arrangement has not yet been found for other endogenous pararetroviruses (BSV and TVCV) that can trigger a viral infection. As an alternative, one might consider that tandemly arranged DNA is created prior to the integration event. A rolling circle replication mechanism (e.g. as for geminiviruses) is unlikely, since there is no indication for the required enzymatic functions (Pfeiffer and Hohn, 1983); however, one could imagine that readthrough transcription could have led to the generation of dimeric pregenomic RNA leading to tandemly arranged DNA units upon reverse transcription. However, such a possibility would require a much higher packaging capacity of pararetroviruses than available.
Clusters of integrated PVCV DNA were identified by FISH in five of the seven P.hybrida chromosomes. The fluorescent signals generated were high in both abundance and strength. This high-copy-number PVCV integration pattern in petunia is distinct from that of the BSV/banana system (Harper et al., 1999). In the latter case, only two integration loci were identified, comprising 150 and 50 kb. Although interspersion with other sequences possibly contributes to the large in situ hybridization signals of at least 50 kb, some of these clusters probably contain multiple copies in tandem arrays. In fact, one case of tandemly arranged copies was found in a P.hybrida genome library. The tandem array would provide an appropriate mechanism for escape of a pararetroviral genome by recombination or, more likely, by transcription: from the promoter of the upstream copy and termination at the polyadenylation site of the consecutive one (Figure 3B). Such a mechanism corresponds to the transcription from LTR to LTR of a true retrovirus or LTR transposon. Redundant sequences upstream of the primer binding site are probably lost through recombination during replication (reviewed by Hohn, 1994) and are absent in the viral progeny (Figure 4). In other cases, escape of viral sequences might involve a more complex pathway of transcription, complementation and recombination of (fragmented) viral sequences.
From the frequency of occurrence (72 positive clones) in the genomic library, we estimate that ∼50–100 copies of integrated viral sequences exist in the haploid petunia genome. Despite this high frequency, spontaneous release of viruses is rare. However, this release can be induced by wound stress. For retrotransposons, it has been shown that stress conditions induce transcriptional activation (Grandbastien, 1998). Wound stress would cause callus formation accompanied by chromatin replication and transient hypomethylation. PVCV sequences appear to be clustered with metaviridae-like sequence in the petunia genome, and most likely the same mechanism applys for the control of these retroelements by the host. Indeed, integrated PVCV sequences of healthy plants appear to be methylated (Figure 5). Upon induction, where only a short period of demethylation in a single plant cell at one of the ∼100–200 integration loci would be enough to release the virus, a distinct pattern appears dominated by episomal forms of PVCV. Most of those are unmethylated, with the exception of a minor fraction that is methylated. Methylation of episomal virus has also been shown for CaMV (Tang and Leisner, 1998).
Other integrated pararetrovirus sequences are known (Harper et al., 2002, 2003). However, due to the well-known exclusion of these viruses from the meristem, integrants will usually not become manifested in the reproductive cells and would therefore be found only rarely. However, the common practice of vegetative propagation and tissue culturing of petunia might have allowed callus tissue, which, like meristem, represents a region of actively dividing, dedifferentiated cells, to be derived from somatic cells harbouring integrated proviruses. The same situation applies to the BSV/banana system, since cultivated bananas are generally sterile and are propagated vegetatively. In fact, all plants with high regenerative capacity, which are therefore easy to propagate vegetatively, are probably candidates for host endogenous pararetrovirus invasion. In analogy, endogenous retro- and pararetro-viruses could accumulate in other eukaryotes when clonal propagation technology is applied.
Our investigation of this endogenous pararetrovirus in petunia may provide insights into the evolution of retroelements in general and the genomic organization and transfer mechanisms of pararetroviruses in particular. Such knowledge will be fundamental to the development of strategies to control their release from the host genome. Understanding and evaluating the impact of endogenous, but still infectious, pararetroviruses on the host genome will be essential in the production of genetically stable and disease-free plant material.
Materials and methods
Plant material and grafting
Greenhouse plants were grown in 12–14 h light at 22–25°C and fertilized with ‘Osmocote Exact’ (Scotts International, B.V. Heerlen, The Netherlands) for long growth periods. The plants were trimmed every 4 weeks. For tissue culture, petunia seeds were surface sterilized. Plants were then grown in 16 h light at 25°C and 60% humidity. The apical part of plant shoots was transferred to fresh medium every 4–6 weeks.
Seeds of P.hybrida cv. Himmelsröschen were purchased from NL Chrestensen (Erfurt, Germany). Infected P.hybrida cv. Himmelsröschen leaves were harvested from induced plants (Lesemann and Casper, 1973). The infected plant material was vegetatively propagated by cuttings and further amplification and maintained via seeds. PVCV was transmitted from infected plants to N.glutinosa and P.parodii using cleft grafting.
Genomic DNA extraction and PCR
Genomic plant DNA was extracted using the Nucleon PhytoPure extraction kit (Amersham Life Science) and used as template in PCR mixtures containing Pfu polymerase (Promega) and for blotting analyses. For PCR analysis using Taq polymerase (Invitrogen), leaf material was added directly to the recommended reaction mixture.
All oligonucleotides (Table III) for PCR were produced by the FMI DNA synthesis facility. For PCR cloning of episomal PVCV, the Expand Long Template PCR system (Roche) was used according to the manufacturer’s recommendations. Abutting primers 32463 and 32462, which have overlapping complementary sequences at the single SacI restriction site, were used to generate a full-length PVCV genome from a circularized DNA template. The PCR products of 10 different reactions were gel isolated and combined for cloning into pBluescript II KS (+) (Stratagene).
Table III. Oligonucleotides used for PCR.
| Primer | Orientation | Nucleotide sequence (5′–3′) |
|---|---|---|
| 32463 | Forward | CAA GGA GCT CCC CTT ACA AAA GAC TCC |
| 32462 | Reverse | AGG GGA GCT CCT TGG ATT TGG ACT TGG |
| 16a | Forward | CGC ATT GGA GCA GAT GG |
| 16b | Reverse | GTG AGA GAA GAG TGT GAG |
| 35817 | Forward | GAG TCA TAG AGA TTG CCT ATA TAA GG |
| 37215 | Forward | GTG CCG TGT TAA AGC CCA GTA G |
| 38955 | Reverse | GAT ACC AAT ACT TAG GAA AGA |
| 38956 | Forward | TCT TTC CTA AGT ATA GGA TG |
| 36828 | Reverse | CGA GAA CTC TGA TAA GAC CTT G |
| 36191 | Forward | CAG TCT AGC AGT CAC CTT GG |
| 34961 | Reverse | TGC TCT CAT GTC CAT TTC AAC C |
| 34926 | Forward | TTG CTG ATT TCC TAT CAA GGC C |
Bold italic letters indicate mismatches compared with the clone λ5 sequences.
Plants were directly tested for the presence of PVCV sequences using primers 16a and 16b as described in Richert-Pöggeler and Shepherd (1997). PCR analysis of QTRs in viral progeny after bombardment with episomal or chromosomal PVCV sequences was conducted using Pfu polymerase. Some primers (37215, 35817) used for sequencing the PVCV full-length clone p72-2 were also used for the analysis of the chromosomal sequences, despite mismatches in clone λ5 (indicated in bold italic in Table III). A fragment of 1100 bp was generated using primers 35817 and 32462 (Figure 3B). An initial denaturation at 94°C for 2 min was followed by 30 cycles of 45 s denaturation, 45 s annealing at 58°C and 2 min extension at 72°C. The final extension step was continued for 10 min.
The QTR sequence was determined using primers 37215 and 38955 as indicated in Figure 3B. Primer 38956, which is a unique sequence in λ5 outwith the QTR1 region, and primer 38955 were used to identify QTR1 in W138 genomic DNA preparations.
Screening of a genomic petunia library and Southern hybridization
A genomic DNA library of P.hybrida cv. W138, cloned in the λ vector GEM11 (Promega) after partial digestion with Sau3A, was provided by Dr R.van Blokland (University of Amsterdam). Bacteriophage λ growth, purification and DNA extraction was performed according to Sambrook and Russell (2001). The viral probe of 1600 bp (p16; Richert-Pöggeler and Shepherd, 1997) comprised the consensus sequences for reverse transcriptase and Rnase H (Figure 3A). One microgram of biotinylated probe produced using the BIO PRIME DNA labelling system (Gibco-BRL) was used per filter assay. Blotting, hybridization, high stringency washing and detection steps followed the protocol of Photogene Nucleic Acid Detection System Version 2.0 (Gibco-BRL). For Southern hybridization of genomic DNA, probe labelling and detection were performed with the DIG High Prime DNA Labeling and Detection Starter Kit II (Roche).
The primary screen with viral probe p16 resulted in 72 positive plaques, of which three were randomly selected and confirmed in a secondary screen. The DNAs of the three positive λ clones (referred to as clones λ3, λ4 and λ5) were then digested with SacI and ligated into the Bluescript vector. The subclones obtained were sequenced directly by primer walking.
Biolistic delivery
Transfections were performed under sterile conditions with the Bio-Rad PDS-1000/He particle delivery system according to the manufacturer’s recommendations. For each series of five shots, 5 µg (linearized PVCV genome) or 13 µg (λ5) of DNA were precipitated on gold particles (1 or 1.6 µm in size). The recombinant PVCV construct was digested with SacI to release the linearized full-length molecule from the vector prior to bombardment. The λ clone DNA was used directly without further digestion. Macro-carriers, rupture disks and stopping screens were sterilized with 70% ethanol (EtOH). Gold particles (60 mg) were washed with EtOH and sterile water and resuspended in 1 ml of 50% glycerol (sterile). For macro-carrier preparation, the following was combined under continuously mixing: 25 µl of resuspended gold particles, 5–13 µg of plasmid (1 µg/µl), 25 µl of 2.5 M CaCl2 and 10 µl of 0.1 M spermidine. The mixture was washed with 200 µl of 70% EtOH followed by a wash with 200 µl of EtOH and finally resuspended in 40 µl of EtOH. Per macro-carrier, 6 µl out of this mixture were added and used for biolistic inoculation as soon as the EtOH had been evaporated.
Plants used for biolistic inoculation were 5–6 weeks old. As a control, 5–10 plants were bombarded with vector DNA alone. Three to five plants with an average leaf size of 1 cm2 were placed in a Petri dish and bombarded at a target distance of 8 cm, applying helium pressure of 1100 p.s.i. After biolistic delivery, plants were transferred to separate tissue culture boxes. Upon appearance of vein clearing, symptomatic leaves were tested by PCR for the presence of virus-specific sequences. Positive samples were further tested by Dr D.-E.Lesemann at the Biologische Bundesanstalt für Land- und Forstwirtschaft (BBA, Institute for Plant Virology, Microbiology and Biosafety in Braunschweig, Germany) for the presence of virions using immunosorbent electron microscopy.
Petunia hybrida plants (cvs Himmelsröschen and W138) were bombarded with gold particles alone to test whether a single injury would cause activation of endogenous PVCV sequences. After biolistic delivery, plants were monitored for 6 months with regular transfers for elevated symptom expression compared with plants that had not been bombarded.
Sequence analysis
Sequence comparisons, assembly and analysis were performed using the Wisconsin Package Version 10.2 (Genetics Computer Group, Madison, WI). For similarity searches, nucleotide or deduced amino acid sequences were submitted to the nr database of the National Center for Biotechnology Information using different BLAST programs (Altschul et al., 1990).
In situ hybridization
Probe labelling, chromosome preparation and in situ hybridization followed the procedure of Schwarzacher and Heslop-Harrison (2000).
The 5S rDNA probe was a 410 bp fragment from the clone pTa794 (Gerlach and Dyer, 1980) containing the 5S rDNA repeat unit of Triticum aestivum. It was labelled with digoxigenin-11-dUTP (Roche) by PCR using M13 forward and reverse sequencing primers.
Three viral probes that, in combination, cover most of the sequence of an infectious chromosomal PVCV copy were produced using the SacI subclone λ5-7 as template for PCR. The following primer combinations (Figure 3B) were chosen: 32463 and 36828 to cover the left part of the genome (nt 658–1794, PVCV-L); 36191 and 34961 to cover the middle part (nt 2236–5322, PVCV-M); and 34926 and 32462 to cover the intergenic region and surrounding sequences (nt 5444–5671, PVCV-R). Viral probes were labelled with 0.4 mM biotin-11-dUTP (Roche) and 0.2 mM TTP.
Root tips from young plants were treated with 0.2 M hydroxychinolin for 3–4 h and fixed with alcohol:acetic acid (3:1). Chromosome preparations were made following digestion with cellulase and pectinase, treated with RNAs and postfixed in 4% paraformaldehyde. Labelled probe DNAs (100–200 ng) were applied to the slides. Chromosomes and probe were denatured together and allowed to hybridize overnight at 37°C. Post-hybridization washes were at 42°C in 20% formamide and 0.1 × SSC, giving a stringency of 80–85%. Detection of hybridization sites was carried out with fluorescein-conjugated anti-digoxigenin (Roche; 2.5 µg/ml) and Alexa 495-conjugated streptavidin (Molecular Probes; 1 µg/ml), and chromosomes were counterstained with DAPI.
Slides were analysed with a Zeiss Axioplan2 fluorescent microscope and images captured with an Optronix S97790 cooled CCD camera. Overlays of hybridization signal and DAPI pictures of 10 metaphases were prepared with Adobe Photoshop 6 using only those functions that are applied to all pixels of the image.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
The authors are very grateful to E.Souer, R.van Blokland and J.Kooter (University of Amsterdam) for supplying seeds of P.hybrida cv. W138 and P.axillaris and P.integrifolia, as well as for the generous gift of a genomic library of P.hybrida cv. W138. Furthermore, we are thankful to Dr J.-P.Renou (INRA-CR Angers) for seeds of P.parodii and Dr R.J.Shepherd (University of Kentucky) for seeds of N.glutinosa. We appreciate many valuable discussions with all members of our laboratory. We offer special thanks to S.van Eeden for assistance in the grafting experiments and M.Müller for the maintenance of tissue culture plants. We are very grateful to D.-E.Lesemann for performing the immunosorbent electron microscopy studies, and to B.Hohn, U.Klahre and H.Rothnie for critical reading of the manuscript and many stimulating discussions. The excellent services for oligonucleotide synthesis by P.Müller and for sequencing by H.Angliker and M.Pietrzak are gratefully acknowledged. K.R.R.-P. was supported by the German Research Foundation (Ri992/1-1). This work was supported in part by EU grant QLK3-CT-2002-02098.
References
- Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Dallot S., Acuna,P., Rivera,C., Ramirez,P., Cote,F., Lockhart,B.E.L. and Caruana,M.L. (2001) Evidence that the proliferation stage of micropropagation procedure is determant in the expression of Banana streak virus integrated into the genome of FHIA 21 hybrid (Musa AAAB). Arch. Virol., 146, 2179–2190. [DOI] [PubMed] [Google Scholar]
- Danilevskaya O.N., Arkhipova,I.R., Traverse,K.L. and Pardue,M.L. (1997) Promoting in tandem: the promoter for telomere transposon HeT-A and implications for the evolution of retroviral LTRs. Cell, 88, 647–655. [DOI] [PubMed] [Google Scholar]
- Dietrich A.J.J., de Jong,J.H. and Mulder,R.J.P. (1981) Location and variation of the constitutive heterochromatin in Petunia hybrida. Genetica, 55, 85–91. [Google Scholar]
- Fransz P.F., Stam,M., Montijn,B., Hoopen,R.T., Wiegant,J., Kooter,J.M., Oud,O. and Nanninga,N. (1996) Detection of single-copy genes and chromosome rearrangements in Petunia hybrida by fluorescence in situ hybridization. Plant J., 9, 767–774. [Google Scholar]
- Gerlach W.L. and Dyer,T.A. (1980) Sequence organization of the repeating units in the nucleus of wheat, which contain 5S rRNA genes. Nucleic Acids Res., 8, 4851–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbanova V. and Levy,A.A. (1997) Non-homologous DNA end joining in plant cells is associated with deletions and filler DNA insertions. Nucleic Acids Res., 25, 4650–4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandbastien M.-A. (1998) Activation of plant retrotransposons under stress conditions. Trends Plant Sci., 3, 181–187. [Google Scholar]
- Harper G., Osuji,J.O., Heslop-Harrison,J.S. and Hull,R. (1999) Integration of banana streak badnavirus into the Musa genome: molecular and cytogenetic evidence. Virology, 255, 207–213. [DOI] [PubMed] [Google Scholar]
- Harper G., Hull,R., Lockhart,B. and Olszewski,N. (2002) Viral sequences integrated into plant genomes. Annu. Rev. Phytopathol., 40, 119–136. [DOI] [PubMed] [Google Scholar]
- Harper G., Richert-Pöggeler,K.R., Hohn,T. and Hull,R. (2003) Detection of petunia vein clearing virus: model for the detection of episomal viruses with endogenous pararetrovirus sequence counterparts. J. Virol. Methods, 107, 177–184. [DOI] [PubMed] [Google Scholar]
- Hohn T. (1994) Recombination of a plant pararetrovirus: cauliflower mosaic virus. In Paszkowski,J. (ed.), Homologous Recombination in Plants. Kluwer Academic, Dordrecht, The Netherlands, pp. 25–38. [Google Scholar]
- Jakowitch J., Mette,M.F., van der Winden,J., Matzke,M.A. and Matzke,A.J.M. (1999) Integrated pararetroviral sequences define a unique class of dispersed repetitive DNA in plants. Proc. Natl Acad. Sci. USA, 96, 13241–13246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalendar R., Tanskanen,J., Immonen,S., Nevo,E. and Schulman,H. (2000) Genome evolution of wild barley (Hordeum spontaneum) by BARE-1 retrotransposon dynamics in response to sharp microclimatic divergence. Proc. Natl Acad. Sci. USA, 97, 6603–6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A. and Bennetzen,J.L. (1999) Plant retrotransposons. Annu. Rev. Genet., 33, 479–532. [DOI] [PubMed] [Google Scholar]
- Lesemann D.-E. and Casper,R. (1973) Electron microscopy of petunia vein clearing virus, an isometric plant virus associated with specific inclusions in petunia cells. Phytopathology, 63, 1118–1124. [Google Scholar]
- Lheureux F., Carreel,F., Jenny,C., Lockhart,B.E.L. and Iskra-Caruana,M.L. (2003) Identification of genetic markers linked to Banana streak disease expression in inter-specific Musa hybrids. Theor. Appl. Genet., 106, 594–598. [DOI] [PubMed] [Google Scholar]
- Lockhart B.E.L. and Lesemann,D.-E. (1997) Occurrence of petunia vein clearing virus in the U.S.A. Plant Dis., 82, 262. [DOI] [PubMed] [Google Scholar]
- Lockhart B.E., Menke,J., Dahal,G. and Olszewski,N.E. (2000) Characterization and genomic analysis of tobacco vein clearing virus, a plant pararetrovirus that is transmitted vertically and related to sequences integrated in the host genome. J. Gen. Virol., 81, 1579–1585. [DOI] [PubMed] [Google Scholar]
- McClure M.A., Donaldson,E. and Corro,S. (2002) Potential multiple endonuclease functions and a ribonuclease H encoded in retroposon genomes. Virology, 296, 147–158. [DOI] [PubMed] [Google Scholar]
- Mette M.F., Kanno,T., Aufsatz,W., Jakowitsch,J., van der Winden,J., Matzke,M.A. and Matzke,A.J.M. (2002) Endogenous viral sequences and their potential contribution to heritable virus resistance in plants. EMBO J., 21, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishiba K.-I., Ando,T., Mii,M., Watanabe,H., Kokubun,H. Hashimoto,G. and Marchesi,E. (2000) Nuclear DNA content as an index character discriminating taxa in the genus Petunia sensu Jussieu (solanaceae). Ann. Bot., 85, 665–673. [Google Scholar]
- Ndowora T., Dahal,G., LaFleur,D., Harper,G., Hull,R., Olszewski,N.E. and Lockhart,B. (1999) Evidence that badnavirus infection in Musa can originate from integrated pararetroviral sequences. Virology, 255, 214–220. [DOI] [PubMed] [Google Scholar]
- Orend G., Linkwitz,A. and Doerfler,W. (1994) Selective sites of adenovirus (foreign) DNA integration into the hamster genome: changes in integration patterns. J. Virol., 68, 187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardue M.L. and De Baryshe,P.G. (1999) Drosophila telomeres: two transposable elements with important roles in chromosomes. Genetica, 107, 189–196. [PubMed] [Google Scholar]
- Pfeiffer P. and Hohn,T. (1983) Involvement of reverse transcription in the replication of cauliflower mosaic virus: a detailed model and test of some aspects. Cell, 33, 781–789. [DOI] [PubMed] [Google Scholar]
- Pringle C.R. (1999) Virus taxonomy—1999. Arch. Virol., 144, 421–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pröls F. and Meyer,P. (1992) The methylation patterns of chromosomal integration regions influence gene activity of transferred DNA in Petunia hybrida. Plant J., 2, 465–475. [DOI] [PubMed] [Google Scholar]
- Richert K.R. (1992) Untersuchungen zur Charakterisierung des petunia vein clearing virus (PVCV), ein samenübertragbares Pararetrovirus. PhD thesis, University of Göttingen, Göttingen, Germany.
- Richert-Pöggeler K.R. and Shepherd,R. (1997) Petunia vein clearing virus: a plant pararetrovirus with core sequences for an integrase function. Virology, 236, 137–146. [DOI] [PubMed] [Google Scholar]
- Salomon S. and Puchta,H. (1998) Capture of genomic and T-DNA sequences during double-strand break repair in somatic plant cells. EMBO J., 17, 6086–6095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J. and Russell,D.W. (2001) Molecular Cloning: A Laboratory Manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Schwarzacher T. and Heslop-Harrison,J.S. (2000) Practical in situ Hybridization. Bios, Oxford. [Google Scholar]
- Song S.U., Gerasimova,T., Kurkulos,M., Boeke,J.D. and Corces,V.G. (1994) An Env-like protein encoded by a Drosophila retroelement: evidence that gypsy is an infectious retrovirus. Genes Dev., 8, 2046–2057. [DOI] [PubMed] [Google Scholar]
- Svitashev S.K., Pawlowski,W.P., Makarevitch,I., Plank,D.W. and Somers,D.A. (2002) Complex transgene locus structures implicate multiple mechanisms for plant transgene rearrangement. Plant J., 32, 433–446. [DOI] [PubMed] [Google Scholar]
- Tang W. and Leisner,S. (1998) Methylation of nonintegrated multiple copy DNA in plants. Biochem. Biophys. Res. Commun., 245, 403–406. [DOI] [PubMed] [Google Scholar]
- Tsuei D.-J., Chang,M.-H., Chen,P.-J., Hsu,T.-Y. and Ni,Y.-H. (2002) Characterization of integration patterns and flanking cellular sequences of hepatitis B virus in childhood hepatocellular carcinomas. J. Med. Virol., 68, 513–521. [DOI] [PubMed] [Google Scholar]
- Varmus H.E. (1982) Form and function of retroviral proviruses. Science, 216, 812–820. [DOI] [PubMed] [Google Scholar]
- Vicient C.M., Kalendar,R. and Schulman,A.H. (2001) Envelope-class retrovirus-like elements are widespread, transcribed and spliced, and insertionally polymorphic in plants. Genome Res., 11, 2041–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijsman H.J.W. (1982). On the interrelationships of certain species of Petunia. I. Taxonomic notes on the parental species of Petunia hybrida. Acta Bot. Neerl., 31, 477–490. [Google Scholar]
- Yoshikawa N., Sasaki,E., Kato,M. and Takahashi,T. (1992) The nucleotide sequence of apple stem grooving capillovirus genome. Virology, 191, 98–105. [DOI] [PubMed] [Google Scholar]
- Zeidan M., Sikron,N., Cohen,J. and Gera,A. (2001) Improved detection of petunia vein clearing caulimovirus. HortScience, 35, 1279–1282. [Google Scholar]
- zur Hausen H. (1991) Viruses in human cancers. Science, 254, 1167–1173. [DOI] [PubMed] [Google Scholar]