

Abstract
The nuclear receptor heterodimers of liver X receptor (LXR) and retinoid X receptor (RXR) are key transcriptional regulators of genes involved in lipid homeostasis and inflammation. We report the crystal structure of the ligand-binding domains (LBDs) of LXRα and RXRβ complexed to the synthetic LXR agonist T-0901317 and the RXR agonist methoprene acid (Protein Data Base entry 1UHL). Both LBDs are in agonist conformation with GRIP-1 peptides bound at the coactivator binding sites. T-0901317 occupies the center of the LXR ligand-binding pocket and its hydroxyl head group interacts with H421 and W443, residues identified by mutational analysis as critical for ligand-induced transcriptional activation by T-0901317 and various endogenous oxysterols. The topography of the pocket suggests a common anchoring of these oxysterols via their 22-, 24- or 27-hydroxyl group to H421 and W443. Polyunsaturated fatty acids act as LXR antagonists and an E267A mutation was found to enhance their transcriptional inhibition. The present structure provides a powerful tool for the design of novel modulators that can be used to characterize further the physiological functions of the LXR–RXR heterodimer.
Keywords: crystal structure/lipid metabolism/mutational analysis/nuclear receptor/transcription
Introduction
Liver X receptors, LXRα (NR1H3) and LXRβ (NR1H2), are nuclear receptors (NRs) that regulate a number of genes involved in cholesterol homeostasis (Lu et al., 2001). The LXRs are activated by certain oxysterols in the cholesterol biosynthetic pathway and promote transcriptional activation by binding as heterodimers with retinoid X receptors (RXRα, β and γ) to LXR-responsive elements (DR4/LXRE) of target gene promoters (Willy et al., 1995; Janowski et al., 1996). The gene encoding 7α-hydroxylase was the first target gene described, and implicated a role for LXRs in cholesterol catabolism via bile acid synthesis (Lehmann et al., 1997; Peet et al., 1998). Recent studies establish LXRs as regulators of several aspects of cholesterol biology, including reverse cholesterol transport, intestinal absorption and lipoprotein remodeling (Costet et al., 2000; Laffitte et al., 2001; Zhang et al., 2001; Repa et al., 2002). In the liver, activation of LXRs induce de novo fatty acid biosynthesis, which has led to the suggestion that LXRs are sensors of the balance between cholesterol and fatty acid metabolism (Peet et al., 1998; Repa et al., 2000). The fact that unsaturated fatty acids can function as LXR antagonists, and thereby create a feedback mechanism, supports this suggestion further (Ou et al., 2001).
In peripheral tissues, including macrophages, LXRs seem to orchestrate a response to cholesterol loading by inducing genes involved in cholesterol efflux, e.g. ApoE, ABCA1 and ABCG1 (Laffitte et al., 2001; Repa et al., 2002). In addition, a recent study demonstrates that LXRs are implicated in negative regulation of macrophage inflammatory gene expression (Joseph et al., 2003). Although the exact mechanism needs to be elucidated further, the observed reciprocal regulation of macrophage immune response and cholesterol efflux give hope for the development of LXR agonists as a new strategy for intervention in human cardiovascular disease. In accordance, administration of the synthetic LXR agonist, T-0901317 (referred to below as T-17), has been shown to reduce foam cell formation and development of atherosclerosis in mice (Joseph et al., 2002).
Based on several functional and structural studies, a simple mechanism for ligand dependent activation of nuclear receptors, including LXRs, has been proposed. As the agonist binds in the core of the ligand-binding domain (LBD), a conformational change involving the C-terminal helix 12 (also known as AF2) takes place, introducing a binding site for coactivators in a groove formed mainly by helices 3, 4 and 12. The subsequent coactivator binding induces a multitude of activities, including recruitment of the basal transcriptional machinery. The complexity of NR activation has increased considerably with the discovery of selective NR agonists inducing distinct receptor conformations, allowing selectivity in the recruitment of cofactors and thereby also selectivity in biological signaling (Lonard and Smith, 2002). In addition, with ligands spanning a functional range from full agonism to full antagonism, NR signaling can be modulated considerably and has therefore become attractive for pharmacological intervention.
By binding both oxysterols and fatty acids, LXRs are functionally related to both the group of steroid hormone receptors (PR, GR, MR, AR and ER of the NR3 subfamily), and the more divergent group of receptors binding fatty acids and related compounds (PPAR, RAR and ROR of the NR1 subfamily, and RXR and HNF4 of the NR2 subfamily). Although promiscuous, the LXRs display an interesting selectivity. Whereas oxysterols activating LXRs include 22(R)-,24(S)-hydroxy cholesterol (HC), their enantiomers are either an LXR antagonist or a non-binder, respectively (Janowski et al., 1999; Spencer et al., 2001). The importance of a strong hydrogen acceptor at either carbon 22, 24 or 27 is illustrated by 24(S),25-epoxy cholesterol (24,25-EC) being an efficient LXR activator, whereas cholesterol, which lacks the functional group, does not affect the receptor at all.
In order to gain more insights into the structural determinants for ligand-dependent activation of LXRs, we decided to determine the structure of the LXR LBD, and here we report the crystal structure of the LXRα LBD as a heterodimer with RXRβ LBD. Both LBDs are in agonist conformation, complexed with two coactivator peptides and the synthetic agonist T-17 bound to LXRα and the pesticide metabolite methoprene acid (MPA) bound to RXRβ.
Results and discussion
Structure determination
Heterodimers of LXR and RXR were produced in Escherichia coli by recombinant coexpression and subsequent isolation. Coexpression with RXRs increased the yields and solubility of both LXRα and LXRβ considerably. Contaminating tetrameric apo-RXR could be removed by gel filtration, whereas no homodimeric LXR could be detected (data not shown). Initial crystallization conditions were screened for a number of LXR–RXR heterodimers, but crystals were only obtained with the LXRα–RXRβ combination. Crystals were grown in the presence of the synthetic LXR agonist T-17, the RXR agonist MPA and a GRIP-1-derived peptide. The structure was solved by molecular replacement using the RARα–RXRα heterodimer as search model. None of the two protomers in the search model have helix 12 (H12) in an agonistic conformation, thus the correctness of the solution could be confirmed by the appearance of continuous electron density for H12 of both LXRα and RXRβ at their expected agonistic positions. Ligands and GRIP-1 peptides could be modeled into unambiguous electron densities in the Fo – Fc map. Despite the limited resolution, 2.9 Å, the quality of the electron density map is good and all atoms are well defined, with the exception of some atoms of side chains of exposed residues. The model allows detailed investigation of protein–ligand interactions, although it should be noted that the estimated overall coordinate error is 0.37 Å. The final model comprises LXRα (219 residues), RXRβ (214 residues), two GRIP-1 peptides (nine and 10 residues, respectively), two ligands and 12 water molecules (Figure 1). The final Rcryst and Rfree values are 21.9% and 32.6%, respectively (Table I). The Ramachandran plot shows no residues in the disallowed regions, as defined by the program Procheck (Morris et al., 1992).
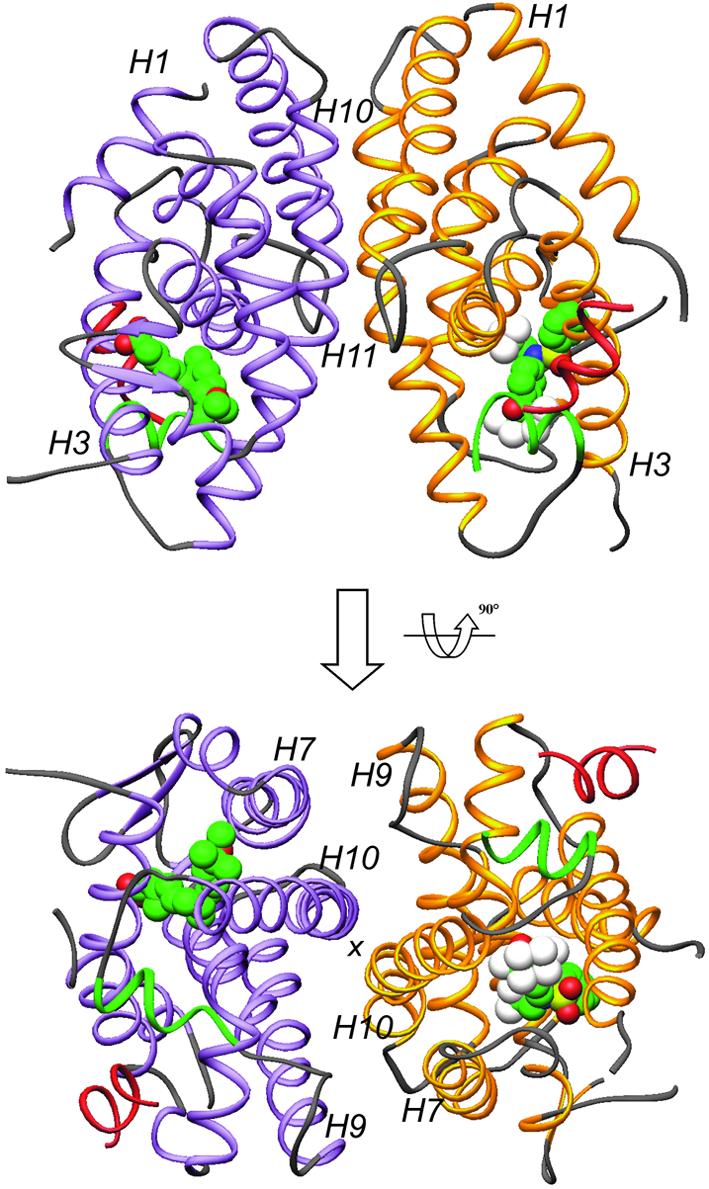
Fig. 1. Overall structure of the LXRα–RXRβ LBD-heterodimer presented in two views separated by 90°. The LXRα LBD is shown in yellow and the RXRβ LBD in purple, except for AF2 helices which are depicted in green. GRIP-1 peptides are colored red. T-17 and MPA are in space-filling representation, with carbon, oxygen, nitrogen, sulfur and fluoride atoms colored in green, red, blue, yellow and white, respectively. Selected secondary elements are annotated with numbers positioned at their N-terminal ends.
Table I. Data collection and refinement statistics.
| Wavelength (Å) | 1.11 |
| Resolution (last shell) | 20–2.9 (3.0–2.9) |
| Unique reflections | 12 550 |
| Redundancy | 6.4 |
| Completeness (%) | 99.9 (99.8) |
| Rsym (%) | 9.3 (42) |
| I/σ | 9.2 (1.9) |
| Rcryst (%) | 21.9 |
| Rfree (%) | 32.6 |
| R.m.s.d. bonds (Å) | 0.029 |
| R.m.s.d. angles (°) | 2.78 |
| Average B factor (Å2) | |
| RXRβ/LXRα/GRIP1 peptides | 26.5/36.7/54.3 |
| MPA/T-17/water | 25.3/48.8/22.8 |
| Ramachandran (%) | |
| Favored | 83.7 |
| Additional | 12.6 |
| Generous | 3.7 |
| Disallowed | 0 |
R.m.s.d., root mean square deviation from ideal geometry.
The overall LXRα LBD structure
The LXRα LBD adopts the canonical three-layered α-helical sandwich structure seen in all NR structures reported (Figure 2A). The LBD contains 10 α-helices with H2 missing, and helices 10 and 11 contiguous according to RXR nomenclature (Bourguet et al., 1995). LXR is structurally closely related to RAR and superimposes well on the retinoic acid RARγ complex, with a root-mean-square deviation (r.m.s.d.) of 1.3 Å over 199 Cα atoms [Protein Data Bank (PDB) accession code 2LBD]. Compared with RAR, H1 in LXR is extended by one turn at the C-terminal end, with the main chain continuing in the direction of H1, and does not fold back onto the two-stranded β-sheet. The major part of the ω-loop connecting H1 and H3 (residues 230–247) was not possible to model due to lack of interpretable electron density. This is a variable and loosely structured region for many NRs. More surprising is the lack of electron density for the loop after strand 2 in the β-sheet (residues 315–317). Compared with RAR, additional differences are found in the loops connecting H6 and H7 (L6–7), and H11 and H12 (L11–12). Instead, the L6–7 loop resembles that of the vitamin D receptor (PDB entry 1DB1), whereas the L11–12 loop is most closely related to that of PXR (PDB entry 1ILG). Among the NRs structurally determined thus far, these two receptors are also those most closely related to LXR in a phylogenetic context (Nuclear Receptor Nomenclature Committee, 1999). In the crystal, LXRα adopts the typical agonist-bound conformation, with H12 (AF2) forming a lid over the ligand-binding pocket, a conformation generally accepted as representing the transcriptionally active state of the receptor. The GRIP-1 coactivator peptide is bound in a helical conformation in the predominantly hydrophobic groove formed by H3, H4–5 and H12, a binding mode identical to that first seen for the SRC-1 peptide in the PPARγ–rosiglitazone complex (Nolte et al., 1998).

Fig. 2. Molecular basis of T-17 recognition by LXRα. T-17 bound in the core of the LBD with helix12 (AF2) capping the ligand-binding pocket and the GRIP-1-derived peptide bound at the coactivator-binding site. T-17 is shown in space-filling representation, and the coactivator peptide and AF2 helix are depicted in red and green, respectively. Secondary elements are annotated with numbers positioned at their N-terminal ends. Stereo diagram showing interactions between T-17 and LXRα. The difference electron density map used for modeling of T-17 is depicted in blue and contoured at 2.9 σ. Possible hydrogen bonds are indicated with broken lines (purple). Schematic representation of LXRα/T-17 interactions. Broken lines and arrows represent van der Waals contacts and hydrogen bonds, respectively. Topography of the ligand-binding pocket of LXR. T-17 is centrally positioned in the ligand-binding pocket. The ligand-accessible void is shown in blue.
Ligand recognition of LXRα
The ligand-binding pocket is stretched out between H12 and the β-sheet, and is shaped as a relatively straight cylinder with a length of ∼17 Å. Depending on the position of the unordered loop after the second strand in the β-sheet, the accessible volume is estimated to be in the range 700–800 Å3. As expected for an NR that binds oxysterols, the ligand-binding pocket is predominantly hydrophobic, with only a few possible hydrogen bond interactions. Oxysterols, with molecular volumes just below 400 Å3, can easily fit the pocket and are presumably not as tightly enclosed by the receptor as the steroid hormones are by their receptors. T-17, with a molecular volume of 304 Å3, is centrally positioned, with the hydroxyl head group coordinated by a strong hydrogen bond to the Nε of H421 (Figure 2). The Nε of W443, at 3.7 Å from the hydroxyl oxygen of the agonist, can possibly serve as a hydrogen donor. W443 together with L439 are the only residues of H12 that interact directly with the agonist. The hydrogen bond between T-17 and H421 presumably represents an important indirect interaction of T-17 with H12, as it will lower the pKa of the H421 imidazole side-chain, which in turn can interact electrostatically with π-electrons in the indole side-chain of W443. Taken together, these interactions are likely to stabilize the active conformation of H12 (AF2), and may serve as a molecular basis for the ligand-dependent activation of LXR.
The binding of T-17 is further stabilized by interactions with residues from H3, H5 and H7 (Figure 2C). The trifluoro ethyl group of T-17 completely fills a subpocket lined with residues from H5 and H7, and functions as a hydrogen acceptor for T302. The central sulfonamide moiety of T-17 does not contribute significantly to binding, except for a van der Waals interaction with A261. However, the sulfonamide is essential for producing the correct geometry between the three arms of the molecule. The third arm of the molecule is the phenyl group, which reaches towards the β-sheet side of the pocket but leaves a part of the pocket unoccupied. The unoccupied β-sheet side of the pocket is lined with the polar residues S264 (H3), E267 (H3) and R305 (H5). Another unoccupied void is present at the other end of the pocket close to the hydroxyl head group of T-17, i.e. towards H12. This void, shaped by the loop connecting H11 and H12 (L11–12), is entirely hydrophobic and lined by V425 (H11), L428 (H11), R429 (L11–12), P436 (L11–12), L439 (H12) and F254 (H3).
Docking of oxysterols demonstrates possible determinants of selectivity
In order to gain insights into the determinants for binding of endogenous ligands, a number of oxysterols were docked into the present structure. As a control, T-17 was docked, which resulted in a preferred binding mode essentially identical to that seen in the crystal structure. Top-scoring docking results for the most potent agonistic oxysterols [22(R)-HC, 24(S)-HC, 27-HC or 24,25-EC] suggested a common anchoring of the 22-, 24- or 27-hydroxyl/epoxy group to H421 and W443, essentially identical to that seen for the hydroxyl head group of T-17 (Figure 3). In this binding mode, the aliphatic chain (C23–27) of 22-HC fitted nicely in the hydrophobic void, described above as not occupied by T-17 (Figure 3). The isopropyl moiety of 24(S)-HC (C25–27) made similar contacts with the receptor as the two triflouromethyl groups of T-17, whereas 27-HC made few contacts in this part of the ligand-binding pocket. The docking exercise further suggested that neither 22(S)- nor 24(R)-hydroxy cholesterols can, due to steric hindrance, form the same interactions with H421 and W443 as their agonistic enan tiomers. However, the antagonistic 22(S)-HC fitted the ligand-binding pocket equally as well as the agonistic 22(R)-HC, in a conformation where the 22(S)-hydroxyl group interacted with Q424 instead of H421 (Figure 3). Thus, docking analysis indicates that interaction with H421 and W443 is critical for establishing a transcriptionally active complex.

Fig. 3. Docking of oxysterols into the ligand-binding pocket of LXRα. Docking of agonistic oxysterols suggests a common binding of their 22-, 24- or 27-hydroxyl group to H421 and W443 [exemplified by 22(R)-HC 24,25-EC and 27-HC], interactions that cannot be formed by the antagonistic oxysterol 22(S)-HC due to steric hindrance. By allowing conformational flexibility in the side chain of R305, all oxysterols docked with the 3-hydroxyl group hydrogen bonded to this residue. The original position of R305 is shown in gray. Possible hydrogen bonds are indicated with broken lines (purple).
By analogy with the binding mode of steroid hormones to their nuclear receptors, the oxysterols preferentially docked with the A ring of the steroid ring system located at the β-sheet side of the ligand-binding pocket. Due to the fixed anchoring of the 22-, 24- or 27-hydroxyl group in the pocket and the differences in intra-molecular distance between the two hydroxyls of the oxysterols, no consensus with respect to binding of the 3-hydroxyl group was initially achieved. The recognition of the 3-oxygen of steroid hormones by their receptors is conserved and involves hydrogen bonding to the guanidinium group of an arginine from H5 and, in the case of 3-keto steroids, hydrogen bonding to the side-chain amido group of a Gln from H3 (Williams and Sigler, 1998). In the estrogen receptor, the only steroid hormone receptor binding 3-hydroxyl steroids (estradiol and its homologs), the glutamine amido group is replaced by a carboxylate of a glutamate that accepts a hydrogen bond from the 3-hydroxyl group of the hormone (Brzozowski et al., 1997). In the LXRs, the conserved arginine is present (R305), whereas the Gln/Glu is substituted by serine (S264). By allowing conformational flexibility in the side chain of R305, the residue could hydrogen bond the 3-hydroxyl group of all oxysterols docked, suggesting a conserved function for this residue also in the LXRs. In the present structure, R305 forms a salt bridge with E267, which is positioned one turn further along H3 compared with S264 (Figure 2). Interestingly, 27-HC was docked with the 3-hydroxyl group anchored to E267 and R305, analogous to the 3-hydroxyl group recognition described for the estrogen receptor, suggesting a role for E267 in 3-hydroxyl group recognition by the LXRs.
Mutational analysis identifies key residues for transcriptional activation
In order to test our binding hypothesis for oxysterols we decided to generate LXR receptors mutated at one of the three positions, E267, H421 and W443, and evaluate the ability of ligands to transcriptionally activate these receptors in a cell-based reporter assay. A strong ligand-dependent activation of the wild-type receptor was seen for 24,25-EC, T-17, 22(R)-HC and 24(S)-HC (Figure 4; data not shown), correlating well with previously reported data (Janowski et al., 1999). In contrast, the H421A and W443A mutants did not respond to any of the agonists. For the oxysterols, the structurally more conserved H421N and W443F replacements also abolished transcriptional activation. T-17, on the other hand, could activate these two mutants although exhibiting both lower potency and efficacy than observed for the wild-type receptor (Figure 4). Apparently oxysterols are strictly dependent on H421 and W443 for transcriptional activation, whereas T-17 is not. This discrepancy could be due to the fact that T-17 is a more potent agonist and that the head-group of T-17 is more efficient in recruiting AF2 and therefore can accept also the suboptimal interactions offered by the introduced N421 and F443.

Fig. 4. Transcriptional activation of LXRα by T-17, 22(R)-HC and 24,25-EC. CaCo-2/TC7 cells were transiently transfected with a 4× GAL4-RE luciferase reporter and a GAL4-LXR wild-type or mutant LBD fusion construct, and subsequently treated with T-17, 22(R)-HC or 24,25-EC in optimized serial dilutions, as indicated in the figures. Data are shown as fold induction of agonist-induced luciferase activity divided by luciferase activity of vehicle.
Replacing E267 for alanine did not affect activation by T-17. Considering that this ligand does not occupy the far end of the β-sheet side of the ligand-binding pocket, acceptance of this mutation could be expected. Moreover, the unaffected activation by T-17 is a good indication that the mutation does not cause any major perturbation propagated to other parts of the receptor. The E267A mutation caused a 2- to 3-fold reduction in efficacy for the oxysterols examined. In addition, 24,25-EC showed a 2-fold reduction in potency. These results show that an altered pattern of hydrogen bonding at the β-sheet side of the ligand-binding pocket negatively affects transcriptional activation and prompts us to suggest that E267 is important for binding of the 3-hydroxyl group of oxysterols. For 22(R)-HC, not reaching E267, modeling suggests that an interaction could be mediated via a water molecule bridging the 3-hydroxyl group and the carboxylate of E267. Evaluation of the exact nature of the binding of the different oxysterols must await the determination of their LXRα complexes.
The E267A mutation sensitizes LXR towards arachidonic acid inhibition
Polyunsaturated fatty acids are natural antagonists of the LXRs that are able to inhibit oxysterol-dependent activation by competing for binding in the LBD of LXR (Ou et al., 2001). In transient transfection experiments, T-17-induced transcriptional activation was inhibited by micromolar concentrations of arachidonic acid, the fatty acid previously reported as the most potent LXR antagonist (Figure 5). The same experiment using the E267A mutant resulted in an even more pronounced inhibition of transcriptional activiation. Thus, the substitution of E267 into alanine results in a receptor more sensitive towards arachidonic acid inhibition. This effect could be explained by assuming that R305 is the coordinator of the carboxylate group of fatty acids. In the absence of E267, R305 could then interact more strongly with the fatty acid. The corresponding arginine is not only present in the steroid receptors, but is also conserved in the RXRs and the RARs, where it coordinates the carboxylic head-group of retinoic acid (Renaud et al., 1995). In this context it is interesting to note that RXRs bind the agonistic fatty acid docosa hexanoic acid in an analogous way (Egea et al., 2002).

Fig. 5. Inhibition of T-17-induced transcriptional activation of the LXRα wild-type and E267A mutant by arachidonic acid. CaCo-2/TC7 cells were transiently transfected with a 4× GAL4-RE luciferase reporter and a GAL4-LXR wild-type or mutant LBD fusion construct, and subsequently treated with T-17 and arachidonic acid in the concentrations indicated. Data are shown as fold induction where luciferase activity was divided by reporter activity of pCMXGal4 lacking an insert. For statistical analysis, the E267A mutant was compared with wild-type LXR (Student’s t-test). *p ≤ 0.05; **p ≤ 0.01.
The RXRβ LBD in agonistic conformation
The first structure of RXRβ was recently solved in complex with the RXR-specific agonist LG100268 (Love et al., 2002). In contrast to the typical activated state, with H12 (AF2) capping the ligand-binding pocket, H12 was found to adopt a novel position. In our complex with MPA this is not the case. It has been suggested that RXR agonists only weakly recruit AF2 and that coactivator binding is required to stabilize the helix in the active conformation. It is therefore likely that the difference in AF2 position in the two crystal structures is due to the absence or presence of coactivator peptide.
By analogy with previously described agonist–RXR complexes, MPA does not interact with AF2 directly, an observation that could be taken to account for the poor agonist-driven AF2 recruitment. MPA adopts an L-shaped conformation to fit the ligand-binding pocket by two consecutive 90° bond rotations, and thereby mimics the sharp cis-bend of 9-cis retinoic acid (9-cis RA) (Figure 6). The carboxylate is anchored by ion–ion interactions with R387 (H5) and hydrogen bonding to the amide nitrogen of A398. The methoxy iso-butyl moiety is smaller than the β-ionone ring of 9-cis RA, and therefore makes less extensive contacts with I339 (H3), V413 (H7), I416 (H7), C503 (H11) H506 (H11) and L507 (H11), and in addition lacks contacts with V420, F510 (4.5 Å cut-off). These differences in interactions with the receptor most likely account for the lower affinity observed for MPA in comparison with 9-cis RA (Harmon et al., 1995). The MPA–RXRβ complex shows a narrower ligand-binding pocket and although MPA is a smaller ligand (267 Å3 compared with 293 Å3 for 9-cis RA), both ligands occupy about half of the accessible volume (MPA, 52%; 9-cis RA, 53%). The reduced pocket size is not primarily due to α/β isoform differences, but is rather caused by subtle changes in the position of side chains lining the ligand-binding pocket. Similar changes are observed when comparing different RXRα complexes, e.g. the BMS-649 complex (Egea et al., 2002).
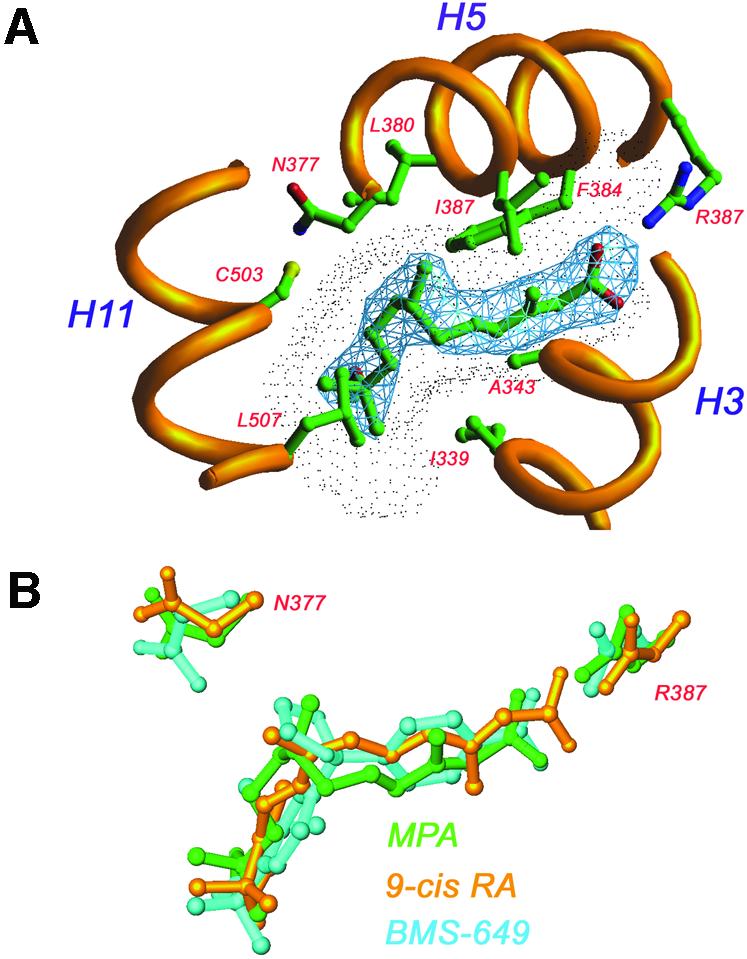
Fig. 6. Molecular basis of MPA recognition by RXRβ. In the RXR complex MPA adopts an l-shaped conformation resembling that of 9-cis RA. MPA interacts mainly with residues from helices 3, 5 and 11. The 2Fo – Fc electron density map (blue) is contoured at 1.2 σ. The ligand-accessible void is shown as a dotted surface. Superimposition of three RXR–ligand complexes: MPA (green), 9-cis RA (yellow) and BMS-649 (blue). Differences in pocket size are mainly due to different conformations of N377 and R387.
MPA is a major metabolite of the juvenile hormone mimetic pesticide methoprene, and was originally identified as a transcriptional activator of RXR in a screen of natural and synthetic isoprenoid compounds (Harmon et al., 1995). MPA is an RXR-selective agonist, i.e. unlike 9-cis RA it does not activate the RARs, although simple docking indicates that it could fit the straight ligand-binding pocket of RAR without strain. Again, absence of the β-ionone ring found in retinoic acid may be an important determinant for the RXR selectivity. In the RAR–9-cis RA complex, where 9-cis RA is forced into a straight conformation, the β-ionone ring is the part of the molecule interacting with AF2 (Klaholz et al., 1998).
The asymmetric LXRα–RXRβ interface
The overall topography of the LXRα–RXRβ interface resembles that of other RXR dimers, with residues from H9 and H10 forming its core. The interface is asymmetric by involving residues from H7 of RXRβ, but not from H7 of LXRα. Conversely, the loop in LXRα connecting H8 and H9 contributes to binding, while the same loop in RXRβ does not. This asymmetry is more pronounced than that observed for other RXR dimers and as a consequence the dimerization interface is ∼20% smaller (1115 Å2). The two protomers contribute equally to the hydrophobic core of the interface and a comparison with the RXR homo dimer shows a conserved topography. In contrast, the pattern of salt bridges and other polar interactions lining the hydrophobic core of the interface is significantly different in the LXR–RXR heterodimer (Figure 7). Key changes are found in H9 where H383, E387 and H390 in LXR correspond to E465, A469 and E472 in RXR, resulting in novel salt bridges. These differences affect the salt bridge pattern at the L8–9 loop/H7 part of the interface as well. Taken together, it is likely that the observed differences in dimer interactions influence the relative stability of the LXR–RXR heterodimer compared with other RXR dimers. It has long been known that LXR requires RXR to form a functional dimer (Willy et al., 1995), but it should also be noted that LXRα as a monomer can function as a cAMP-responsive transcriptional regulator of a number of genes, including rennin and c-myc (Tamura et al., 2000; Anderson et al., 2003). By sedimentation velocity analysis we could show that the isolated LXRα LBD is exclusively monomeric, whereas an equimolar mixture of LXRα and RXRβ LBDs is exclusively dimeric (data not shown). Superimposition of LXRα onto RXRβ in the present structure to constitute a hypothetical LXR homodimer results in a non-optimal interface along the 2-fold dimer axis. The bulky L402 and P405, corresponding to glycine and alanine in RXR, cause steric hindrance at the N-terminal part of H10, while the cluster of basic residues around the L9–10 loop (H383, H390, R401, R406) results in unfavorable electrostatic interactions. We suggest that these are determinants excluding the formation of strong homodimers of LXR.

Fig. 7. Comparison of the dimer interaction surfaces of RXRβ in the RXRβ homodimer (left; PDB accession code 1H9U) and in the LXRα–RXRβ heterodimer (right). The central hydrophobic core of the interface is conserved, whereas the polar interactions differ significantly between the two dimers. The area of the dimer interaction surface in the homodimer is 1443 Å2, while in the heterodimer it is 1115 Å2. Green, hydrophobic surface; blue, hydrogen bond donating groups; red, hydrogen bond accepting groups.
A novel ligand-induced conformational change?
As mentioned previously, the conformation of the loop following strand 2 in the β-sheet of LXRα could not be determined in the present structure, and the lack of interpretable electron density for this structural element indicates that it is highly flexible. Further, modeling suggests that in the absence of ligand it could form a β-turn identical to that seen for RARγ. This conformation would bury the hydrophobic side chains of F315 and L316 and reduce the accessible volume of the ligand-binding pocket, which we predict would stabilize the apo-form of the receptor. The hypothetical closed conformation causes a clash between the phenyl ring of T-17 and the side chain of F315 and, according to modeling, the closed conformation would also cause steric hindrance to oxysterol binding. We therefore suggest that ligand binding could involve a displacement of the loop following strand 2 in the β-sheet, from a closed state to a more exposed and flexible one where the loss of entropy caused by exposure of the hydrophobic residues F315 and L316 could be compensated for by increased mobility in this structural segment. Evaluation of such a binding mechanism will have to await structure determination of the apo-LXR.
LXR isoform variability
The conservation of amino acid sequence is relatively high for the LXR LBDs, with a sequence identity of 77% for the two human isoforms. The conservation is also reflected in ligand-binding characteristics where the α- and β-isoforms show similar binding affinities for a range of substances (Janowski et al., 1999). As for the T-17 complex, none of the residues within van der Waals distance of the ligand differ between LXRα and LXRβ. In this respect LXR may be as challenging in terms of selectivity as RXR, for which isoform-selective agonists have proven difficult to design.
In conclusion, the present LXRα–RXRβ structure provides important insight to the structural basis for its biological activity, including aspects on heterodimerization, selectivity and transcriptional activation. The LXR–RXR heterodimer is a key transcriptional regulator of genes related to lipid homeostasis, and the design of selective modulators of this functional unit is anticipated to yield new means of therapeutic intervention for a number of lipid-related disorders.
Materials and methods
Isolation of the LXRα–RXRβ LBD heterodimer
The ligand-binding domains of LXRα [amino acids (aa) 207–447] and RXRβ (aa 295–533) were subcloned in-frame with the N-terminal His tag sequence of pET15 and pET28 expression vectors (Novagen), respectively. The proteins were produced separately or by coexpression in cultivations of E.coli BL21 (DE3) strain at 20°C using 0.2 mM isopropyl-β-d-thiogalactopyranoside according to suppliers recommendations. Cells were disrupted by sonication in 50 mM Tris–HCl, 2 mM TCEP, 5 mM imidazole, 300 mM NaCl, 5% (w/v) glycerol, pH 8.0. After 48 000 g centrifugation, soluble protein was separated by Ni2+-Sepharose chromatography with a stepwise imidazole elution procedure, where the LXR–RXR heterodimer eluted at 300 mM imidazole. In order to remove the N-terminal His tags, the buffer was changed by G-25 Sepharose chromatography to 50 mM Tris–HCl, 2 mM tris carboxyethyl phosphine, 2 mM CaCl2, 300 mM NaCl, 5% (w/v) glycerol, pH 8.0. The protein was subsequently subjected to Thrombin digestion (10 U/mg at 24°C for 16 h). Further purification involved a second Ni2+-Sepharose chromatography step and a subsequent molecular sieving step (Superdex75) in the buffer used for thrombin digestion. Isolated LXR–RXR heterodimer was dialyzed against 20 mM Tris–HCl, 2 mM TCEP, pH 8.0, and concentrated to 9.5 mg/ml before use in crystallization trials.
Crystallization of the LXRα–RXRβ LBD heterodimer
The LXRα–RXRβ LBD heterodimer was crystallized in complex with MPA, T-17 and a synthetic peptide (KEKHKILHRLLQDS) with a sequence derived from the GRIP-1 coactivator. Crystals were grown by the hanging drop diffusion method at 32°C and appeared in 3–5 days. The well solution contained 0.1 M MES buffer pH 6.5, 16% (w/v) polyethylene glycol 12 000. Typically, 2 µl of the precipitant was mixed with 2 µl of a solution containing 9.5 mg/ml LXRα–RXRβ, 1 mM GRIP-1 coactivator peptide and 0.5 mM of each ligand in the drop. Microseeding techniques were used in order to improve crystal size. The crystals belong to the orthorhombic space group P212121, having unit cell lengths of 67.6, 89.0 and 90.8 Å, containing one dimer per asymmetric unit and a solvent content of 47%.
Data collection and structure determination
Data were collected at 100 K using a MAR CCD at beam line BL711, MAX laboratory, Lund, Sweden. The crystals were soaked in crystallization buffer supplemented with 20% (v/v) glycerol before flash-freezing in a nitrogen stream. A complete data set was collected on one crystal, which diffracted to 2.9 Å. The diffraction data were integrated and scaled using DENZO and SCALEPACK programs (Otwinowski and Minor, 1997). The structure was solved by molecular replacement using the program MOLREP (Vagin and Teplyakov, 1997). The HsRARα–MmuRXRα heterodimer served as search model (PDB accession code 1DKF). The correct solution in MOLREP had the highest score, with a correlation coefficient of 0.372 and an R-factor of 52%. After rigid body refinement in Refmac (CCP4), the R-factor was 48%. Models were built using O (Jones et al., 1991) and refined using Refmac (Murshudov et al., 1997) and Buster (Bricogne, 1993). The atomic coordinates have been deposited in the PDB (accession code 1UHL).
Docking analysis
Oxysterols were docked to LXRα using the ICM software (version 2.8; Molsoft Inc.) (Abagyan et al., 1994). The automatic docking was implemented as a Monte Carlo minimization of the ligand in a set of protein-derived grid potentials plus the internal energy of the ligand. The non-ring rotatable bonds as well as the location and orientation of the ligand were sampled in a molecular mechanics force field with the interaction energy between the receptor and ligand calculated using five different pre-calculated grids. This procedure results in a stack of possible ligand-binding conformations, sorted according to energy. In this study we analyzed the first 20 conformations for each ligand to find frequent binding modes with high consistency between the different oxysterols.
DNA constructs for GAL4-LBD fusion analysis
The LBD of LXRα (aa 163–447) was generated by PCR amplification using PFU polymerase (Stratagene) and gene-specific primers flanked with restriction enzymes KpnI and BamHI, respectively (LXRα LBD 5′, 5′-GGTACCATGCGGGAGGAGTGTGTCCT; LXRα LBD 3′, 5′-GGA TCCTCATTCGTGCACATCCCAGATCTC). The LBD was subcloned into the pCMXGal4 vector, containing the GAL4 DBD (Perlmann and Jansson, 1995). Point mutations at three different amino acid positions, E267, H421 and W443, were introduced. The following mutants were created: E267A, H421N, H421A, W443F and W443A. The mutations were introduced by PCR mutagenesis in a two-step reaction using the following primers (mutations are shown in lower case letters): E267A (5′), 5′-CTCTGTGCAGGcGATAGTTGACT; E267A (3′), 5′-AGTC AATATCgCCTGCACAGAG; H421N (5′), 5′-GAGCAGCGTCaAC TCAGAGCAA; H421N (3′), 5′-TTGCTCTGAGTtGACGCTGCTC; H421A (5′), 5′-GAGCAGCGTCgcCTCAGAGCAA; H421A (3′), 5′-TTGCTCTGAGgcGACGCTGCTC; W443F (3′), 5′-GGATCCTCATT CGTGCACATCaaAGATCTC; W443A (3′), 5′-GGATCCTCATTCGT GCACATCCgcGATCTC.
Cell-based reporter assay
Transient transfection experiments for the functional analysis of LXRα wild-type and mutant receptors were performed in CaCo-2 subclone TC7 (CaCo-2/TC7, a colon adenocarcinoma cell line) cells in 96-well plates. For batch transfections, cells were seeded at a concentration of 6.3 × 106 cells per 225 cm2 and incubated for 24 h at 37°C in medium containing Dulbecco’s modified Eagle’s medium (DMEM; SVA, Sweden), 10% charcoal/dextran-treated fetal bovine serum (Hyclone), non-essential amino acids (NEA) (10 ml/l) and glutamine (20 ml/l) (Life Technologies). The medium was replaced with transfection medium containing Optimem (Invitrogen) and 10% charcoal/dextran-treated fetal bovine serum. The cells were cotransfected with 5 µg of receptor plasmid and 50 µg of reporter plasmid using FuGENE-6 (Roche) according to the manufacturer’s instructions. After 20–24 h, cells were seeded at a concentration of 0.25 × 105 cells/well in induction medium (Optimem, 5% charcoal/dextran-treated fetal bovine serum), incubated for ∼5 h and subsequently treated with T-17 (Alexis), 22(R)- (Sigma), 24(S)-HC or 24,25-EC (BIOMOL Research Laboratories) in optimized serial dilutions, as indicated in the figures. Following 24 h incubation, cells were harvested in lysis buffer (0.1 M Tris–HCl, 2 mM EDTA, 0.25% Triton X-100) and the cell lysates were analyzed for luciferase activity with Luciferase assay kit (BioThema AB, Sweden). All experiments were performed at least three times in triplicate. Uniform expression levels of wild-type LXRα and mutant proteins were confirmed by western blot analysis, using an anti-Gal4 DBD antibody (sc-577; Santa Cruz Biotechnology) and a horse radish peroxidase conjugated goat-anti-rabbit secondary antibody (12-348; Upstate Biotechnology).
Transient transfection experiments determining the effect of polyunsaturated fatty acids on wild type LXRα and the E267A mutant were essentially performed as described previously (Ostberg et al., 2002). CaCo2/TC7 cells were seeded in 6-well plates (2 × 105 cells/well), transfected with 0.2 µg of receptor and 2 µg of reporter, treated with T-17 and arachidonic acid in the indicated concentrations, and harvested following 24 h incubation. Culture media were as described above. The arachidonic acid was loaded on albumin as described previously (Ou et al., 2001). The experiment was repeated on three separate occasions in triplicate.
Analytical ultracentrifugation
The size distribution of LXRα preparations was obtained from sedimentation velocity experiments using a Beckman Optima-XLI with interference optical detection system. Epon double-sector centrepieces were filled with 400 µl of 0.5 mg/ml protein in 50 mM Tris–HCl, 2 mM TCEP, 300 mM NaCl, 5% (w/v) glycerol, pH 8.0. Experiments were performed at a rotor speed of 50 000 r.p.m. at 20°C. The data were analyzed applying the Lamm equation on sedimentation using the program SEDFIT (Schuck, 2000).
Acknowledgments
Acknowledgements
We thank Jonas Uppenberg for critical review of the manuscript, Yngve Cerenius for assistance using the MAX-lab beamline I711, Elisée Wiita and Mona Sydow-Bäckman for technical assistance with cell-based assays, and Sven-Åke Franzén, Andrea Varadi and Marianne Israelsson for DNA sequence analysis. The TC7 subclone of CaCo-2 cells was kindly provided by Dr Monique Rousset, Institut National de la Santé et de la Recherche Médicale U178, Villejuif, France. Tove Östberg was supported by a predoctoral fellowship from Biovitrum to the Karolinska Institute.
References
- Abagyan R.A., Totrov,M.M. and Kuznetsov,D.N. (1994) ICM—a new method for protein modeling and design. Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem., 15, 488–506. [Google Scholar]
- Anderson L.M., Choe,S.E., Yukhananov,R.Y., Hopfner,R.L., Church,G.M., Pratt,R.E. and Dzau,V.J. (2003) Identification of a novel set of genes regulated by a unique liver X receptor-α-mediated transcription mechanism. J. Biol. Chem., 278, 15252–15260. [DOI] [PubMed] [Google Scholar]
- Bourguet W., Ruff,M., Chambon,P., Gronemeyer,H. and Moras,D. (1995) Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-α. Nature, 375, 377–382. [DOI] [PubMed] [Google Scholar]
- Bricogne G. (1993) Direct phase determination by entropy maximization and likelihood ranking: status report and perspectives. Acta Crystallogr. D, 49, 37–60. [DOI] [PubMed] [Google Scholar]
- Brzozowski A.M. et al. (1997) Molecular basis of agonism and antagonism in the oestrogen receptor. Nature, 389, 753–758. [DOI] [PubMed] [Google Scholar]
- Costet P., Luo,Y., Wang,N. and Tall,A.R. (2000) Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. J. Biol. Chem., 275, 28240–28245. [DOI] [PubMed] [Google Scholar]
- Egea P.F., Mitschler,A. and Moras,D. (2002) Molecular recognition of agonist ligands by RXRs. Mol. Endocrinol., 16, 987–997. [DOI] [PubMed] [Google Scholar]
- Harmon M., Boehm,M., Heyman,R. and Mangelsdorf,D. (1995) Activation of mammalian retonid X receptors by the insect growth regulator methoprene. Proc. Natl Acad. Sci. USA, 92, 6157–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowski B.A., Willy,P.J., Devi,T.R., Falck,J.R. and Mangelsdorf,D.J. (1996) An oxysterol signalling pathway mediated by the nuclear receptor LXRα. Nature, 383, 728–731. [DOI] [PubMed] [Google Scholar]
- Janowski B.A., Grogan,M.J., Jones,S.A., Wisely,G.B., Kliewer,S.A., Corey,E.J. and Mangelsdorf,D.J. (1999) Structural requirements of ligands for the oxysterol liver X receptors LXRα and LXRβ. Proc. Natl Acad. Sci. USA, 96, 266–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Joseph S.B. et al. (2002) Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc. Natl Acad. Sci. USA, 99, 7604–7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph S.B., Castrillo,A., Laffitte,B.A., Mangelsdorf,D.J. and Tontonoz,P. (2003) Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med., 9, 213–219. [DOI] [PubMed] [Google Scholar]
- Klaholz B.P., Renaud,J.P., Mitschler,A., Zusi,C., Chambon,P., Gronemeyer,H. and Moras,D. (1998) Conformational adaptation of agonists to the human nuclear receptor RARγ. Nat. Struct. Biol., 5, 199–202. [DOI] [PubMed] [Google Scholar]
- Laffitte B.A., Repa,J.J., Joseph,S.B., Wilpiltz,D.C., Kast,H.R., Mangelsdorf,D.J. and Tontonoz,P. (2001) LXRs control lipid-inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc. Natl Acad. Sci. USA, 98, 507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann J.M. et al. (1997) Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J. Biol. Chem., 272, 3137–3140. [DOI] [PubMed] [Google Scholar]
- Lonard D.M. and Smith,C.L. (2002) Molecular perspectives on selective estrogen receptor modulators (SERMs): progress in understanding their tissue-specific agonist and antagonist actions. Steroids, 67, 15–24. [DOI] [PubMed] [Google Scholar]
- Love J.D., Gooch,J.T., Benko,S., Li,C., Nagy,L., Chatterjee,V.K.K., Evans,R.M. and Schwabe,J.W.R. (2002) The structural basis for the specificity of retinoid-X receptor-selective agonists: new insights into the role of helix H12. J. Biol. Chem., 277, 11385–11391. [DOI] [PubMed] [Google Scholar]
- Lu T.T., Repa,J.J. and Mangelsdorf,D.J. (2001) Orphan nuclear receptors as eLiXiRs and FiXeRs of sterol metabolism. J. Biol. Chem., 276, 37735–37738. [DOI] [PubMed] [Google Scholar]
- Morris A.L., MacArthur,M.W., Hutchinson,E.G. and Thornton,J.M. (1992) Stereochemical quality of protein structure coordinates. Proteins, 12, 345–364. [DOI] [PubMed] [Google Scholar]
- Murshudov G., Vagin,A.A. and Dodson,E.J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Nolte R.T. et al. (1998) Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-γ. Nature, 395, 137–143. [DOI] [PubMed] [Google Scholar]
- NRNC (1999) A unified nomenclature system for the nuclear receptor superfamily. Cell, 97, 161–163. [DOI] [PubMed] [Google Scholar]
- Ostberg T., Bertilsson,G., Jendeberg,L., Berkenstam,A. and Uppenberg,J. (2002) Identification of residues in the PXR ligand binding domain critical for species specific and constitutive activation. Eur. J. Biochem., 269, 4896–4904. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Macromol. Crystallogr., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Ou J.F., Tu,H., Shan,B., Luk,A., DeBose-Boyd,R.A., Bashmakov,Y., Goldstein,J.L. and Brown,M.S. (2001) Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. Proc. Natl Acad. Sci. USA., 98, 6027–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peet D.J., Turley,S.D., Ma,W.Z., Janowski,B.A., Lobaccaro,J.M.A., Hammer,R.E. and Mangelsdorf,D.J. (1998) Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXRα. Cell, 93, 693–704. [DOI] [PubMed] [Google Scholar]
- Perlmann T. and Jansson,L. (1995) A novel pathway for vitamin A signaling mediated by RXR heterodimerization with NGFI-B and NURR1. Genes Dev., 9, 769–782. [DOI] [PubMed] [Google Scholar]
- Renaud J.P., Rochel,N., Ruff,M., Vivat,V., Chambon,P., Gronemeyer,H. and Moras,D. (1995) Crystal structure of the RAR-γ ligand-binding domain bound to all-trans retinoic acid. Nature, 378, 681–689. [DOI] [PubMed] [Google Scholar]
- Repa J.J. et al. (2000) Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRα and LXRβ. Genes Dev., 14, 2819–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repa J.J., Berge,K.E., Pomajzl,C., Richardson,J.A., Hobbs,H. and Mangelsdorf,D.J. (2002) Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors α and β. J. Biol. Chem., 277, 18793–18800. [DOI] [PubMed] [Google Scholar]
- Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J., 78, 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer T.A. et al. (2001) Pharmacophore analysis of the nuclear oxysterol receptor LXRα. J. Med. Chem., 44, 886–897. [DOI] [PubMed] [Google Scholar]
- Tamura K. et al. (2000) LXRα functions as a cAMP-responsive transcriptional regulator of gene expression. Proc. Natl Acad. Sci. USA, 97, 8513–8518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A. and Teplyakov,A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr., 30, 1022–1025. [Google Scholar]
- Williams S.P. and Sigler,P.B. (1998) Atomic structure of progesterone complexed with its receptor. Nature, 393, 392–396. [DOI] [PubMed] [Google Scholar]
- Willy P.J., Umesono,K., Ong,E.S., Evans,R.M., Heyman,R.A. and Mangelsdorf,D.J. (1995) LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev., 9, 1033–1045. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Repa,J.J., Gauthier,K. and Mangelsdorf,D.J. (2001) Regulation of lipoprotein lipase by the oxysterol receptors, LXRα and LXRβ. J. Biol. Chem., 276, 43018–43024. [DOI] [PubMed] [Google Scholar]