

Abstract
The genetic inactivation of the atypical protein kinase C (aPKC) inhibitor, Par-4, gives rise to increased NF-κB activation and decreased stimulation of JNK in embryo fibroblasts. Here we have characterized the immunological phenotype of the Par-4–/– mice and found that the loss of this gene leads to an increased proliferative response of peripheral T cells when challenged through the TCR. This is accompanied by a higher increase in cell cycle entry and inhibition of apoptosis, with enhanced IL-2 secretion but normal CD25 synthesis. Interestingly, the TCR-triggered activation of NF-κB was augmented and that of JNK was severely abrogated. Consistent with previous data from knock outs of different JNKs, NFATc1 activation and IL-4 secretion were augmented in the Par-4-deficient CD4+ T cells, suggesting that the loss of Par-4 drives T-cell differentiation towards a Th2 response. This is compelling evidence that Par-4 is a novel modulator of the immune response through its ability to impact aPKC activity, which translates into lower JNK signaling.
Keywords: atypical PKCs/B lymphocytes/NF-κB/Par-4/T lymphocytes
Introduction
Adequate regulation of cell growth and apoptosis is central to the control of the immune response. Par-4 is a novel cell death modulator that interacts and inhibits the atypical protein kinase C (aPKC) isoforms (λ/ιPKC and ζPKC), which leads to reduced cellular NF-κB activation and increased apoptosis (Diaz-Meco et al., 1996, 1999). Consistently, embryo fibroblasts (EFs) from Par-4- deficient mice display enhanced aPKC activity as well as NF-κB transcriptional activation (Garcia-Cao et al., 2003). Importantly, this produces an inhibition of c-Jun N-terminal kinase (JNK) and p38 stimulation in response to TNFα, which correlates with an increased resistance to TNFα-induced apoptosis (Garcia-Cao et al., 2003). Interestingly, EFs from ζPKC–/– mice show an opposite phenotype, consisting of a more pronounced and prolonged activation of JNK, inhibition of NF-κB transcriptional activity and increased cell death (Garcia-Cao et al., 2003), similar to what has been reported for IKKβ–/– and RelA–/– cells (De Smaele et al., 2001; Tang et al., 2001). These results are consistent with a model whereby JNK and p38 are downstream in vivo targets of Par-4/ζPKC actions in the control of cell apoptosis/survival.
The phenotype of ζPKC knockout (KO) mice has been recently characterized in our laboratory (Leitges et al., 2001). Although the T-cell compartment of these mice was normal, they display a significant delay in the development of secondary lymphoid organs, which was particularly evident in young (2-week-old) animals; this is characterized by alterations in the spleen, which have defects in the marginal zone and smaller B-cell follicles while the overall structure of the organ is preserved (Leitges et al., 2001). Alterations were also detected in the peripheral and mesenteric lymph nodes as well as in Peyer’s patches, which displayed an impaired segregation between B- and T-cell zones and a decrease in the number of follicular dendritic cells (Leitges et al., 2001). These alterations were much less apparent in adult ζPKC–/– mice and, in fact, the B-cell subpopulations of these mutant mice were indistinguishable from those of wild-type (WT) animals (Martin et al., 2002). However, the loss of ζPKC impairs signaling through the B-cell receptor (BCR), resulting in decreased proliferation and increased apoptosis of B cells and a defect in the ability of these mice to mount an optimal T-dependent immune response (Martin et al., 2002). In addition, the expression of a series of NF-κB-dependent genes was severely inhibited in the ζPKC–/– B cells (Martin et al., 2002), consistent with the role that this kinase plays in the control of NF-κB transcriptional activity (Leitges et al., 2001). However, the lack of ζPKC does not affect T-cell proliferation in response to anti-CD3 challenge in either the absence or presence of CD28 (Martin et al., 2002). Therefore, ζPKC appears to play a critical and non-redundant role in B-cell function that cannot be fully compensated for by the presence of the closely related λ/ιPKC.
Since the loss of ζPKC produces alterations in the immune system in vivo, together with the fact that Par-4 antagonizes aPKC signaling and function in EFs, we have investigated in this study potential alterations caused by the loss of Par-4 in the immune system in vivo and in vitro. These results are particularly relevant because Par-4 inhibits both aPKCs, and therefore they will be useful to understand the potential role played by not only Par-4 but also by λ/ιPKC in vivo.
Results
Splenomegaly but normal development of T and B lymphocytes in Par-4–/– mice
The generation of Par-4-deficient mice has been described recently (Garcia-Cao et al., 2003). Homozygous Par-4-deficient mice were born at the expected Mendelian ratio. The only gross apparent pathological abnormality observed in these mice was a moderate splenomegaly (Figure 1A) due to a general increase of spleen cells (69 ± 15 × 106 in WT versus 94 ± 14 × 106 in Par-4–/–). However, the percentage of B cells and of CD4+ and CD8+ T cells was not significantly different in the spleens of Par-4-deficient mice as compared with WT controls. Thus, flow cytometric analysis of cells from spleen, thymus and lymph nodes of Par-4–/– mice show that the loss of this gene does not produce alterations in T-cell or B-cell populations in mice of 2 (not shown) or 4 (Figure 1B–D) weeks of age. The frequency of pro-B (B220intCD43hi), pre-B (B220intCD43int) and mature B (B220+CD43–) cells in bone marrow of Par-4–/– mice was indistinguishable from that of WT mice (Figure 1B). Also, three-color flow cytometric analysis of splenocytes from Par-4-deficient mice demonstrates that the percentages of mature (CD21intIgMlowCD23+), marginal zone (CD21hiIgMhi CD23–), T1 (CD21lowIgMhiCD23–) and T2 (CD21hi IgMhiCD23+) B-cell subpopulations were not significantly different from those of the WT mice (Figure 1C). Analysis of thymus demonstrates that Par-4–/– mice have normal numbers and proportions of CD4–CD8–, CD4+CD8–, CD4–CD8+ and CD4+CD8+ thymocytes (Figure 1D). Spleen (Figure 1D), peripheral (Figure 1D) and mesenteric (not shown) lymph nodes from Par-4–/– show normal percentages of mature CD4+ and CD8+ T cells. Therefore, similar to the ζPKC–/– mice (Leitges et al., 2001; Martin et al., 2002), Par-4 deficiency does not affect thymocyte differentiation. However, in contrast to the ζPKC-deficient mice, which show a delay in the development of secondary lymphoid organs (Leitges et al., 2001), this parameter is intact in the Par-4 KO mice.

Fig. 1. Immunological phenotypic analysis of Par-4-deficient mice. (A) Representative spleens (of >20 mice) from WT and Par-4–/– mice. (B) Single cell suspensions of bone marrow cells from WT and Par-4–/– mice were stained with anti-CD43 versus anti-B220. R2, pro-B; R3, pro-B and pre-B; R4, mature B cells. Values in parentheses correspond to the percentage of each subpopulation. (C) Three-color flow cytometric analysis of splenocytes from WT and Par-4-deficient mice were carried out by staining with antibodies to IgM, CD21 and CD23. Cells were separated into CD23-positive and -negative; MZ (CD21hiIgMhiCD23–), marginal zone B cells; mature B cells are characterized by CD21intIgMlowCD23+; T1 (CD21lowIgMhiCD23–), transitional 1 immature B cells; T2 (CD21hiIgMhiCD23+), transitional 2 immature B cells. Values in parentheses correspond to the relative percentage of each subpopulation. (D) Single cell suspensions of thymuses, spleen and peripheral lymph nodes from WT and Par-4-deficient mice were stained with anti-CD4 versus anti-CD8. Results in (B–D) are representative of four experiments with similar results.
Enhanced proliferation of Par-4-deficient B and T cells
As the loss of ζPKC inhibits B-cell proliferation in response to the triggering of the BCR, we next determined the impact that Par-4 deficiency has on this parameter. Results of these experiments demonstrate that the loss of Par-4 significantly increases the proliferative response of B cells to activation through the BCR but not in response to activation by LPS or CD40 (Supplementary figure 1, available at The EMBO Journal Online). This would be consistent with the role that ζPKC plays in BCR signaling, but not in LPS- or anti-CD40-activated B cells (Martin et al., 2002). Of note, the activation of ERK in response to the BCR challenge, which is inhibited in ζPKC-deficient B cells (Martin et al., 2002), is severely enhanced in the Par-4–/– B cells (Supplementary figure 2). This would be consistent with the notion that Par-4 is a negative regulator of ζPKC in B cells, which is the major, if not the only, aPKC activated following BCR stimulation (Martin et al., 2002).

Fig. 2. T-cell proliferation. T cells from either WT (empty bars) or Par-4- deficient (black bars) mice were incubated for 48 h (for T cells) with different concentrations of anti-CD3 antibody with or without anti-CD28 antibody. Afterwards, the amount of [3H]thymidine incorporated was determined (A). In the experiment in (B), T cells were stimulated as above and the percentage of cells undergoing proliferation (S + G2/M) was determined by flow cytometry. (C) Mixed lymphocyte reactions were performed with T cells from WT (empty bars) and Par-4 KO (black bars) mice and irradiated splenic cells from allogeneic WT mice. (D) In another set of experiments, proliferation of non-activated (empty bars) or activated (10 µg/ml anti-CD3; black bars) purified CD4+ (upper panel) or CD8+ (lower panel) T cells from WT or Par-4–/– mice was determined as described above for total T cells. The results are mean ± SD of three independent experiments with incubations carried out in duplicate.
Unexpectedly, when proliferation was measured in peripheral T cells after stimulation with plate-bound anti-CD3 monoclonal antibody in the absence or in the presence of anti-CD28, it became apparent that the Par-4–/– T cells displayed an enhanced proliferation in response to anti-CD3, determined as the amount of thymidine incorporation (Figure 2A). In the presence of CD28 co-stimulation, the differences between the WT and the Par-4–/– T cells were still significant, although less apparent (Figure 2A). Consistent with these observations, flow cytometric analyses revealed that the loss of Par-4 enhances the cell cycle entry of T cells activated through the T-cell receptor (TCR) (Figure 2B). Of note, proliferation of Par-4–/– T cells was also significantly enhanced compared with WT controls in mixed lymphocyte reactions (Figure 2C). Collectively, these results indicate that the loss of Par-4 enhances the ability of T cells to proliferate. Also, B-cell proliferation is positively impacted by the loss of Par-4. This suggests that Par-4/ζPKC are critical players in B cells, whereas Par-4 modulates T-cell proliferation through a mechanism independent of ζPKC, which is not implicated in T-cell function (Martin et al., 2002), but possibly through the other aPKC isoform, λ/ιPKC. We next determined how the lack of Par-4 affected cell proliferation parameters of purified CD4+ and CD8+ T cells. The results shown in Figure 2D demonstrate that proliferation in response to anti-CD3 stimulation was enhanced in both purified CD4+ (upper panel) and CD8+ (lower panel) T-cell populations from Par-4-deficient mice compared with controls.
Interleukin (IL)-2 production is a hallmark of T-cell activation. Therefore, we next determined the effect of the loss of Par-4 in IL-2 synthesis. Similar to the proliferative response, the production of IL-2 is reproducibly enhanced in Par-4-deficient T cells activated with anti-CD3 (Figure 3A). Again, in the presence of CD28 co-stimulation, the differences between the WT and the Par-4–/– T-cells were still significant, although less apparent (Figure 3A). The expression of CD25 (IL-2Rα) in response to anti-CD3 stimulation (Figure 3B) or to anti-CD3 plus anti-CD28 (not shown) in Par-4-deficient cells was indistinguishable from that in WT cells. Interestingly, no differences in proliferation were detected between WT and Par-4-deficient T cells when stimulated with different concentrations of murine recombinant IL-2 (Figure 3C). Together, these results demonstrate that Par-4 plays a critical role in the negative regulation of the TCR signaling at the level of IL-2 production, which accounts for the higher proliferative activity of the Par-4–/– T cells. In keeping with the data for total T cells, the secretion of IL-2 was also enhanced in both CD4+ and CD8+ T-cell subsets from the mutant mice (Figure 3D), whereas expression of CD25 (IL-2Rα) in CD4+ and CD8+ Par-4-deficient T cells was indistinguishable from those of WT (Figure 3E). In the presence of CD28 co-stimulation, the differences between the WT and the Par-4–/– T-cell subsets were still significant, although much less important (not shown).

Fig. 3. IL-2 secretion and proliferation in Par-4-deficient T cells. (A) IL-2 production in supernatants of T cells from WT and Par-4–/– mice activated (black bars) or not (empty bars) with 10 µg/ml of anti-CD3. (B) Surface expression of CD25 was analyzed by flow cytometry 24 h after stimulation of WT or Par-4 KO T cells with different concentrations of anti-CD3. (C) T cells from WT (empty bars) or Par-4 KO (black bars) mice were stimulated with 0.1 µg/ml of anti-CD3 antibody in the absence or in the presence of increasing concentrations of recombinant murine IL-2 (rmIL2) for 72 h. (D) In another set of experiments, IL-2 production by non-activated (empty bars) or activated (10 µg/ml anti-CD3; black bars) purified CD4+ (upper panel) or CD8+ (lower panel) T cells from WT or Par-4–/– mice was determined as described above for total T cells. (E) CD25 surface expression of non-activated or activated (10 µg/ml anti-CD3) purified CD4+ (upper panel) or CD8+ (lower panel) T cells from WT or Par-4–/– mice was determined as described above for total T cells. The experiments in (B) and (E) are representative of another two with similar results and those in (A), (C) and (D) are the mean ± SD of three independent experiments with incubations carried out in duplicate.
Because Par-4 is a pro-apoptotic protein the genetic inactivation of which reduces cell death in EFs, we sought to determine whether the Par-4 deficiency affects T-cell apoptosis. To address this possibility, annexin V staining was measured in WT and Par-4–/– T cells that had been stimulated with anti-CD3 (10 µg/ml) for 24 h. Anti-CD3-induced annexin V staining is significantly reduced in the Par-4-deficient T cells compared with the WT (Figure 4A and B). To further determine cell death in T lymphocytes we also followed another protocol more similar to the activation-induced cell death (AICD) phenomenon. T cells from either WT or Par-4-deficient mice were cultured for 48 h at 37°C with soluble anti-CD3 (3 µg/ml) and murine rIL-2. Pre-activated T cells were harvested, washed and replated, followed by the addition of anti-CD3 or antiCD3 plus anti-CD28. Cell viability was determined as above at 36 h post-stimulation. Again, the Par-4-deficient T cells were less sensitive to the apoptotic challenge than their WT controls (Figure 4C).
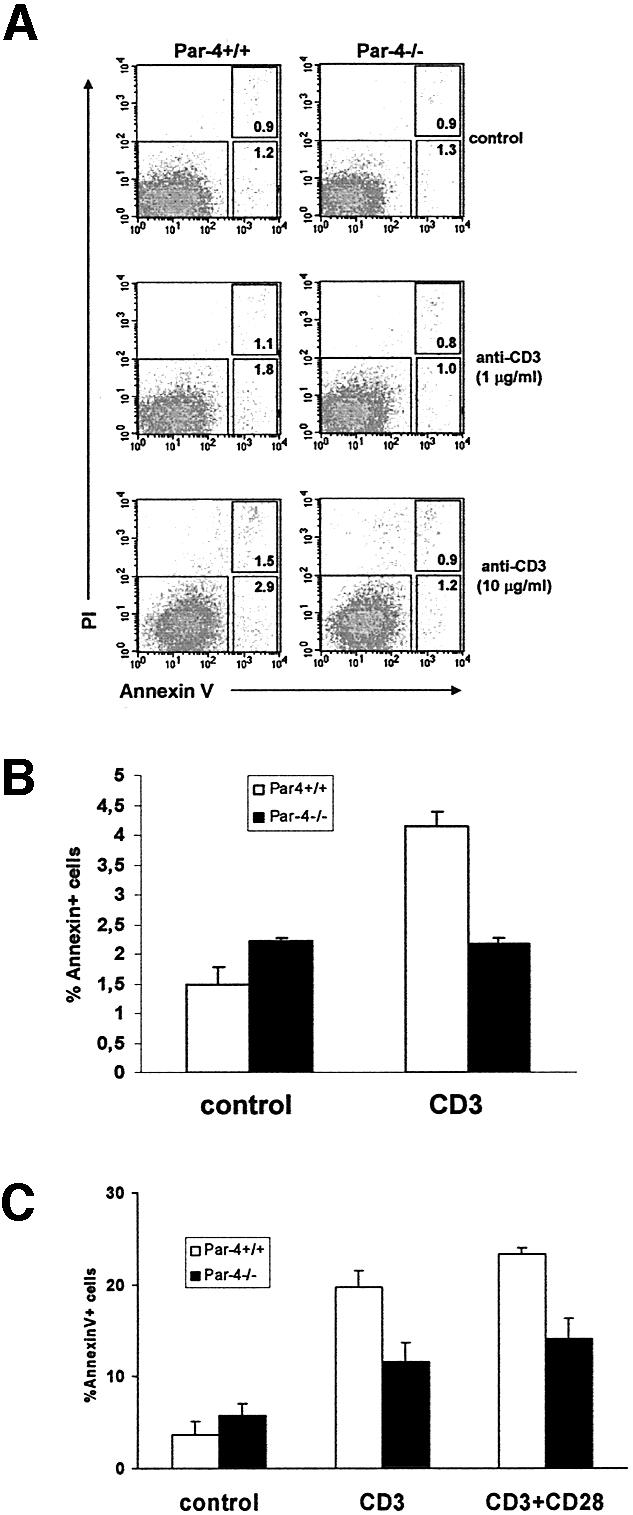
Fig. 4. T-cell apoptosis. (A) T cells from WT and Par-4 KO mice were stimulated with different concentrations of anti-CD3 antibody for 24 h, stained for the binding of annexin V and analyzed by flow cytometry gated for viable cells; the right upper inset corresponds to cells undergoing early apoptosis whereas the right lower inset corresponds to cells at a more advanced stage of the apoptotic process. Shown is a representative experiment of another three experiments with incubations carried out in duplicate. (B) The mean ± SD of the total (early + late) apoptosis of these experiments. (C) In another set of experiments, T cells, either WT or Par-4-deficient, were stimulated with anti-CD3 (3 µg/ml) in the presence of rIL-2 for 48 h, after which activated T cells were harvested, washed and re-plated, followed by the addition of anti-CD3 or anti-CD3 plus anti-CD28. Cell viability was determined as above at 2 days post-stimulation.
It could be argued that the reduced apoptosis observed in the Par-4-deficient T cells could account, at least in part, for the increased proliferation observed in these mutant cells (Figure 2A). However, the fact that CD25 induction is not affected (Figure 3B), together with the observation that the proportion of Par-4–/– cells entering the cell cycle is higher than that of the WT cells and that they respond normally to IL-2-induced proliferation (Figure 2B), suggest that in addition to its role as a pro-apoptotic protein, Par-4 also modulates cell cycle progression.
The enhanced proliferation observed in the Par-4-deficient T cells is not due to differences in the percentage of memory/effector or regulatory T cells, as the proportions of both types of cells are identical in spleens and lymph nodes of WT and Par-4–/– mice (Figure 5). Therefore, the effects on cell proliferation in Par-4-deficient T cells reported here are most likely due to intrinsic alterations in the signaling cascades.

Fig. 5. Normal proportions of memory/effector and regulatory T cells. Naïve and memory T-cell populations were determined from spleen and mesenteric lymph nodes of 4-week-old WT and Par-4-deficient mice. (A) Cells were stained with CD4, CD44 and CD62L. Three-color flow cytometry analysis was performed on gated CD4+ T cells. Percentage of memory/effector T cells is shown as CD4+CD44+ CD62L–. (B) Cells were stained with CD4 and CD25 and two-color flow cytometry analysis was performed on gated CD4+ T cells. Percentage of regulatory T cells is shown as CD4+CD25+. Experiments shown are representative of at least another five with very similar results.
Alterations of signaling pathways in Par-4-deficient T cells
Par-4–/– EFs show enhanced activation of aPKC activity, which leads to a dramatic reduction in the activation of JNK activity (Garcia-Cao et al., 2003). As Par-4 impacts TCR-mediated T-cell proliferation, we next determined whether T-cell activation leads to increased aPKC activity and whether the loss of Par-4 enhances that activation. Interestingly, anti-CD3 activation of WT T cells leads to the stimulation of aPKC activity, determined as increased phosphorylation of the kinase T-loop site (Figure 6A). Of note, the presence of anti-CD28 does not further increase the activation of the aPKCs promoted by anti-CD3 (Figure 6A), indicating that the aPKCs are selectively located in the TCR signaling pathway. Consistent with Par-4 being a negative regulator of the aPKCs, their activation in CD3-stimulated T cells was significantly enhanced in Par-4-deficient T cells compared with WT T cells (Figure 6A). Control immunoblots show that no detectable differences exist in the levels of ζPKC or λ/ιPKC in mutant T cells compared with WT cultures (Figure 6A). The aPKCs have been implicated in the control of NF-κB nuclear translocation at least in some cell systems. Interestingly, the loss of Par-4 leads to a dramatic increase in the amount of nuclear NF-κB in anti-CD3-activated T cells (Figure 6B). In the presence of CD28 co-stimulation the differences are less apparent, although still significant (Figure 6B). This observation is particularly relevant since the lack of ζPKC in KO mice revealed that this aPKC isoform is not required for NF-κB nuclear translocation in EFs or B cells, but seems to be necessary in lungs in in vivo experiments (Leitges et al., 2001). Because the loss of ζPKC does not affect T-cell proliferation (Martin et al., 2001), it is unlikely that the actions of Par-4 on NF-κB activation are mediated through ζPKC. Since both ζPKC and λ/ιPKC can promote the activation of the canonical NF-κB pathway upstream of its nuclear translocation (Lallena et al., 1999), the results of Figure 6B may suggest that Par-4 actions could be mediated in T cells by λ/ιPKC.
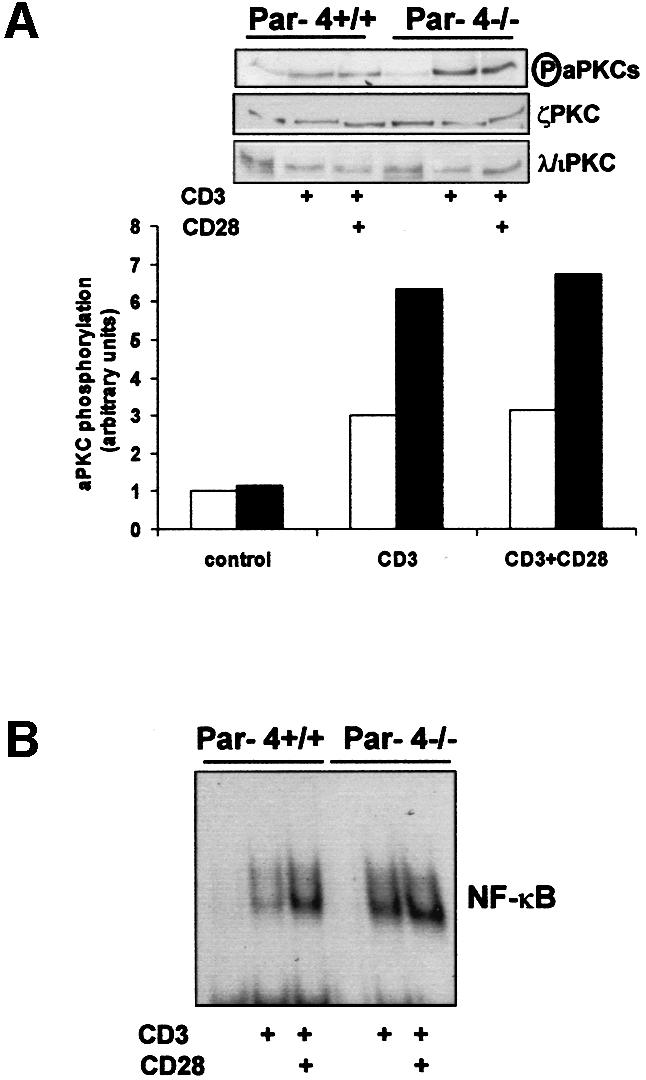
Fig. 6. aPKC and NF-κB signaling in Par-4–/– T cells. (A) Extracts from T cells activated with anti-CD3 antibody either in the absence or in the presence of anti-CD28 antibody for 24 h were analyzed by immunoblotting with anti-phospho-aPKC, anti-ζPKC and anti-λ/ιPKC. The phospho-aPKC immunoblot gels were quantitated by densitometry. (B) Similar experiments were performed in which T cells were stimulated for 16 h and nuclear extracts were prepared, followed by analysis of NF-κB by EMSA using a κB oligonucleotide probe.
As the hyperactivation of the aPKCs observed in Par-4–/– EFs leads to a decreased JNK activation (Garcia-Cao et al., 2003), most likely due to the enhanced stimulation of NF-κB (De Smaele et al., 2001; Tang et al., 2001), we determined in the next series of experiments whether a similar reduction in JNK activation would also be observed in Par-4–/– T cells. Therefore, T cells from either WT or Par-4-deficient mice were stimulated or not with anti-CD3 in the absence or in the presence of anti-CD28, after which JNK activity was determined. The results shown in Figure 7A demonstrate that JNK is only fully stimulated by the presence of anti-CD3 plus anti-CD28. Interestingly, the activation of JNK is severely abrogated in Par-4-deficient T cells compared with the WT controls (Figure 7A). However, in contrast to EFs, the activation of p38 is not affected in Par-4–/– T cells (not shown). Of note, the protein levels of JNK (Figure 7A) in the Par-4-deficient cells were indistinguishable from those of the WT controls. From the data shown in Figure 6B it seems that although Par-4–/– T cells displayed an increased NF-κB activation, as determined by electrophoretic mobility-shift assay (EMSA), in the presence of anti-CD3 the co-stimulation of CD28 minimizes the differences between WT and Par-4–/– T cells. The fact that JNK activation by CD3+CD28 is inhibited in Par-4-deficient T cells although no large differences are observed in NF-κB nuclear levels suggests that in addition to controlling NF-κB nuclear translocation, Par-4 also regulates RelA transcriptional activity, as has been shown in EFs (Garcia-Cao et al., 2003).
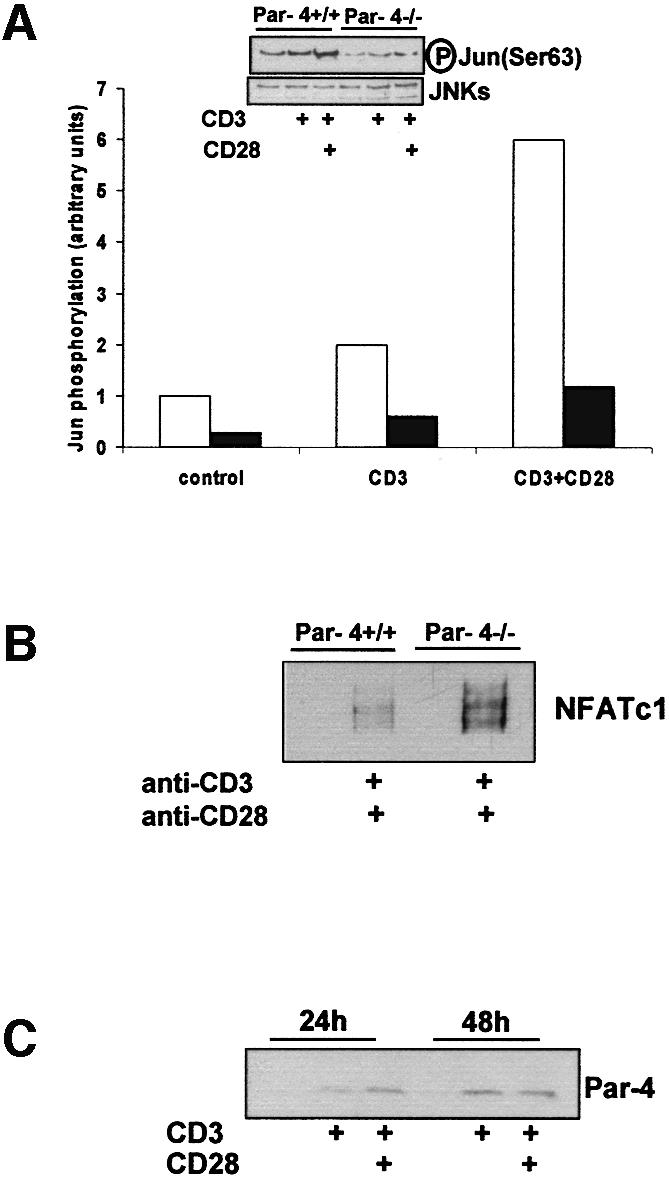
Fig. 7. JNK and NFATc1 activation. (A) Extracts from T cells activated with anti-CD3 antibody either in the absence or in the presence of anti-CD28 antibody for 48 h were used to measure JNK activity (determined by the level of Ser63 phosphorylation of c-Jun in anti-JNK immunoprecipitates). These extracts were also blotted with anti-JNK to verify the levels of the enzyme. (B) In another set of experiments, T cells either WT or Par-4–/– were stimulated as above and the amount of nuclear NFATc1 was determined by immunoblotting. (C) Parallel extracts were analyzed by immunoblotting using an anti-Par-4 antibody. Experiments shown are representative of another three with similar results.
JNK activation, particularly that of the JNK1 isoform, has been demonstrated to inhibit NFATc1 stimulation by phosphorylation of a residue in the calcineurin targeting domain (Chow et al., 2000). The phosphorylation of this site impedes calcineurin–NFATc1 interaction and the subsequent dephosphorylation and nuclear translocation of NFATc1. This has important functional repercussions, since this is a critical transcription factor for T-cell activation and differentiation (Dong et al., 1998; Crabtree and Olson, 2002). We reasoned that the inhibition of JNK activity in Par-4-deficient T cells should give rise to an increased activation of NFATc1 nuclear translocation similar to that reported in JNK1–/– T cells (Dong et al., 1998). The results of Figure 7B show that this is actually the case, as the stimulation of Par-4–/– T cells produced a more robust activation of NFATc1 than in control WT cells. As NFAT has been reported to play a relevant role in IL-2 synthesis (Crabtree and Olson, 2002), these results suggest strongly that the loss of Par-4 leads to JNK inhibition, which unleashes NFATc1 activation, driving a higher IL-2 synthesis that accounts for the increased T-cell proliferation in the absence of Par-4. Interestingly, the stimulation with anti-CD3 or anti-CD3 plus anti-CD28 promotes a reproducible increase in Par-4 levels (Figure 7C), suggesting that activated T cells trigger the accumulation of Par-4 as a mechanism to limit the activation of the aPKCs. The loss of Par-4 allows these kinases to be more active than in the WT cells.
In EFs, the loss of ζPKC leads to the opposite alterations to those of Par-4–/– cells, showing increased JNK activity (Garcia-Cao et al., 2003). When we analyzed TCR-activated T cells from ζPKC–/– mice, there was a reproducible, although modest, increase in JNK activation that correlates with an also modest decrease in NFATc1 stimulation (data not shown). As the Par-4 deficiency provokes a stronger phenotype in T cells than that of ζPKC, in terms of NF-κB, JNK, NFATc1 activation and cell proliferation, these results suggest that the loss of ζPKC is compensated for by λ/ιPKC in T cells.
Effect of the loss of Par-4 in the secretion of Th1 and Th2 cytokines
It has been proposed that JNK2 is required for the synthesis of the Th1 cytokine IFNγ (Yang et al., 1998), whereas JNK1 represses the synthesis of the Th2 cytokine IL-4, by inhibiting NFATc1 activation (Dong et al., 1998). Since the loss of Par-4 abrogates JNK activation and hyperactivates NFATc1, we determined in the next experiments the synthesis of IFNγ and IL-4 in CD4+ T-cell cultures. From these experiments it become apparent that whereas the synthesis of IFNγ is not affected by the loss of Par-4 (Figure 8A), there is a reproducible enhancement in the production of the Th2 cytokine IL-4 in the Par-4-deficient CD4+ T cells (Figure 8B). Collectively, these results suggest that the loss of Par-4 leads to the hyperactivation of the aPKCs, which provokes the inhibition of JNK leading to an overstimulation of IL-4 synthesis and Th2 differentiation.

Fig. 8. Effect of the lack of Par-4 in the secretion of Th1 and Th2 cytokines. Purified CD4+ T cells from WT and Par-4-deficient mice were untreated (empty bars) or stimulated (black bars) with 10 µg/ml of anti-CD3 and anti-CD28 (5 µg/ml) for 96 h, after which the release of (A) IFNγ and (B) IL-4 to the supernatant was determined. Results are the mean ± SD of one experiment representative of another two with similar results, and with incubations carried out in duplicate.
Discussion
The aPKCs have long been implicated in the control of key signaling events. Using dominant-negative mutants it has been shown that inhibition of ζPKC or λ/ιPKC leads to the blockade of NF-κB activation in overexpression experiments (for a review see Moscat et al., 2003). Recently, the characterization of ζPKC KO mice has demonstrated that in fact, ζPKC is important in the activation of NF-κB transcription in EFs and, accordingly, in cell survival (Leitges et al., 2001). In addition, B cells from ζPKC–/– mice show a significant impairment in the ability to grow and proliferate when activated through the BCR (Martin et al., 2002). Although NF-κB activation determined by EMSA is not affected by the loss of ζPKC in B cells, the synthesis of a number of NF-κB-dependent genes is severely inhibited, consistent with the evidence in EFs that ζPKC plays a non-redundant role in the control of NF-κB transcriptional activity, most likely by direct phosphorylation of RelA (Leitges et al., 2001). These signaling defects may explain why ζPKC-deficient young mice show a delay in the development of secondary lymphoid organs and why adult ζPKC KO mice are unable to mount an optimal T-dependent immune response (Martin et al., 2002).
The λ/ιPKC KO has a lethal phenotype at very early embryonary stages (J.Moscat and M.T.Diaz-Meco, unpublished observations), which precludes any studies on the physiological role of this aPKC in vivo. However, as Par-4 binds both ζPKC and λ/ιPKC (Diaz-Meco et al., 1996), the study of the Par-4 KO may shed light on the role played by λ/ιPKC in a whole organism. We found here that the loss of Par-4, in addition to enhancing the ability of B cells to proliferate when challenged through the BCR (due to increased aPKC activity; see Supplementary figures 1 and 2), also leads to an increase in the ability of T cells to respond to activation through the TCR, producing higher proliferation and cell cycle entry. These observations correlate with an elevated production of IL-2, a hallmark of T-cell activation. Interestingly, the loss of Par-4 does not affect the expression of CD25 (IL-2Rα), and consequently cells respond identically to WT T cells to the exogenous addition of recombinant IL-2. Consequently, it seems that Par-4 is a negative modulator of the signaling pathways that control IL-2 synthesis. It is noteworthy that, like in EFs, the aPKCs can be hyperactivated in response to the TCR challenge in Par-4–/– T cells, demonstrating that these kinases are physiologically relevant targets of Par-4 in vivo in several cell systems. As the ζPKC–/– mice have intact T-cell proliferation, it is tempting to speculate that the other aPKC, λ/ιPKC, may be the one that is critically involved in the modulation of T-cell function, whereas ζPKC actions appear to be restricted to B cells. However, other potential unknown targets of Par-4 may be involved in this T-cell function in an aPKC-independent manner. In addition, the fact that the ζPKC KO has an intact T-cell response may be due to the presence of λ/ιPKC, which may provide some redundancy in T cells but not in B cells.
The loss of Par-4 does not affect the nuclear accumulation of NF-κB as determined by EMSA in EFs (Garcia-Cao et al., 2003). In marked contrast, this parameter is significantly enhanced in Par-4-deficient T cells (see above), suggesting that λ/ιPKC may mediate in this cell type the activation of NF-κB at a step upstream of the control of its nuclear translocation. In addition, the measurement of JNK activation, which is inhibited by NF-κB stimulation in several cell systems (see above), re veals that it is severely abrogated in the activated Par-4–/– T cells. Therefore, all these results would be consistent with the notion that the aPKCs (and Par-4) are critical for the control of NF-κB activity. Interestingly, we show here that T-cell activation correlates with increased Par-4 levels, suggesting the this protein is induced as a mechanism to restrain T-cell stimulation that is lost in the Par-4 KO cells. Of note, previous reports have demonstrated that T cells from c-Rel KO mice are impaired in their ability to proliferate and synthesize IL-2 in response to TCR stimulation (Kontgen et al., 1995). The exogenous addition of IL-2 restores the proliferative capacity of the c-Rel–/– T cells, indicating that the proliferation defect caused in T cells by the absence of c-Rel can be accounted for by a poor induction of IL-2 (Kontgen et al., 1995). However, thymocyte differentiation is not affected in the c-Rel KO mice (Kontgen et al., 1995). Therefore, it seems that the T-cell proliferative phenotype of the Par-4-deficient mice is the opposite of that of the c-Rel KO mice, whereas both KOs display intact thymocyte differentiation (Kontgen et al., 1995). These results would be in keeping with the notion that the aPKCs and Par-4 are critical modulators of NF-κB also in T cells. It remains to be determined whether c-Rel-deficient T cells display a higher activation of JNK in response to TCR stimulation and whether the Par-4–/– phenotype can be reverted in double KO mice of Par-4 and c-Rel. Experiments underway in our laboratory will attempt to solve these questions.
The dramatic inhibition of TCR-triggered JNK activation produced by the loss of Par-4 is consistent with our earlier data indicating that in EFs Par-4 modulates JNK activation in response to TNFα and IL-1 (Garcia-Cao et al., 2003). The role played by JNKs during the T-cell response is complex (Arbour et al., 2002; Conze et al., 2002). Thus, for example, it has been reported that JNK1-deficient CD8+ T cells are unable to proliferate even in the presence of exogenous IL-2 due to an inhibition of CD25 expression (Conze et al., 2002). In contrast, the loss of JNK2 leads to enhanced proliferation and IL-2 production in CD8+ T cells (Conze et al., 2002). Our data show that Par-4 deficiency leads to enhanced proliferation and IL-2 production in both CD4+ and CD8+ T cells, with no changes in CD25 expression. In that respect the phenotype of the Par-4 KO mice in CD8+ T cells is similar to that of the JNK2-deficient mice. Interestingly, the loss of Par-4 leads to an increase in the ability of CD4+ T cells to proliferate and to produce IL-2, which is similar to what has been reported in mice in which both JNKs have been genetically inactivated in vivo (Dong et al., 2000). Furthermore, like in these mice, the Par-4–/– CD4+ T cells produce more IL-4 but not more IFNγ than the WT controls, indicating that the loss of Par-4 most likely drives T-cell differentiation towards a Th2 response. This very interesting phenotype could be explained at least in part by the fact that Par-4 inactivation provokes JNK inhibition in T cells, which gives rise to higher activation of NFATc1. In this regard, it should be noted that CD4+ T cells from mice in which both JNKs have been genetically inactivated preferentially develop along the Th2 lineage (Dong et al., 2000). Of note is the fact that, in contrast to what has been observed in EFs, T cells from ζPKC–/– mice show only a limited alteration in JNK activation. This is consistent with the lack of defects in proliferation in the ζPKC–/– T cells and reinforces the notion of tissue specificity for the aPKC actions. In this regard, although the B cells from Par-4-deficient mice hyperproliferate, these KOs have intact development of the secondary lymphoid organs. This may indicate that whereas the inhibition of B-cell function impacts the development of spleen, lymph nodes and Peyer’s patches, its hyperactivation, at least to the levels observed in the Par-4–/– mice, produces no effects on this parameter.
In summary, we present here compelling evidence that Par-4 is a physiologically relevant regulator of lymphocyte function in vivo. The aPKCs, like many other kinases, are tremendously promiscuous enzymes in vitro, which leads us to propose that some kind of cellular constraints must exist in vivo to confer specificity to the aPKCs actions during cell signaling (Moscat and Diaz-Meco, 2000). In addition to the existence of adapters, scaffolds and selective regulators, like Par-4, another layer of selectivity may reside in the tissue specificity of some signaling proteins. The data presented here are a good example of this. Thus, the fact that the ζPKC KO mice have significant alterations in the function of B cells but not of T cells, whereas the Par-4 KO mice show a phenotype in both cell types, suggest that the aPKCs play different roles in different cells types of the immune system, which may be executed through different aPKC isotypes. On the other hand, the observation that the lack of Par-4 enhances the production of IL-4, a Th2 cytokine, may be relevant from the point of view of inflammation therapeutics, particularly asthma, because it is believed that this pathological situation arises by an aberrant Th2 immune response to the inhalation of common antigens (Liu, 2002). Therefore, we can speculate that inhibitors of the aPKCs could be considered potentially novel anti-asthmatic drugs. In addition, Par-4 expression has been shown to be decreased in Ras-transformed fibroblasts (Barradas et al., 1999; Nalca et al., 1999), which is essential for tumor progression in vivo (Barradas et al., 1999). More recently, and of particular interest to the data reported in the present study, Par-4 levels have also been reported to be reduced in cells of patients of acute lymphatic leukemia and chronic lymphocytic leukemia (Boehrer et al., 2002), suggesting that Par-4 down-regulation contributes to the pathogenesis of lymphatic malignancies.
Materials and methods
Isolation of T and B cells
Spleens and mesenteric lymph nodes were removed from 4- to 6-week-old mice. B cells were isolated from spleens as follows. Single cells suspensions were made by crushing organs between glass slides. Red blood cells were lysed with an NH4Cl buffer. Afterwards, splenocytes were incubated with anti-mouse CD43 (Ly-48) microbeads (Miltenyi Biotec). T cells were isolated from mesenteric lymph nodes by incubation with biotin-conjugated anti-B220, anti-CD11b and anti-IgM antibodies. For CD4+ and CD8+ T-cell preparations, cells were incubated also with anti-CD8 and anti-CD4 antibodies, respectively, followed by incubation with anti-biotin microbeads (Miltenyi Biotec). B and T cells were separated by using an autoMACS magnetic cell sorter (Miltenyi Biotec). The purity of B and T cells, determined by flow cytometry, was >90%.
Flow cytometric analysis
Single cell suspensions were made from the indicated organs and washed in phosphate-buffered saline (PBS) supplemented with 0.5% bovine serum albumin. Afterwards, cells were incubated for 20 min at 4°C with the following antibodies: FITC-conjugated anti-CD8 (clone 53-6.7), PE-conjugated anti-CD4 (clone GK1.5), PE-conjugated anti-CD25 (clone PC61.5), FITC-conjugated anti-B220 (clone RA3-6B2) and PE-conjugated anti-CD43 (clone S7). For the three fluorescent staining of B-cell subpopulations, cells were incubated with FITC-conjugated anti-CD21 (clone 7G6), PE-conjugated anti-CD23 (clone B3B4) and biotin-conjugated anti-IgM (clone R6-60.2) followed by streptavidin-tricolor. For the analysis of effector/memory T-cell population, cells were incubated with FITC-conjugated anti-CD44 (clone IM7), PE-conjugated anti-CD4 (clone GK 1.5) and biotin-conjugated anti-CD62L (clone MEL-14), followed by streptavidin-tricolor. For the analysis of pre-activated apoptotic T cells, purified T cells were incubated for 48 h with soluble anti-CD3 (3 µg/ml) and rmIL-2 (R&D Systems). Activated T cells were harvested using Ficoll (Pharmacia), washed and replated in 96-well plates with anti-CD3 (10 µg/ml) alone or in combination with anti-CD28 (1 µg/ml).
Cell cycle analysis was performed incubating cells in a buffer containing 0.1% sodium citrate, 0.6% NP-40, 20 µg/ml RNAse and 50 µg/ml of propidium iodide for 30 min in the dark and analyzing the cells by flow cytometry. Analysis of apoptotic cells was performed by staining with FITC-conjugated annexin V (R&D Systems), after 24 h of culture with or without 1 or 10 µg/ml anti-CD3. Analyses were performed on a FACS-calibur flow cytometer (Becton Dickinson) with CELL-Questpro software. Viable cells were analyzed by excluding dead cells from the analysis on the basis of low forward-light scatter.
Proliferation assays
Purified B cells were cultured in 96-well plates at 2 × 105 cells/well in 100 µl RPMI 1940 medium supplemented with 10% fetal calf serum, 2 mM l-glutamine, 50 U/ml penicillin/streptomycin and 50 µm 2-mercaptoethanol, and were stimulated for 72 h with anti-mouse IgM antibody (Jackson Immunoresearch Laboratories), LPS (Escherichia coli; Sigma) or anti-CD40 (clone HM40-3). Purified total T cells, CD4+ and CD8+ cells were cultured at 0.5 × 105 cells/well in 160 µl of medium and were stimulated for 48 h on plates precoated with anti-CD3ε antibody (clone 145-2C11; PharMingen) for 1 h. For co-stimulation of T cells, soluble anti-CD28 (clone 37.51; PharMingen) was also added at 1 µg/ml. Recombinant murine IL-2 (R&D Systems) was also added in the indicated experiments for 72 h. Mixed lymphocyte reactions assays were performed by mixing purified T cells and splenocytes, from BALB/c mice γ-irradiated with 3000 rads, in 200 µl of medium at the indicated relations for 4 days. Proliferation was assessed by the incorporation of [3H]thymidine added at 1 µCi/well during the last 12 h of culture in triplicate wells. Cells were collected using a cell harvester and [3H]thymidine incorporation was quantified by scintillation counting (Wallac Oy 1450; Microbeta).
ELISA assay for cytokines
Purified total T cells (2 × 105), CD4+ or CD8+ cells (1.85 × 104) were cultured at in 96-well plates with or without 10 µg/ml of anti-CD3ε and the supernatants were harvested at 24 h for measurement of IL-2, and at 72 h for IL-4 and IFN-γ. Cytokine levels were determined by enzyme-linked immumosorbent assay (ELISA) (PharMingen) according to the manufacturer’s instructions.
Inmunoblot analysis
T cells either untreated or incubated with anti-CD3 antibody either in the absence or in the presence of anti-CD28 for different times were extracted with lysis buffer and cellular extracts were resolved in SDS–polyacrylamide gels. Afterwards, they were electrophoretically transferred into a nitrocellulose membrane (Hybond ECL; Amersham Pharmacia Biotech) and incubated with anti-JNK and anti-phospho-aPKC (Cell Signaling), anti-λ/ιPKC (Becton Dickinson) and anti-ζPKC (H-1; Santa Cruz Biotechnology). In some experiments, nuclear extracts were analyzed by immunoblotting with anti-NFATC1 antibody (Santa Cruz Biotechnology). The bands were visualized with the enhanced chemiluminescence system (Amersham).
Nuclear extract preparation and EMSA
Nuclear extracts from T cells were prepared as described previously (Leitges et al., 2001). Binding reactions for EMSA were performed as described previously using 3 µg of nuclear protein and the following end-labeled double-stranded oligonucleotide NF-κB probe: 5′-GCCTG GGACTTCCCCCTCAACT-3′.
MAP kinase assays
Purified T cells from WT and Par-4-deficient mice were stimulated or not with immobolized anti-CD3 antibody (5 µg/ml) in the absence or in the presence of soluble anti-CD28 antibody (5 µg/ml) for 48 h, after which cell extracts were prepared and JNK activity was measured as decribed (Garcia-Cao et al., 2003), using a commercially available kit (Cell Signaling) according to the manufacturer’s instructions. JNK cellular levels were determined by immunoblotting using an anti-JNK antibody (Santa Cruz Biotechnology).
Purified B cells were stimulated or not with 20 or 50 µg/ml of affinity-purified goat anti-mouse IgM (Jackson Immune Research) for 10 min, after which cell extracts were analyzed by immunoblotting with anti-phospho-ERK and anti-ERK antibodies.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
J.M. is recipient of the Ayuda Investigación Juan March 2001. This work was supported by grants SAF1999-0053 (to M.T.D.-M.), SAF2002-03402 (to M.S.) and SAF2002-0187 (to J.M.) from MCYT, and by an institutional grant from Fundación Ramón Areces to the CBMSO.
References
- Arbour N., Naniche,D., Homann,D., Davis,R.J., Flavell,R.A. and Oldstone,M.B. (2002) c-Jun NH2-terminal kinase (JNK)1 and JNK2 signaling pathways have divergent roles in CD8+ T cell-mediated antiviral immunity. J. Exp. Med., 195, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barradas M., Monjas,A., Diaz-Meco,M.T., Serrano,M. and Moscat,J. (1999) The downregulation of the pro-apoptotic protein Par-4 is critical for Ras-induced survival and tumor progression. EMBO J., 18, 6362–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehrer S., Chow,K.U., Ruthardt,M., Hoelzer,D., Mitrou,P.S. and Weidmann,E. (2002) Expression and function of prostate-apoptosis-response-gene-4 in lymphatic cells. Leuk. Lymphoma, 43, 1737–1741. [DOI] [PubMed] [Google Scholar]
- Chow C.-W., Dong,C., Flavell,R.A. and Davis,R.J. (2000) c-Jun NH2-terminal kinase inhibits targeting of the protein phosphatase calcineurin to NFATc1. Mol. Cell. Biol., 20, 5227–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conze D. et al. (2002) c-Jun NH2-terminal kinase (JNK)1 and JNK2 have distinct roles in CD8+ T cell activation. J. Exp. Med., 195, 811–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree G.R. and Olson,E.N. (2002) NFAT signaling: choreographing the social lives of cells. Cell, 109, S67–S79. [DOI] [PubMed] [Google Scholar]
- De Smaele E., Zazzeroni,F., Papa,S., Nguyen,D.U., Jin,R., Jones,J., Cong,R. and Franzoso,G. (2001) Induction of gadd45β by NF-κB downregulates pro-apoptotic JNK signalling. Nature, 414, 308–313. [DOI] [PubMed] [Google Scholar]
- Diaz-Meco M.T., Municio,M.M., Frutos,S., Sanchez,P., Lozano,J., Sanz,L. and Moscat,J. (1996) The product of par-4, a gene induced during apoptosis, interacts selectively with the atypical isoforms of protein kinase C. Cell, 86, 777–786. [DOI] [PubMed] [Google Scholar]
- Diaz-Meco M.T., Lallena,M.J., Monjas,A., Frutos,S. and Moscat,J. (1999) Inactivation of the inhibitory κB protein kinase/nuclear factor κB pathway by Par-4 expression potentiates tumor necrosis factor α-induced apoptosis. J. Biol. Chem., 274, 19606–19612. [DOI] [PubMed] [Google Scholar]
- Dong C., Yang,D.D., Wysk,M., Whitmarsh,A.J., Davis,R.J. and Flavell,R.A. (1998) Defective T cell differentiation in the absence of Jnk1. Science, 282, 2092–2095. [DOI] [PubMed] [Google Scholar]
- Dong C., Yang,D.D., Tournier,C., Whitmarsh,A.J., Xu,J., Davis,R.J. and Flavell,R.A. (2000) JNK is required for effector T-cell function but not for T-cell activation. Nature, 405, 91–94. [DOI] [PubMed] [Google Scholar]
- Garcia-Cao I., Lafuente,M., Criado,L., Diaz-Meco,M., Serrano,M. and Moscat,J. (2003) Genetic inactivation of Par4 results in hyperactivation of NF-κB and impairment of JNK and p38. EMBO rep., 4, 307–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontgen F., Grumont,R.J., Strasser,A., Metcalf,D., Li,R., Tarlinton,D. and Gerondakis,S. (1995) Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity and interleukin-2 expression. Genes Dev., 9, 1965–1977. [DOI] [PubMed] [Google Scholar]
- Lallena M.J., Diaz-Meco,M.T., Bren,G., Pay,C.V. and Moscat,J. (1999) Activation of IκB kinase β by protein kinase C isoforms. Mol. Cell. Biol., 19, 2180–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitges M. et al. (2001) Targeted disruption of the zetaPKC gene results in the impairment of the NF-κB pathway. Mol. Cell, 8, 771–780. [DOI] [PubMed] [Google Scholar]
- Liu A.H. (2002) Endotoxin exposure in allergy and asthma: reconciling a paradox. J. Allergy Clin. Immunol., 109, 379–392. [DOI] [PubMed] [Google Scholar]
- Martin A.G., San-Antonio,B. and Fresno,M. (2001) Regulation of nuclear factor kappa B transactivation. Implication of phosphatidylinositol 3-kinase and protein kinase C zeta in c-Rel activation by tumor necrosis factor alpha. J. Biol. Chem., 276, 15840–15849. [DOI] [PubMed] [Google Scholar]
- Martin P., Duran,A., Minguet,S., Gaspar,M.L., Diaz-Meco,M.T., Rennert,P., Leitges,M. and Moscat,J. (2002) Role of ζPKC in B-cell signaling and function. EMBO J., 21, 4049–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat J. and Diaz-Meco,M.T. (2000) The atypical protein kinase Cs. Functional specificity mediated by specific protein adapters. EMBO rep., 1, 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat J., Diaz-Meco,M.T. and Rennert,P. (2003) NF-κB activation by protein kinase C isoforms and B-cell function. EMBO rep., 4, 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalca A., Qiu,S.G., El-Guendy,N., Krishnan,S. and Rangnekar,V.M. (1999) Oncogenic Ras sensitizes cells to apoptosis by Par-4. J. Biol. Chem., 274, 29976–29983. [DOI] [PubMed] [Google Scholar]
- Tang G., Minemoto,Y., Dibling,B., Purcell,N.H., Li,Z., Karin,M. and Lin,A. (2001) Inhibition of JNK activation through NF-κB target genes. Nature, 414, 313–317. [DOI] [PubMed] [Google Scholar]
- Yang D.D., Conze,D., Whitmarsh,A.J., Barrett,T., Davis,R.J., Rincon,M. and Flavell,R.A. (1998) Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity, 9, 575–585. [DOI] [PubMed] [Google Scholar]