

Abstract
Replication of simple retroviruses depends on the recruitment of a single large primary transcript toward splicing, transport/packaging and translation regulations. In this respect, we studied the novel SD′ 4.4 kb RNA of murine leukemia retroviruses (MLV) which results from alternative splicing of the primary transcript. We showed that SD′ RNA was required for optimal replication since expression of a pre spliced SD′ RNA trans-complemented the impaired infectivity of a SD′-defective mutant. We monitored the fate of this novel transcript throughout early and late events of the viral life cycle. SD′ RNA was specifically incorporated into virions demonstrating that the unspliced RNA was not the unique viral RNA present in virions. Furthermore, SD′ RNA was reverse transcribed and its DNA copy integrated into the host genome, thus constituting a new splice donor-associated retroelement (SDARE) in infected cells. Finally, we showed that SD′ mRNA encoded a 50 kDa polyprotein, and to a lower extent an additional 60 kDa polyprotein, which harbored Gag and integrase domains.
Keywords: defective retrovirus/Gag translation/RNA encapsidation/viral RNA splicing/virion assembly
Introduction
Murine leukemia viruses (MLV) are infectious retroviruses which have been invaluable in furthering our understanding of eukaryotic gene expression and viral pathology, and more recently in achieving therapeutic trials with MLV-based gene transfer vectors. MLV oncoviruses belong to the simple retrovirus family distinguishable by their elementary genomic organization with gag, pol and splice-dependent expression env genes: gag directs the synthesis of the proteins that form internal virion structures [matrix (MA), p12, capsid (CA) and nucleocapsid (NC) proteins]; pol encodes the protease (PR), reverse transcriptase (RT) and integrase (IN) enzymes; env contains the sequence coding for the surface (SU) and transmembrane (TM) components of viral envelope glycoprotein (Leis et al., 1988).
After nuclear export, the primary viral transcript has two essential roles, an mRNA template for protein synthesis and a genomic RNA packaged into progeny virions. Post-transcriptional regulatory systems recruit viral and cellular partners during these processes. However, simple retroviruses lack the viral ‘accessory’ proteins which derive from singly or multiply alternative spliced messages of complex retroviruses such as lentiviruses (e.g. HIV, SIV) or complex oncoviruses (e.g. HTLV, BLV). Accessory proteins regulate and coordinate viral gene expression, virion assembly and adaptation to the host. Nevertheless, we have recently shown that an alternative splicing event takes place in MLV and generates a previously undescribed subgenomic 4.4 kb RNA (Dejardin et al., 2000). This RNA derives from splicing between an alternative splice donor site, designated SD′, located within the capsid sequence of the gag gene, and the canonical envelope splice acceptor site. Conservation of the SD′ site among simple mammalian retroviruses (Dejardin et al., 2000) strongly suggests a role in viral replication. Indeed, reduction in infectivity of two prototypic MLV viruses (Friend-MLV and Moloney-MLV) was observed upon inactivation of SD′ without change in the gag amino acid sequence (Dejardin et al., 2000). We also reported an SD′-suppression mutation which extends Moloney-MLV leukemogenic properties in vivo. Thus, while wt Moloney MLV induces exclusive development of T lymphomas when inoculated in newborn mice, SD′-defective Moloney MLV leads to the rapid development of a broad panel of leukemic cell types, including cells from the thymic, erythroid and myelomonocytic lineages (Audit et al., 1999).
To evaluate the effects of this subgenomic 4.4 kb transcript in MLV infection ability and its fate during the viral cycle, we tested whether the expression of a prespliced SD′ RNA could complement in trans the infectivity of the SD′-defective mutant. We then studied the SD′ RNA and potential derived product(s) throughout early and late events of the viral life cycle. For this purpose, we undertook extensive quantitative studies of each viral RNA species (unspliced and spliced) in infected cells and viral particles. Production of subsequently integrated proviral DNA was also precisely quantitated. The alternatively spliced SD′ RNA was found to be specifically incorporated into MLV particles along with the genomic unspliced RNA. In addition, our results demonstrate that SD′ RNA acts as a defective retroelement since it was reverse transcribed and integrated into the host genome as new ‘proviral’ DNA copies. Finally, we showed that the SD′ RNA leads to synthesis of new MLV p50 and p60 polyproteins.
Results
The novel subgenomic transcript SD′ RNA is required for MLV optimal infectivity
Inactivation of SD′ by site-directed mutagenesis which did not change the gag coding ability led to a reduction in infectivity of replication-competent MLV (Dejardin et al., 2000). Because inactivation of the SD′ site also reduced the formation of env mRNA, it remains to be demonstrated that the loss of SD′ RNA accounts for the observed mutant infectivity reduction. To evaluate these observations separately, we designed experiments based on reconstitution of virus particles from three distinct plasmids allowing the separate production of the Gag/Pol, Env and genomic RNA components (Figure 1A). A mutant version of the genomic vector was derived from our previous Friend SD′ inactivated mutant virus F1 displaying three-point mutations in SD′ site (Dejardin et al., 2000) (Figure 1A). 293T cells were transiently transfected with the three-plasmid expression system. Virus preparations were titrated on NIH3T3 target cells by staining for alkaline phosphatase (AP) activity carried by the packaged genomic RNA. While reconstituted wt virions displayed an average titer of 1.7 × 104 FFU/ml, the SD′ mutations in the context of the three-vector system maintained a 10-fold lower viral titer despite the trans-production of Env and Gag/Pol proteins (Figure 1C). To ensure that reduced infectivity arised from the loss of SD′ RNA, we measured the effect of introduction in trans of a fourth vector expressing the prespliced 4.4 kb SD′ RNA (Figure 1B). Co-introduction of SD′ cDNA restored wt infectivity levels of the SD′-defective mutant, whereas it did not change the viral titer of wt particles (Figure 1C). Taken together, these results demonstrated the importance of SD′ RNA in optimal infection ability. We then quantitated the presence of SD′ RNA throughout early and late stages of life cycle of replication-competent MLV.


Fig. 1. Effect of SD′ mutation on virus replication in vector production system and trans-complementation assay. (A) Schematic maps of plasmids used in the virus vector production system and expressing the viral components independently. Location of splice donor (SD) and acceptor (SA) sites is shown. Mutations are indicated by asterisks and inactivated functions are crossed. The two versions of the genomic vector are represented: wt, wild type; mutant, inactivated SD′ site (referred to F1 mutation; Dejardin et al., 2000). (B) Structure of plasmid allowing prespliced SD′ RNA and used in trans-complementation assays. (C) Titration of virus stocks on NIH3T3 target cells. The vector pseudotypes of retroviruses shared common capsid and envelope proteins; only the nature of the genomic vector varied. In trans-complementation assays, the additional vector is noted as +SD′ for the p57cDNASD′ vector. Titers are given in AP focus-forming units per milliliter. Experiments were performed at least three times for each virus in parallel and each test was performed in quadruple. The bars indicate the standard error of the mean of each series.
The subgenomic SD′ RNA is encapsidated into wt MLV particles
Alternative splicing maintains the non-coding 350-nucleotide-long Psi packaging sequence downstream of the canonical 5′ splice donor site of MLV (Mann et al., 1983). This region contains the four hairpin structures that we previously described as the minimal elements required for specific encapsidation of Moloney-MLV RNA into MLV particles (Mougel et al., 1996). Since alternative splicing maintains Psi in SD′ RNA, we asked whether the latter was packaged into MLV particles and with what efficiency. Supernatant from Mus dunni cells, either infected with wt replication-competent Friend-MLV or uninfected, were collected. RNA contents of MLV virions and infected M.dunni cells were monitored by quantitative RT-PCR analysis. Four different real-time PCRs were conducted from a unique RT reaction to separately detect and quantitate the presence of full-length RNA, SD′ RNA, env mRNA and control GAPDH mRNA (Figure 2). As expected, no viral RNA was detected in either mock-infected cells or pelleted medium samples from mock M.dunni cells (Figure 2A, lanes 1 and 3). Despite the low level of SD′ RNA present in infected cells (63 copies/cell) compared with that of viral genomic RNA (813 copies/cell) and env RNA (1177 copies/cell) (Figure 2 B), SD′ RNA was efficiently incorporated into progeny virions (2.5% compared with a 4% incorporation for genomic RNA) (Figure 2C). Although the env RNA level was 18 times higher in infected cells than that of the SD′ RNA, env RNA packaging efficiency (0.002%) was closer to that observed with the GAPDH negative control (0.0006%) (Figure 2C). These data showed for the first time that two different RNA species were preferentially packaged into MLV virions: full-length genomic RNA and alternatively spliced SD′ RNA.

Fig. 2. Measure of SD′ RNA content in replication-competent MLV infected M.dunni cells and virions by real-time RT-PCR. (A) Analysis of RT-PCR products by agarose gel electrophoresis. Viral RNA components were extracted from either infected cells or virions, and were analyzed by quantitative RT-PCR. From a unique RT reaction, four different PCRs were performed specific to each RNA species: genomic (FL), alternatively spliced (SD′), canonically spliced (SD) and control GAPDH mRNA. Approximate positions of the primers used to amplify the different RNA species are indicated by the numbered arrows. Mock samples correspond to similar experiments conducted with non-infected cells. Amplified samples were loaded on agarose gel and stained by ethidium bromide. Each RNA species detected is indicated on the left side of the gel, with corresponding oligonucleotide pairs identified by numbers on the right. (B) Results of quantitative RT-PCR experiments. Relative levels of FL (fixed to 100), SD′ and SD RNA in both chronically infected cells and virions were determined as described in Materials and methods. Values represent average of at least three independent RT-PCR assays with standard deviations. Absolute quantification values (parentheses) are expressed as RNA copy number per infected cell. (C) RNA encapsidation efficiency of each viral RNA species. Final encapsidation levels were calculated as the ratio of viral to cellular RNA values obtained in (B) and expressed as a percentage.
SD′ RNA is present throughout early events of infection
Assuming that SD′ RNA is also packaged in infectious particles, we postulated that it might be present in the early stages of the viral life cycle which consist of virus entry, uncoating, reverse transcription and nuclear entry of viral DNA followed by integration of proviral DNA in the host genome.
SD′ RNA is copied in an SD′ DNA by the reverse transcriptase. SD′ RNA harbors all the cis-signals required for reverse transcription including a tRNA primer-binding site and polypurine tract for initiation of first- and second-strand DNA synthesis as well as a repeated (R) region at both ends of the viral RNA required for transfer of DNA synthesis between templates. Reverse transcription initiated in the virions is completed in the infected cells within the first hour after infection. Thus total cellular DNA of M.dunni cells either infected with Friend-MLV or uninfected was extracted 4 h post-infection for analysis of non-integrated proviral DNA content. Using the real-time PCR conditions described above, without a prior reverse transcription step, two viral DNA species were detected which corresponded to the expected full-length proviral DNA and to the additional reverse transcribed subgenomic SD′ DNA (Figure 3). Four hours post-infection, we found that the level of non-integrated SD′ DNA production was 24-fold lower than that of proviral DNA. Not surprisingly, no DNA copy of env mRNA was significantly detected, since env mRNA is marginally detected in virions. Control amplifications with each primer combination performed with samples from mock-infected M.dunni cells gave no background signal (data not shown).

Fig. 3. Analysis of proviral DNA in the MLV-infected M.dunni cells by real-time PCR. Cellular DNA was extracted from 4 h infected or from chronically or mock-infected M.dunni cells. The same amount of cellular DNA (50 ng) was submitted to quantitative PCR amplifications using the same specific primers as used in previous RT-PCR experiments (see Materials and methods). Results were normalized to canonical full-length proviral DNA arbitrarily fixed to 100. Results are presented as means ± SD (error bars) of three independent measurements. Absolute quantification values (parentheses) are expressed as proviral DNA copy number per cell.
Reverse transcript from SD′ RNA is integrated in the host genome. Investigations of integrated forms of viral DNA were conducted on chronically infected cells in which only integrated forms are present. This absence of de novo reverse transcription is due to superinfection resistance of infected cells. Quantitative PCR assays showed that SD′ DNA copy was present among integrated DNAs. It is noteworthy that 17 copies of FL proviral DNA were integrated into the host genome and only 0.5 copy for SD′ proviral DNA (Figure 3). These values correlated with the relative RNA contents measured in viral particles since viral FL RNA level was 20 times higher than that of viral SD′ RNA. In addition, no significant effect of Zidovudine (AZT), an inhibitor of the reverse transcriptase, was observed on FL and SD′ proviral DNA copy numbers, confirming the integrated form of the two proviral DNA (data not shown).
Proviral DNA from SD′ RNA is a novel splice donor-associated retroelement (SDARE). To demonstrate that proviral DNA from SD′ RNA was integrated by viral mechanism, we analyzed its integration sites in M.dunni cell genome. To carry out integration, the virus-encoded integrase first cleaves two nucleotides from each 3′ viral DNA end and then joins the newly exposed 3′ hydroxyl groups to phosphodiester bonds in the target host DNA (Brown et al., 1989). Completion of integration leads to a duplication of a short sequence from the target site, which flanks the integrated provirus (Van Beveren et al., 1980). Thus we isolated specific integration sites and looked for these characteristics at the integrant termini. We used the inverse-PCR approach, initially applied to identification of clonal MLV integration sites in mouse tumors (Lund et al., 2002; Suzuki et al., 2002), to analyze MLV integrants. Inverse PCR and nested PCR were adapted in order to selectively amplify the novel retroelement against the abundant full-length proviral DNA. Strategy and positions of used primers are provided in the Supplementary data (available at The EMBO Journal Online). Amplified products were cloned, integration junctions were sequenced and sequences were subjected to Blast analysis against the mouse genome database from Ensembl. Sequencing of several termini and adjacent cellular sequences showed that SDARE harbored the dinucleotide deletion resulting from integrase terminal cleavage and a 4 bp flanking short direct repeat from the cellular target which also resulted from integration pathway (Supplementary table I). Therefore the SD′ RNA acted as a bona fide retroelement, sharing all the essential steps with the full-length genomic RNA such as encapsidation, reverse transcription and viral integration.
The subgenomic SD′ RNA is translated into novel Gag/Pol polyproteins
Coupled transcription/translation in reticulocyte lysate systems. As alternative splicing is a common event for increasing translational capacity of a primary transcript, it was interesting to evaluate the coding capability of SD′ RNA. Indeed, the coding potential of SD′ RNA is large since the transcript includes the translation initiation codons of the glyco-gag and gag used in the FL, as well as several putative initiation codons in the two other open reading frames (ORFs) and the intact env ORF (Figure 4A). Translation of SD′ RNA was first investigated in the rabbit reticulocyte lysate in vitro system (RRL). Linearized p57cDNASD′ plasmid was used as template to perform a coupled transcription/translation reaction. As shown in Figure 4B, two major products were synthesized with apparent molecular masses of 50 kDa and 60 kDa, respectively (lane 2). In parallel, the molecular clone of Friend-MLV was used as positive control (lane 1). MLV genomic RNA directs the synthesis of two Gag-related proteins: Pr65gag, the precursor Gag with MA, p12, CA and NC, and Pr75glyco–gag, the precursor of glycosylated Gag. Translation of Pr65gag starts at an AUG (position 619) while Pr75glyco–gag is initiated at a CUG (position 355) in frame with AUGgag (Evans et al., 1977; Edwards and Fan, 1979; Saris et al., 1983; Prats et al., 1989) (Figure 4A). Based on the sizes of the two bands observed with the SD′ vector (50 and 60 kDa), SD′ translation was likely to correspond to the recruitment of the CUGglyco–gag and the AUGgag codons and the use of the natural pol stop codon. Similar experiments performed with both templates linearized by PvuII which cuts upstream of the SD′ site confirmed the positions of initiation codons (Figure 4A). Thus identical patterns were obtained for both templates with two bands at 43 and 33 kDa (lanes 3 and 4). These results indicated that SD′ translation recruited the same initiation codons as those used to genomic MLV translation.

Fig. 4. In vitro transcription/translation of cDNASD′. (A) Schematic representation of p57(2LTR) and p57cDNASD′ plasmids used for in vitro transcription of FL and SD′ RNA, respectively. In both cases, transcription starts at the T7 promotor (bold broken arrow) located upstream at the 5′ viral LTR (rectangular boxes) and ends either at the XhoI or PvuII restriction sites as noted on the map. The positions of translation initiation codons of Glyco-Gag, Gag and Env proteins are indicated by thin broken arrows. Putative additional initiation sites of p57cDNASD′ are indicated by arrowheads. (B) Translated products from SD′ mRNA in rabbit reticulocyte lysate. Template plasmids described in (A) were used for one-step in vitro transcription/translation assays after linearization with XhoI (lanes 1 and 2) or PvuII (lanes 3 and 4) and resulting [35S]methionine-labeled proteins were electrophoresed on polyacrylamide gel. The background of the reactions was controlled by the missing DNA vector (lane 5).
SD′ expression in murine and human cells. Because in vitro translation may hide such particular translational features as readthrough, cap-independent initiation or usage of suboptimal initiation codons, all of which are commonly reported in retroviral translation, the usage of CUG and AUG Gag initiation codons was also evaluated in murine NIH3T3 cells, either infected with Friend-MLV or uninfected, and in human 293T cells commonly used for their high transfectable efficiency.
First, the SD′ ORF detected upon in vitro assays was cloned into pGFP-N1, pGFP-N2 and pGFP-N3 vectors carrying enhanced fluorescent protein gene in different frames (Figure 5A). The three p57SD′/GFP constructs were transiently transfected into murine NIH3T3 and 293T cells and protein analysis was performed by immunoblotting using an anti-GFP antibody (Figure 5B). Control transfections were performed with each corresponding empty vector harboring GFP alone (lanes 1, 3, 5, 7, 9 and 11). Fusion proteins were observed only when SD′ cDNA was inserted so that the CUGglyco–gag and the AUGgag codons were in frame with the GFP ORF (lanes 4 and 10). This approach did not reveal any other functional initiation codon of SD′ mRNA in the other reading frames (lanes 2, 6, 8 and 12). The major band obtained in both murine and human cells corresponded to a protein of approximately 77 kDa, which corresponded to the fusion product of the 50 kDa protein (p50) seen in the in vitro translation assay (Figure 4) with the 27 kDa GFP protein. A minor band corresponding to an 87 kDa apparent molecular mass was also observed in NIH3T3 cells (lane 4). This product most likely corresponded to the usage of the CUGglyco–gag. Interestingly, the alternative CUG initiation codon did not appear to be as efficiently used in the 293T cells (lane 10).

Fig. 5. Search for SD′ mRNA ORF ex vivo. (A) Schematic representation of p57SD′/GFP construct. SpeI-SacII fragment from p57cDNASD′ was fused to the NheI-SacII cloning sites of vectors expressing GFP in three frames. Arrows indicate initiation codon positions of Glyco-Gag, Gag and Env. (B) Analysis of proteins fused to the N-terminus of GFP by immunoblotting. The three constructs were transfected into NIH3T3 (lanes 1–6) or 293T (lanes 7–12) cells, and cellular protein extracts were analyzed by western blot with an anti-GFP antibody as described in Materials and methods. Reading frame and GFP plasmids, fused (+) or not fused (–) to SD′, are noted at the top.
We further characterized the SD′ gene encoded products in murine cells, infected or not, with different anti-MLV antibodies. The main SD′-derived protein p50, as produced by the SD′ cDNA clone (p57cDNASD′), was detected by an anti-Pol antibody which also detected IN, RT and Gag-Pol precursor as described elsewhere (Tanese et al., 1986) (Figure 6A, left panel). Closer epitope analysis was performed using a polyclonal anti-integrase antibody which allowed detection of p50 alone (lanes 4 and 5) or p50 fused to GFP (77 kDa) as produced by p57SD′/GFP vector (lane 3) (Figure 6A, right panel). While the anti-IN did not allow detection of Gag-Pol precursor, integrase (45 kDa) was visualized in infected cells (lane 5). Control samples performed with mock-transfected NIH3T3 cells (lane 1) or NIH3T3 cells transfected only with the GFP-expressing plasmid (lane 2) confirmed the reaction specificity. Similar results were obtained with a monoclonal anti-matrix antibody (data not shown). In contrast, no signal was obtained when using either the H187 monoclonal antibody directed against the capsid C-terminus or an anti-envelope, indicating that corresponding epitopes were missing in p50 and p60 (data not shown). In summary, these experiments established that the SD′ mRNA encoded a polyprotein, p50, which includes MA, p12, the first 110 aa of CA in frame with the last 116 aa of IN and an additional and less abundant protein p60, which is 10 kDa larger, using the upstream CUGglyco–gag codon (Figure 6B).

Fig. 6. Characterization of SD′ gene product. (A) Western blot analysis of cellular SD′ gene products. The p50 protein was detected with an anti-Pol antibody in infected NIH3T3 cells transfected with p57cDNASD′ (left panel). The right panel shows western blot analysis by using an anti-IN antibody. NIH3T3 cells (lane 1) were transfected with empty GFP vector (lane 2), p57SD′/GFP (lane 3) and p57cDNASD′ (lane 4). Infected NIH3T3 cells were transfected with p57cDNASD′ (lane 5). (B) Structure of p50 and p60 polyproteins encoded by SD′ mRNA.
p50/GFP is associated with cell membranes and virions. We further observed by confocal microscopy the chronically infected cells transiently transfected with p57SD′/GFP. While GFP alone was uniformly distributed throughout the cell (data not shown), fluorescent fusion proteins appeared speckled, distributed in perinuclear area and accumulated along the plasma membrane (Figure 7A) as clearly seen by the three-dimensional visualization (Figure 7B). Similar localizations were observed with the 293T transfected by p57SD′/GFP missing p60 protein expression (data not shown). This distribution indicated that the fusion proteins reached the plasma membrane and might participate in the virus assembly process at the membrane. We further monitored whether fusion proteins could be incorporated into progeny virions. Supernatants from NIH3T3 cells infected with Friend-MLV and transfected with either p57SD′/GFP or control GFP plasmid were collected and filtrated through a 0.22 µm filter. Virions were pelleted on a sucrose cushion and the protein content was analyzed by immunoblotting using an anti-GFP antibody (Figure 8). As expected, no signal was observed with mock-transfected cells (Figure 8, lane 1). Although highly expressed in NIH3T3 cells (lane 2), only a small fraction of GFP was found in viral supernatant pellet (lane 5). In contrast, the p50/GFP protein was found preferentially in the pelleted fraction (comparison of lanes 3 and 6), suggesting its incorporation into virus particles. Interestingly, p60 remained undetectable in the virion preparation.

Fig. 7. Localization of p50/60 protein fused to GFP in NIH3T3 infected with MLV. Infected cells cultured on coverslips were transfected with p57SD′/GFP clone grown for 2 days and analyzed for direct fluorescence localization. (A) Cells were photographed on a Leica scanning microscope using a 100× oil immersion objective with a GFP or Hoechst staining filter. Images were deconvoluted using Huygens 2 software. Arrows indicate plasma membrane (m) and perinuclear (pn) localization of fusion protein. Hoechst nuclear staining appears in blue. (B) Detailed analysis of membrane localization. Three-dimensional visualization of the cell section shown as an open rectangle in (A) was obtained by Imaris software analysis. Fusion protein (green) is localized at the plasma membrane all around the cell.
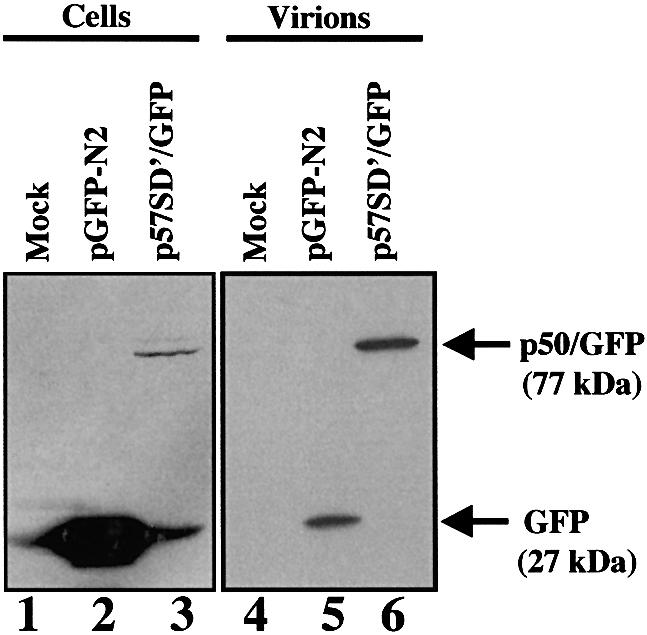
Fig. 8. Detection of p50/GFP protein in MLV particles. Same amount of NIH3T3 infected cells were transfected with p57SD′/GFP or pGFP-N2 vector. To allow quantitative analysis proteins were extracted from 5 × 105 cells (lanes 1–3) and from corresponding cell-free pelleted supernatant (lanes 4–6) and analyzed by western blotting using an anti-GFP antibody. Mock-transfected cells (lanes 1 and 4) and cells transfected with GFP vector (lanes 2 and 5) were used as controls.
Discussion
A new SD′-associated retroelement
As previously reported, inactivation of SD′, a simple oncovirus-conserved alternative splice donor site, leads to reduced replication of replication-competent MLV with viral splicing alterations (Dejardin et al., 2000). To analyze the effects of these SD′ mutations further, a three-plasmid vector system (with dissociated expression of retroviral components) was used. In this vector system, SD′ mutations still maintained a reduction in viral infectivity. Yap and coworkers (Yap et al., 2000) showed that when using these transient vector production systems (where ecotropic envelope is produced from a separate mRNA) virus stock titers produced in 293T are not critically dependent on the balance between the vectors. Thus the effect of the SD′ mutations observed with this system could not have arisen from a defect in env mRNA production, in contrast with results obtained with a replication-competent mutant virus where production of the env mRNA is strictly dependent on production and splicing of the full-length viral RNA (Dejardin et al., 2000). Since SD′ mutations are carried by the genomic RNA vector, and not by the capsid vector, the mutant viral titer essentially resulted from the cis-acting effect of SD′ mutations. This could account for the difference in titer reductions observed for SD′ mutations introduced in the replication-competent MLV (100-fold) or in the three-vector system (10-fold). Importantly, the trans expression of the prespliced SD′ RNA complemented the infectivity of an SD′-defective mutant, thus demonstrating a role of SD′ RNA in viral replication.
In addition to effects of SD′ usage on retroviral splicing (Dejardin et al., 2000), we also decided to search for the presence of SD′ RNA in the different steps of viral replication because this RNA harbors key features of retroviral elements. During expression of the MLV genome in chronically infected cells, we found that SD′ RNA (∼63 copies/cell) was 13-fold less abundant than the full-length RNA, while env mRNA was the most abundant. Full-length newly transcribed RNA assumes at least two essential functions: in addition to acting as mRNAs, unspliced viral transcripts are encapsidated into assembling particles as the genomic viral RNA. Retroviral RNA packaging involves interactions between the Psi sequence (present at the 5′ end of the viral RNA genome) and the Gag polyprotein precursor (Aldovini and Young, 1990; Gorelick et al., 1990; D’Souza et al., 2001; and references cited therein). It was commonly thought that only the unspliced RNA is efficiently incorporated into nascent virions. Here, we found that the alternatively spliced SD′ RNA was also highly selected for encapsidation against a high background of cellular and viral RNAs. Indeed, SD′ RNA and unspliced genomic RNA were encapsidated with similar efficiency. This is the first demonstration that the unspliced genomic RNA is not the unique viral RNA component of virion content. Moreover, the cellular presence of SD′ as a DNA copy 4 h after infection also indicated that SD′ RNA is a functional structural component of infectious MLV virions. Thus it is tempting to consider that constitution of genomic RNA virions includes heterodimeric FL/SD′ forms. Experiments designed to detect the existence of such forms in virion preparations, while not trivial, are in progress.
The concomitant packaging of these two RNA species should influence the reverse transcription which begins early in infection. During reverse transcription, it is known that template switching between heterologous copackaged viral transcripts generates recombinations (Stuhlmann and Berg, 1992). Recombination events allow retroviruses to alter or increase their host range and to escape host defenses and antiretroviral therapy. Integration of proviral SD′ DNA copy may stabilize SD′ DNA against degradation as shown for full-length proviral DNA (Butler et al., 2001).
Finally, SD′ RNA shares all the characteristics of disseminating-defective retroviruses, since it goes through encapsidation, reverse transcription and integration steps. The spreading of such SD′ retroelement derived from alternative usage of splice donors is likely to bear consequences on dissemination and pathogenesis of MLV retroviruses. Indeed, we showed in a previous work that inactivation of the SD′ site leads to extensive modifications of MLV leukemogenic properties (Audit et al., 1999). MLV and other simple oncoretroviruses are implicated in leukemogenic processes through insertional mutagenesis mechanisms, where provirus DNA integrates into the host genome and affects the transcription of the neighbouring genes. Interestingly, the SD′ splice site has been shown to play a key role in c-myb rearrangement in MLV-induced myeloid tumors in adult mice (Shen-Ong, 1987). Rearranged proto-oncogenes often harbor defective proviruses. These defective proviruses were thought to derive from DNA genomic rearrangement or insertion of accidentally produced defective retroviruses (Felder et al., 1994; Fan, 1997; Wolff, 1997; Muriaux and Rein, 2003). We describe an additional source for such inserted retroelements as SDAREs. Another case of such defective provirus generated by pregenome splicing has been described for human foamy retrovirus (HFV) (Saib et al., 1993). This coding-defective HFV was involved in the establishment of HFV chronic infection through interference with lytic properties (Saib et al., 1995).
Proteins encoded by SD′ mRNA
Since alternative splicing is an event commonly described to generate new translational products from a single precursor in eukaryotes, protein encoded by SD′ mRNA could also participate in the viral infectious capability. We found that SD′ RNA directed synthesis of p50 (and to a lesser extent p60) proteins with translation initiations at two initiation codons in the same ORF: AUGgag and CUGglyco–gag, respectively. These two initiation codons were also recruited for synthesis of Gag (Pr65) and Glyco-Gag (Pr75) precursors in full-length RNA via an internal ribosomal entry mechanism (Berlioz and Darlix, 1995; and references cited therein). The new p50 and p60 polyproteins harbored N-terminal Gag domain including MA, p12 and the first 110 amino acids of CA in frame with the last 116 amino acids of integrase. Appending some functional domains of C-term integrase to those from N-term of Gag likely provides particular properties for the novel Gag/Pol polyproteins. The N-terminal part contained signals required for targeting to budding site and virus assembly, which correlate with the membrane location and virion association of the p50/GFP proteins (Wills and Craven, 1991; Yuan et al., 1999). The striking punctate pattern observed around the nucleus and at the plasma membrane by confocal imaging has been related to viral assembly function (Jones et al., 1990; Hermida-Matsumoto and Resh, 2000; Chen et al., 2001). Glyco-Gag was associated with in vivo spreading and pathogenesis of MLV (Corbin et al., 1994; Corbin and Darlix, 1996). Mutagenesis studies also referred to functions of MA, p12 and CA in the early events of the viral life cycle as a synthesis of viral DNA and nuclear imports (Alin and Goff, 1996a,b; Kiernan et al., 1998; Yuan et al., 1999). The C-terminal portion of MLV integrase included in p50 or p60 polyprotein has been functionally characterized to be the site of non-specific DNA binding, and to be required for efficient integration and disintegration activities (Jonsson et al., 1996). Interestingly enough, this domain of MLV integrase contains an insertion specific to MLV-related retroviruses which is not conserved in other retroviruses (Jonsson et al., 1996). In addition to an SD′ RNA role in cis, the p50 and p60 polyproteins could function as regulatory protein for whichever study of precise role is under way. These elucidations will also likely contribute to optimization of the in vivo efficacy of standard MLV-based vectors used for therapeutic purposes.
Materials and methods
Plasmids and viral constructions
Retrovirus vector production is based on the segregation of viral components into three different plasmids that are used in transient transfection (Figure 1A) (Soneoka et al., 1995): (i) retroviral vectors, wt or mutant version producing genomic RNA were derived from the parental Friend 57 or Friend F1 SD′ mutant clone (Dejardin et al., 2000) with three successive stop codons (TAA, TAG and TGA) to prevent Gag/Pol expression [genomic vector also carries the human placental AP reporter gene in place of env whose expression is promoted by an internal ribosome entry site (IRES)]; (ii) a GagPol expression vector (capsid vector), pC57GP, which contains a human CMV immediate early promoter, a β-globin intron upstream of Friend gagpol and a simian virus 40 poly-adenylation signal; (iii) an envelope expression vector FBFSALF (a kind gift from F.Kim), derived from FBMOSALF (Cosset et al., 1995), in which the ecotropic Moloney-MLV env gene was replaced by the Friend 57 gene.
For in vitro transcription/translation assay, the Friend-MLV molecular clone, strain 57 (Sitbon et al., 1990), was cloned downstream of T7 promotor of pSP72 plasmid (Promega) and converted to a two-LTR molecular clone version called p57(2LTR) (Figure 4A). Excision of intronic sequences between SD′ and SA splice sites from p57(2LTR) were performed by PCR mutagenesis to generate the p57cDNASD′ vector which expressed prespliced SD′ RNA (Figures 1B and 4A). A series of SD′ constructs fused to the N-terminus of GFP in the three reading frames was made by inserting the SpeI–SacII fragment (1650 nucleotides long) of p57cDNASD′ into pGFP-N1, pGFP-N2 or pGFP-N3 plasmids (Clontech) cut at NheI and SacII unique sites to generate the p57SD′/GFP vector series (Figure 5A).
Details of plasmid constructions will be provided on request.
Cell culture, transfection and infection
Mus dunni, 293T and NIH3T3 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with glutamine (2 mM), penicillin, streptomycin and 10% heat-inactivated fetal calf serum at 37°C. Transient transfections were performed onto 10 cm diameter plates with cells split the day before. Two micrograms of DNA were commonly transfected by the lipofectamine Plus Reagent™ (Invitrogen) method according to the manufacturer’s instructions, except for transfections performed with the three-vector system where the calcium phosphate precipitation method was used (Graham and van der Eb, 1973). Cells and supernatants were collected 2 days after transfection. Friend-MLV viral stocks used for virus infections displayed an average titer of 105 expressed as infectious focus-forming unit per milliliter. Infections and viral stock preparations were as previously described (Dejardin et al., 2000). Unless indicated otherwise, cells were maintained 2–4 months after infection and considered as chronically infected. AZT treatment of chronically infected cells was performed with 50 µM AZT over a period of 3 days.
Transient three-plasmid expression system
Transient transfections of 293T cells were performed with 2 µg of each genomic, capsid and envelope vector. In trans-complementation assay, 2 µg of the fourth vector, p57cDNASD′, was added. Two days later, transfection efficiencies were monitored by detection of AP activity with a ready-to-use coloration mixture (Sigma). Before staining, cells were fixed in 0.5% glutaraldehyde in phosphate-buffered saline (PBS) for 10 min at room temperature, washed with PBS and incubated for 1 h at 80°C in PBS. Supernatants were collected and filtered through filters with a pore size of 0.20 µm. For virus titration, serial dilutions of supernatant were used to infect NIH3T3 target cells split the day before. Two days later, cells were fixed and stained as described above and virus titers were determined by counting AP-positive cells using binoculars (Leica).
In vitro translation
For analysis of SD′ RNA coding ability, p57cDNASD′ construct or p57(2LTR) control vector were linearized either with XhoI which cuts vector sequences downstream at the viral 3′ LTR or with PvuII upstream of the splice SD′ site (Figure 4A). One hundred nanograms of linearized DNA were used for one-step in vitro transcription/translation using TNT® Coupled Reticulocyte Lysate Systems (Promega) according to the manufacturer′s instructions. [35S]methionine (Amersham) was used for radioactive labeling of synthesized proteins. Translation reactions were prepared for gel loading by adding an equal volume of 2× sample buffer (12.5 mM Tris-HCl pH 6.8), 2% SDS, 20% glycerol, 0.25% bromophenol blue, 5% B-mercaptoethanol, 40 mM dithiothreitol). After boiling for 5 min, samples were subjected to 10% SDS–polyacrylamide gel electrophoresis (SDS–PAGE) and were electroblotted on nitrocellulose filter (Schleicher & Schuell). Then membrane was autoradiographed with Kodak Biomax MR films.
Analysis of viral RNA incorporation in virions
Chronically infected M.dunni cells were cultured in 10 cm diameter dishes. Viral RNA was isolated from viral particles essentially as described previously (Mougel et al., 1996). Cells were washed twice with PBS and total cellular RNA was extracted from cell pellets with TriReagent™ (Sigma) according to the manufacturer’s instructions. The samples were treated with RNase-free DNase (RQ1, Promega) to remove DNA contamination. Cellular RNA concentrations were quantitated by measuring optical absorption at 260 nm and RNA integrity was checked by agarose gel electrophoresis followed by ethidium bromide staining. Reverse transcription was performed with 2 µg of cellular RNA or 1/20 aliquots of viral RNA, the oligo (dT) as primer and the Expand™ reverse transcriptase from Roche under the conditions described (Dejardin et al., 2000). The RT reaction mixture was then inactivated at 95°C for 2 min and chilled at 4°C. Only 5% of the RT product was used for one quantitative PCR. Real-time PCR was performed with QuantiTect™ SYBR® Green PCR Kit (Qiagen) in glass capillary tubes containing a final volume of 20 µl (5 µl cDNA mix + 15 µl PCR mix) with the LightCycler System (Roche). Three combinations of oligonucleotide pairs, where s and a indicate the sense and antisense orientations, respectively, were used to detect specific MLV transcripts. The position of the transcriptional start was as follows, with the size of the amplified products indicated in parentheses: s3350 and a3600 (250 bp) for full-length RNA detection; s76 and a5620 (259 bp) for SD; s1450 and a5620 (276 bp) for SD′. Primers to amplify GAPDH were as follows: (sense) 5′ -GCTCACTGGCATGGCCTTCCGTG-3′ and (antisense) 5′-TGGAA GAGTGGGAGTTG CTGTTGA-3′ (200 bp). Each of 40 PCR cycles consisted of denaturation for 15 s at 95°C, annealing for 15 s at 60°C and extension for 20 s at 72°C. Specificity was determined using melting curve analysis and electrophoresis on 2% agarose gels. For each run, a standard curve ranging from 102 to 106 copies was generated from plasmids p57(2LTR), p57cDNASD′ and pFBFSALF for FL, SD′ and SD RNA quantification, respectively. Dilutions of the plasmids were prepared in a solution containing 20 ng/µl of total RNA from non-infected M.dunni cells and 1:25 RT buffer to mimic RT-PCR experimental conditions. Quantitative analysis was performed using LightCycler™ software version 3 (Roche Molecular Biochemicals). Absolute quantification results were expressed as copy number per cell.
Analysis of proviral DNA in infected cells
Approximately 107 M.dunni cells, either infected or uninfected, were rinsed twice in ice-cold PBS and lysed in 100 mM NaCl, 50 mM Tris pH 7.5, 5 mM EDTA and 0.5% SDS containing 200 µg/ml proteinase K by incubation for 12 h at 37°C under gentle agitation. Cell lysates were extracted twice with 1:1 (by volume) phenol:chloroform and cellular DNA was ethanol precipitated and dissolved in water. Quantitative PCR was performed with 50 ng of cellular DNA using the same oligonucleotide pairs and amplification conditions as described above for RNA analysis. The same standard curves were generated as described above except that dilutions of the control plasmids were performed with cellular genomic DNA (10 ng/µl) from non-infected M.dunni instead of total cellular RNA. I-PCR was adapted from Suzuki and coworkers (Suzuki et al., 2002; Supplementary data). Detailed experimental conditions will be provided on request.
Immunoblotting analysis of viral protein expression
Protein analyses were conducted from cells cultured on 10 cm plates. Extractions of cellular and viral proteins and western blot analysis were performed as previously described (Mougel et al., 1996; Audit et al., 1999). Proteins were detected by immunoblotting with a mouse anti-capsid (p30) monoclonal antibody (H187) (a kind gift from B.Chesebro) used at a 1:300 dilution as the primary antibody and a peroxidase-conjugated (HRP) goat anti-mouse antibody (1:2000) as the secondary antibody (Sigma), or with a mouse anti-matrix (p15) monoclonal antibody (H690) (a kind gift from M.Miyazawa) used at a 1:5 dilution as the primary antibody and an HRP goat anti-mouse antibody (1:1000) as the secondary antibody (Sigma), or with a rabbit anti-Pol polyclonal antibody (a kind gift from M.Roth) used at a 1:2000 dilution and an HRP goat anti-rabbit used at a 1:4000 dilution, or with a rabbit anti-integrase (p45) polyclonal antibody (a kind gift from F.D.Bushman) used at a 1:2000 dilution as the primary antibody and a peroxidase-conjugated goat anti-rabbit antibody (1:2000) as the secondary antibody, or with an anti-envelope polyclonal antibody, anti-gp70 of Raüscher (ViroMED) used at a 1:1000 and a peroxidase-conjugated goat anti-mouse antibody (1:2000) as the secondary antibody. GFP detection required a mouse anti-GFP antibody (Roche) used at 1:1000 as the primary antibody and a peroxidase-conjugated goat anti-mouse antibody (at 1:2000) as the secondary antibody (Sigma).
Fluorescence microscopy
Two days after transfection, cells were washed three times with PBS and fixed for 5 min at room temperature in 3% formaldehyde. Fixed cells were permeabilized with 0.1% Triton X-100 in PBS for 3 min at room temperature and rinsed in PBS. Hoechst staining (2 µg/ml) was performed for 1 min at room temperature before mounting in Mowiol. Cell imaging was carried out at our Institute Integrated Imaging Facility. Briefly, samples were observed with a DMR A Leica microscope using a 100× immersion oil objective (PL APO) attached to a piezoelectric step motor. The images were captured with a black-and-white CCD Captor (MicroMax 1300 Y/HS, Princeton, NJ) using Metamorph (Universal Imaging Corporation, USA). Images were restored using Huygens Professional 2.3.7 (Scientific Volume Imaging, The Netherlands) and viewed with Imaris 3 (Bitplane, Switzerland).
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank F.Carbonell, M.Laffont and N.Vernet for technical assistance. We also thank P.Travo, Head of the Department of Imaging Facility, for support. We acknowledge M.Sitbon (in whose laboratory this work was initiated) for his constant interest and fruitful discussions and J.Lenz, A.Lund and E.Desmarais for their advice on the integration site analysis. This work was supported by grant no. 9521 from the ARC (to M.M.), by grant no. 6889 from the AFM (to M.Sitbon), by grant no. 5989 from the ARC (to M.Sitbon) and by grant no. 03003 from the ANRS (to M.M.). L.H. was supported by a fellowship from the ARC, V.T. by the French government (MENRT) and J.L.B. by INSERM.
References
- Aldovini A. and Young,R.A. (1990) Mutations of RNA and protein sequences involved in human immunodeficiency virus type 1 packaging result in production of non infectious virus. J. Virol., 64, 1920–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alin K. and Goff,S.P. (1996a) Amino acid substitutions in the CA protein of Moloney murine leukemia virus that block early events in infection. Virology, 222, 339–351. [DOI] [PubMed] [Google Scholar]
- Alin K. and Goff,S.P. (1996b) Mutational analysis of interactions between the Gag precursor proteins of murine leukemia viruses. Virology, 216, 418–424. [DOI] [PubMed] [Google Scholar]
- Audit M., Dejardin,J., Hohl,B., Sidobre,C., Hope,T.J., Mougel,M. and Sitbon,M. (1999) Introduction of a cis-acting mutation in the capsid-coding gene of moloney murine leukemia virus extends its leukemogenic properties. J. Virol., 73, 10472–10479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlioz C. and Darlix,J.L. (1995) An internal ribosomal entry mechanism promotes translation of murine leukemia virus gag polyprotein precursors. J. Virol., 69, 2214–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown P.O., Bowerman,B., Varmus,H.E. and Bishop,J.M. (1989) Retroviral integration: structure of the initial covalent product and its precursor and a role for the viral IN protein. Proc. Natl Acad. Sci. USA, 86, 2525–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler S.L., Hansen,M.S. and Bushman,F.D. (2001) A quantitative assay for HIV DNA integration in vivo. Nat. Med., 7, 631–634. [DOI] [PubMed] [Google Scholar]
- Chen B.K., Rousso,I., Shim,S. and Kim,P.S. (2001) Efficient assembly of an HIV-1/MLV Gag-chimeric virus in murine cells. Proc. Natl Acad. Sci. USA, 98, 15239–15244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin A. and Darlix,J.L. (1996) Functions of the 5′ leader of murine leukemia virus genomic RNA in virion structure, viral replication and pathogenesis and MLV-derived vectors. Biochimie, 78, 632–638. [DOI] [PubMed] [Google Scholar]
- Corbin A., Prats,A.C., Darlix,J.L. and Sitbon,M. (1994) A nonstructural gag-encoded glycoprotein precursor is necessary for efficient spreading and pathogenesis of murine leukemia viruses. J. Virol., 68, 3857–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosset F.L., Morling,F.J., Takeuchi,Y., Weiss,R.A., Collins,M.K. and Russell,S.J. (1995) Retroviral retargeting by envelopes expressing an N-terminal binding domain. J. Virol., 69, 6314–6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza V., Melamed,J., Habib,D., Pullen,K., Wallace,K. and Summers,M.F. (2001) Identification of a high affinity nucleocapsid protein binding element within the Moloney murine leukemia virus Psi-RNA packaging signal: implications for genome recognition. J. Mol. Biol., 314, 217–232. [DOI] [PubMed] [Google Scholar]
- Dejardin J., Bompard-Marechal,G., Audit,M., Hope,T.J., Sitbon,M. and Mougel,M. (2000) A novel subgenomic murine leukemia virus RNA transcript results from alternative splicing. J. Virol., 74, 3709–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S.A. and Fan,H. (1979) gag-related polyproteins of Moloney murine leukemia virus: evidence for independent synthesis of glycosylated and unglycosylated forms. J. Virol., 30, 551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans L.H., Dresler,S. and Kabat,D. (1977) Synthesis and glycosylation of polyprotein precursors to the internal core proteins of Friend murine leukemia virus. J. Virol., 24, 865–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H. (1997) Leukemogenesis by Moloney murine leukemia virus: a multistep process. Trends Microbiol., 5, 74–82. [DOI] [PubMed] [Google Scholar]
- Felder M.P., Laugier,D., Yatsula,B., Dezelee,P., Calothy,G. and Marx,M. (1994) Functional and biological properties of an avian variant long terminal repeat containing multiple A to G conversions in the U3 sequence. J. Virol., 68, 4759–4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelick R.J., Nigida,S.M., Bess,J.W., Arthur,L.O., Henderson,L.E. and Rein,A. (1990) Noninfectious human immunodeficiency virus type 1 mutants deficient in genomic RNA. J. Virol., 64, 3207–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham F.L. and van der Eb,A.J. (1973) A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology, 52, 456–467. [DOI] [PubMed] [Google Scholar]
- Hermida-Matsumoto L. and Resh,M.D. (2000) Localization of human immunodeficiency virus type 1 Gag and Env at the plasma membrane by confocal imaging. J. Virol., 74, 8670–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.A., Blaug,G., Hansen,M. and Barklis,E. (1990) Assembly of gag-β-galactosidase proteins into retrovirus particles. J. Virol., 64, 2265–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson C.B., Donzella,G.A., Gaucan,E., Smith,C.M. and Roth,M.J. (1996) Functional domains of Moloney murine leukemia virus integrase defined by mutation and complementation analysis. J. Virol., 70, 4585–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan R.E., Ono,A., Englund,G. and Freed,E.O. (1998) Role of matrix in an early postentry step in the human immunodeficiency virus type 1 life cycle. J. Virol., 72, 4116–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leis J. et al. (1988) Standardized and simplified nomenclature for proteins common to all retroviruses. J. Virol., 62, 1808–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund A.H. et al. (2002) Genome-wide retroviral insertional tagging of genes involved in cancer in Cdkn2a-deficient mice. Nat. Genet., 32, 160–165. [DOI] [PubMed] [Google Scholar]
- Mann R., Mulligan,R.C. and Baltimore,D. (1983) Construction of a retrovirus packaging mutant and its use to produce helper-free defective retrovirus. Cell, 33, 153–159. [DOI] [PubMed] [Google Scholar]
- Mougel M., Zhang,Y. and Barklis,E. (1996) Cis-active structural motifs involved in specific encapsidation of Moloney murine leukemia virus RNA. J. Virol., 70, 5043–5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muriaux D. and Rein,A. (2003) Encapsidation and transduction of cellular genes by retroviruses. Front. Biosci., 8, D135–D142. [DOI] [PubMed] [Google Scholar]
- Prats A.C., DeBilly,G., Wang,P. and Darlix,J.L. (1989) CUG initiation codon used for the synthesis of a cell surface antigen coded by murine leukemia virus. J. Mol. Biol., 205, 363–372. [DOI] [PubMed] [Google Scholar]
- Saib A., Peries,J. and de The,H. (1993) A defective human foamy provirus generated by pregenome splicing. EMBO J., 12, 4439–4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saib A., Koken,M.H., van der Spek,P., Peries,J. and de The,H. (1995) Involvement of a spliced and defective human foamy virus in the establishment of chronic infection. J. Virol., 69, 5261–5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saris C.J.M., van Eenbergen,J., Liskamp,R.M.J. and Bloemers,H.P.J. (1983) Structure of glycosylated and unglycosylated Gag and Gag-Pol precursor proteins of Moloney murine leukemia virus. J. Virol., 46, 841–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen-Ong G.L. (1987) Alternative internal splicing in c-myb RNAs occurs commonly in normal and tumor cells. EMBO J., 6, 4035–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitbon M., Ellerbrok,H., Pozo,F., Nishio,J., Hayes,S.F., Evans,L. and Chesebro,B. (1990) Sequences in the U5-gag-pol region influence early and late pathogenic effects of Friend and Moloney murine leukemia viruses. J. Virol., 64, 2135–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneoka Y., Cannon,P.M., Ramsdale,E.E., Griffiths,J.C., Romano,G., Kingsman,S.M. and Kingsman,A.J. (1995) A transient three-plasmid expression system for the production of high titer retroviral vectors. Nucleic Acids Res., 23, 628–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhlmann H. and Berg,P. (1992) Homologous recombination of copackaged retrovirus RNAs during reverse transcription. J. Virol., 66, 2378–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T., Shen,H., Akagi,K., Morse,H.C., Malley,J.D., Naiman,D.Q., Jenkins,N.A. and Copeland,N.G. (2002) New genes involved in cancer identified by retroviral tagging. Nat. Genet., 32, 166–174. [DOI] [PubMed] [Google Scholar]
- Tanese N., Roth,M.J. and Goff,S.P. (1986) analysis of retroviral pol gene products with antisera raised against fusion proteins produced in E.coli. J. Virol., 59, 328–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Beveren C., Goddard,J.G., Berns,A. and Verma,I.M. (1980) Structure of Moloney murine leukemia viral DNA: nucleotide sequence of the 5′ long terminal repeat and adjacent cellular sequences. Proc. Natl Acad. Sci. USA, 77, 3307–3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills J.W. and Craven,R.C. (1991) Form, function and use of retroviral Gag proteins. AIDS, 5, 639–654. [DOI] [PubMed] [Google Scholar]
- Wolff L. (1997) Contribution of oncogenes and tumor suppressor genes to myeloid leukemia. Biochim. Biophys. Acta, 1332, 67–104. [DOI] [PubMed] [Google Scholar]
- Yap M.W., Kingsman,S.M. and Kingsman,A.J. (2000) Effects of stoichiometry of retroviral components on virus production. J. Gen. Virol., 81, 2195–2202. [DOI] [PubMed] [Google Scholar]
- Yuan B., Li,X. and Goff,S.P. (1999) Mutations altering the moloney murine leukemia virus p12 Gag protein affect virion production and early events of the virus life cycle. EMBO J., 18, 4700–4710. [DOI] [PMC free article] [PubMed] [Google Scholar]