

Abstract
The solution structure of the 33 kDa complex between the dimeric DNA-binding core domain of the transcription factor MEF2A (residues 1–85) and a 20mer DNA oligonucleotide comprising the consensus sequence CTA(A/T)4TAG has been solved by NMR. The protein comprises two domains: a MADS-box (residues 1–58) and a MEF2S domain (residues 59–73). Recognition and specificity are achieved by interactions between the MADS-box and both the major and minor grooves of the DNA. A number of critical differences in protein–DNA contacts observed in the MEF2A–DNA complex and the DNA complexes of the related MADS-box transcription factors SRF and MCM1 provide a molecular explanation for modulation of sequence specificity and extent of DNA bending (∼15 versus ∼70°). The structure of the MEF2S domain is entirely different from that of the equivalent SAM domain in SRF and MCM1, accounting for the absence of cross-reactivity with other proteins that interact with these transcription factors.
Keywords: DNA bending/MADS-box/MEF2/multidimensional NMR/transcription
Introduction
The MEF2 proteins (for myocyte enhancer factor 2) comprise a group of transcription factors that play a key role in myogenesis and morphogenesis of muscle cells (Gossett et al., 1989; Black and Olson, 1998). The MEF2 factors bind specifically to a conserved A⋅T-rich DNA sequence in the control regions of the majority of muscle-specific genes, including several genes that encode myogenic basic helix–loop–helix (bHLH) proteins. Recently, MEF2 proteins have also been shown to have important roles in regulating cell fate in other cell types, including neuronal cell survival (Mao et al., 1999) and T-cell apoptosis (Youn et al., 1999). In addition, the MEF2 proteins have been implicated in gene activation in response to MAP kinase pathway activation (Han et al., 1997; Kato et al., 1997; Ornatsky et al., 1999 ; Zhao et al., 1999) and also T-cell receptor activation where they are thought to function as endpoints for intracellular signaling pathways activated by calcium (Youn et al., 1999).
There are four members of the mammalian MEF2 family, designated MEF2A, MEF2B, MEF2C and MEF2D. The N-terminal ends of the MEF2 proteins (residues 1–85) contain the core DNA-binding domain, which is sufficient for specific DNA binding and dimerization (Sharrocks, 1994; Shore and Sharrocks, 1995). The transcriptional activation domains are located C-terminal to the DNA-binding domain and the activities of these domains are directly regulated by MAP kinase-mediated phosphorylation (Han et al., 1997; Kato et al., 1997; Ornatsky et al., 1999). The DNA-binding core has been divided into two subdomains based on sequence conservation: a so-called MADS-box DNA-binding motif consisting of the first 56 residues, and a MEF2 domain comprising the C-terminal 29 residues (Shore and Sharrocks, 1995). The MADS-box is named after the first four proteins in which this domain was identified: minichromosome maintenance 1 (MCM1), AGAMOUS (AG), DEFICIENS and serum response factor (SRF) (Schwarz-Sommer et al., 1990; Shore and Sharrocks, 1995). There are now a huge number of different MADS-box proteins, found in organisms ranging from yeasts and plants to mammals. In vertebrates, however, there are only two different types of MADS-box proteins: SRF and the MEF2 subfamily. These proteins both contain MADS-boxes within their core DNA-binding domains, but differ within their C-terminal extensions. Indeed, the sequences of the MEF2 domain are highly homologous among members of the MEF2 family, but are very different from those of the analogous region in other MADS-box proteins, such as SRF and MCM1, which have a SAM (SRF, AG and MCM1) domain adjacent to the C-terminus of the MADS-box. Thus, while MEF2 proteins can form both homo- and heterodimers, they do not interact with other MADS-box proteins (Pollock and Treisman, 1991).
Although different MADS-box family members generally recognize A⋅T-rich consensus sequences, they possess distinct DNA-binding specificities. MEF2A recognizes the consensus sequence CTA(A/T)4TAG (Pollock and Treisman, 1991), while the consensus sequence for SRF and MCM1 is (CC(A/T)6GG) and has been termed a (CArG) box (Treisman, 1990). In addition, biochemical and structural studies have demonstrated that MADS-box transcription factors have distinctive DNA-bending propensities. The crystal structures of SRF (Pellegrini et al., 1995) and MCM1 (Tan and Richmond, 1998) bound to DNA show that these MADS-box proteins induce an ∼70° bend in the DNA. These observations are consistent with the conclusions derived from a combination of circular permutation (Gustafson et al., 1989; Smith et al., 1995; West et al., 1997), phasing and ligase-mediated circularization assays (West et al., 1997; West and Sharrocks, 1999). In contrast, biochemical studies show that other MADS-box proteins, including MEF2A, only minimally distort the DNA (West et al., 1997; West and Sharrocks, 1999). Thus, the local architecture of higher order transcription complexes involving MEF2A will clearly be different from those involving SRF and MCM1, which is likely to contribute to their different biological functions. To understand further the structural and molecular basis of DNA recognition and DNA bending by MADS-box proteins, we have solved the three-dimensional (3D) solution structure of MEF2A bound to DNA by multidimensional NMR.
Results and discussion
Structure determination
We have solved the solution structure of a 33 kDa complex between the homodimeric dimeric core domain of MEF2A (residues 1–85) and a self-complementary 20mer DNA duplex comprising the sequence 5′d-(CTCGGCTATTAATAGCCGAG) with the 10 bp consensus sequence (in bold) located in the middle (i.e. bp −5 to +5). Note that the protein sequence starts at Gly1, and the N-terminal methionine is completely excised (Hirel et al., 1989) as judged by mass spectrometry. The structure of the complex was solved by heteronuclear double and triple resonance NMR spectroscopy (Clore and Gronenborn, 1991, 1998a; Bax and Grzesiek, 1993) using both 15N- and 13C-labeled protein and oligonucleotide (Louis et al., 1998). Experiments were carried out on the following 1:1 complexes: [15N]MEF2A and unlabeled DNA, [15N/13C]MEF2A and unlabeled DNA, and [15N]MEF2A and 15N/13C-labeled DNA. The complex is in slow exchange on the chemical shift scale, and is 2-fold symmetric as judged by the presence of only a single set of resonances. The availability of 15N/13C-labeled DNA and protein permitted us to assign with greater confidence intermolecular nuclear Overhauser effects (NOEs) by making use of 13C-separated/12C-filtered NOE experiments to detect specifically NOEs between protons attached to 13C and protons attached to 12C. This was particularly useful for NOEs involving the H4′ and H5′/H5′′ sugar protons of the DNA, which overlap in the 1H spectrum but whose directly bonded carbon atoms (C4′ and C5′) have distinct 13C chemical shifts. An example of the quality of the data is shown in Figure 1, which shows a series of strips taken from a 3D 15N-separated NOE experiment recorded on a 1:1 complex of [15N]MEF2A and unlabeled DNA (Figure 1A), and 3D 13C-separated/12C-filtered NOE experiments recorded on 1:1 complexes of [15N/13C]MEF2A and unlabeled DNA (Figure 1B) and [15N]MEF2A and 15N/13C-labeled DNA (Figure 1C). In addition, one-bond 1DNH and 1DCH residual dipolar couplings were measured for the protein and DNA, respectively, using the 1:1 complex of [15N]MEF2A and [15N/13C]DNA dissolved in a dilute liquid crystalline medium of bicelles. The dipolar couplings provide direct information on the orientation of the protein 15N–H and DNA 13C–H bond vectors with respect to the single alignment tensor and hence yield valuable long-range orientational information (see below). The structure was determined on the basis of 4560 experimental NMR restraints (2280 unique ones since the protein is a homodimer and the DNA palindromic), including 168 intermolecular NOE-derived interproton distance restraints between protein and DNA and 140 residual dipolar coupling restraints. A summary of the structural statistics is provided in Table I, and a superimposition of the final 35 simulated annealing structures is shown in Figure 2A.

Fig. 1. Composite of 15N–H and 13C–H strips taken from (A) a 3D 15N-separated NOE spectrum of a complex of [15N]MEF2A and unlabeled DNA, (B) a 3D 13C-separated/12C-filtered NOE spectrum of a complex of [15N/13C]MEF2A and unlabeled DNA, and (C) a 3D 13C-separated/12C-filtered NOE spectrum of a complex of [15N]MEF2A and [15N/13C]DNA.

Fig. 3. Comparison of the structures of the MEF2A–DNA and SRF–DNA complexes. Two views of the complete MEF2A–DNA and SRF–DNA complexes are shown in (A) and (C) and (B) and (D), respectively; a superimposition of the two complexes best-fitted to the MADS-box is shown in (E) in the same orientation as in (A) and (B); and ribbon diagrams of the MEF2S and SAM domains are shown in (F) and (G), respectively. The electrostatic potential (blue positive and red negative) mapped onto the molecular surface of the proteins is displayed in (A) and (B). The DNA and the path of its long axis are displayed in green and dark blue, respectively, for the MEF2A–DNA complex (A and C), and in red and purple, respectively, for the SRF–DNA complex (B and D). The proteins are displayed as backbone worms in (C), (D) and (E). In (E), the MEF2A–DNA complex is shown in light blue and the SRF–DNA complex in red. Residues 1–74 of MEF2A are displayed in (A), (C) and (E). In (F) and (G) the two subunits are represented in red and blue; the orientation of the MEF2 and SAM domains shown in (F) and (G) is the same as that in (A), (B) and (E). The coordinates of the SRF–DNA complex are taken from Pellegrini et al. (1995) (PDB accession code 1SRS).
Table I. Structural statistics.
| |
<SA>a |
(SA)rb |
| A. R.m.s. deviations from restraints |
0.064 ± 0.001 |
0.060 |
| protein | ||
| inter-residue sequential (|i – j| = 1) (664) | 0.041 ± 0.002 | 0.036 |
| inter-residue medium range (1 < |i – j| ≤ 5) (504) | 0.053 ± 0.003 | 0.042 |
| inter-residue long range (|i – j| > 5) (212) | 0.062 ± 0.006 | 0.062 |
| intraresidue (616) | 0.020 ± 0.002 | 0.020 |
| intersubunit (174) | 0.038 ± 0.005 | 0.034 |
| DNA | ||
| intraresidue (428) | 0.108 ± 0.001 | 0.105 |
| sequential intrastrand (196) | 0.058 ± 0.003 | 0.054 |
| interstrand (24) | 0.097 ± 0.006 | 0.080 |
| protein–DNA (168) | 0.092 ± 0.005 | 0.067 |
| R.m.s. deviations from hydrogen bonding restraints (Å)c protein (138) | 0.030 ± 0.004 | 0.046 |
| DNA (120) | 0.013 ± 0.001 | 0.017 |
| R.m.s. deviations from repulsive restraints (Å) (106)d | 0.011 ± 0.009 | 0.004 |
| R.m.s. deviations from experimental dihedral restraints (°) (708)e | 0.338 ± 0.044 | 0.320 |
| R.m.s. deviations from 3JHNα coupling constraints (Hz)d (72) | 1.00 ± 0.05 | 1.12 |
| R.m.s. deviations from secondary 13C shifts (p.p.m.)d | ||
| 13Cα (140) | 0.98 ± 0.01 | 0.99 |
| 13Cβ (140) | 1.42 ± 0.04 | 1.36 |
| R-factor for residual dipolar couplings (%)f | ||
| protein 1DNH (70) | 1.46 ± 0.04 | 2.32 |
| DNA 1DCH (70) | 3.78 ± 0.16 | 4.58 |
| Deviations from idealized covalent geometry | ||
| bonds (Å) (1946) | 0.008 ± 0.000 | 0.007 |
| angles (°) (3524) | 1.053 ± 0.005 | 1.146 |
| impropers (°) (979) |
1.782 ± 0.026 |
1.791 |
| B. Measures of structure quality | ||
| EL–J (kcal/mol)g | –1136 ± 18 | –1153 |
| % residues in most favorable region of Ramachandran mapg |
88 ± 2 |
87 |
| C. Coordinate precision (Å)h | ||
| Protein backbone + DNA heavy atoms | 0.34 ± 0.06 | |
| Protein heavy atoms + DNA heavy atoms | 0.54 ± 0.05 | |
| Protein backbone | 0.37 ± 0.07 | |
| Protein heavy atoms | 0.65 ± 0.06 | |
| DNA heavy atoms | 0.31 ± 0.09 | |
a<SA> is the final set of 35 simulated annealing structures.
b is the mean structure obtained by averaging the coordinates of the 35 individual SA structures with residues 1–73 of both subunits of the protein and bp –10 to +10 of the DNA best-fitted to each other; ()r is the restrained regularized mean structures obtained by restrained regularization of (Nilges et al., 1988). The total numbers of restraints are given in parentheses. The number of unique restraints is half that since the protein is a symmetric homodimer and the DNA is symmetric palindrome. None of the structures exhibited interproton distance violations >0.5 Å, torsion angle violations >5°, or 3JHNα coupling constant violations >2 Hz.
is the mean structure obtained by averaging the coordinates of the 35 individual SA structures with residues 1–73 of both subunits of the protein and bp –10 to +10 of the DNA best-fitted to each other; ()r is the restrained regularized mean structures obtained by restrained regularization of (Nilges et al., 1988). The total numbers of restraints are given in parentheses. The number of unique restraints is half that since the protein is a symmetric homodimer and the DNA is symmetric palindrome. None of the structures exhibited interproton distance violations >0.5 Å, torsion angle violations >5°, or 3JHNα coupling constant violations >2 Hz.
cBackbone hydrogen bonding restraints (two per hydrogen bond) were added during the final stages of refinement according to standard criteria (Clore et al., 1989). Six distance restraints per base pair are used to represent the Watson–Crick base pairs: for GC base pairs, rN1–N3 = 2.87 ± 0.2 Å, rH1–N3 = 1.86 ± 0.2 Å, rO6–N4 = 2.81 ± 0.2 Å, rN2–O2 = 2.81 ± 0.2 Å, rO6–N3 = 3.58 ± 0.2 Å and rN2–N3 = 3.63 ± 0.2 Å; for AT base pairs, rN1–N3 = 2.92 ± 0.2 Å, rN1–H3 = 1.87 ± 0.2 Å, rN6–O4 = 2.89 ± 0.2 Å, rH2–O2 = 2.94 ± 0.2 Å, rN1–O4 = 3.69 ± 0.2 Å and rN1–O2 = 3.67 ± 0.2 Å. The O6–N3 and N2–N3 distance restraints for GC base pairs and the N1–O4 and N1–O2 distance restraints for AT base pairs serve to prevent unduly large shearing of the base pairs.
dRepulsive distance restraints with a lower bound of 4 Å and no upper bound were introduced in the final stages of the refinement to prevent energetically unfavorable proximity of hydrogen bond donors of the protein (involving Gly1, Arg2, Lys3, Lys4, Arg14 and Lys22) to hydrogen bond donors of the DNA (Omichinski et al., 1997).
eThere are 240 torsion angle restraints for each protein subunit (71 φ, 71 ψ, 56 χ1, 34 χ2 and 8 χ3) and 114 per strand of DNA (see Materials and methods).
fThe dipolar coupling R-factor is defined by the ratio of the r.m.s. deviation between observed and calculated values to the expected r.m.s. deviation if the vectors were randomly oriented. The latter is given by {2Da2[4 + 3R2]/5}1/2 where Da is the magnitude of the alignment tensor and R the rhombicity (Clore and Garrett, 1999). The dipolar couplings per strand of DNA are broken down into 11 for the bases and 24 for the sugars.
gEL–J is the Lennard–Jones van der Waals energy calculated with the CHARMM PARAM19/20 protein and PARNAH1ER1 DNA parameters and is not included in the target function for simulated annealing or restrained regularization. The overall quality of the protein moiety (residues 1–73) was assessed using the program PROCHECK (Laskowski et al., 1993). There are no φ,ψ angles in the disallowed region of the Ramachandran map. The dihedral angle G-factors for φ/ψ, χ1/χ2, χ1 and χ3/χ4 are –0.02 ± 0.03, 0.02 ± 0.07, –0.19 ± 0.11 and –0.32 ± 0.13, respectively.
hThe precision of the coordinates is defined as the average atomic r.m.s.d. between the 35 individual simulated annealing structures and the mean coordinates. The values refer to residues 1–73 of both subunits and bp –10 to +10 of the DNA. Residues 74–85 of the protein are disordered in solution.

Fig. 2. Overall structure of the MEF2A–DNA complex. (A) Stereoview showing a best-fit superimposition of the final 35 simulated annealing structures of the MEF2A–DNA complex (residues 1–74 of each subunit of MEF2A are displayed; note that residues 74–85 are disordered in solution). (B and C) Two views showing ribbon diagrams of the MEF2A–DNA complex. The two protein subunits are shown in red and green, the DNA in blue, and the path of the long axis of the DNA in gray. (D) Summary of intersubunit contacts. Interactions between segments 1 and 2, segments 2 and 2, segments 2 and 3, and segments 3 and 3 are shown as blue, green, purple and red lines, respectively. Residues that have an ASA of <50% relative to a Gly-X-Gly tripeptide in the monomer are indicated by open circles; residues whose ASA is reduced to <50% upon dimerization are indicated by filled circles; the presence of both open and filled circles indicates that the ASA of that residue, which was <50% in the monomer, is reduced by a further 25% or more upon dimerization. Residues whose ASA is reduced to <50% upon complexation with DNA are indicated by asterisks; the presence of both an asterisk and a filled circle indicates that a residue whose ASA is <50% in the dimer is reduced by a further 40% or more upon complexation with DNA.
Description of the structure
The whole complex is 2-fold symmetric. Thus, the molecular 2-fold axis of the MEF2A dimer is coincident with the 2-fold axis relating the two halves (bp 1–10 and bp –1 to –10) of the palindromic 20mer DNA (Figures 2B, 2C and 3C). The 2-fold symmetry axis lies between bp 1 and –1 (Figure 5B) and is orthogonal to the long axis of the DNA at that point (Figure 3C).

Fig. 5. Comparison of protein–DNA interactions in the MEF2A–DNA and SRF–DNA complexes. (A) Sequence alignment of the MEF2A and SRF core DNA-binding domains. Residues interacting with sugars and phosphates are indicated by open circles; residues interacting with bases are indicated by filled circles; residues interacting with A⋅T and G⋅C nucleotides are colored in red and green, respectively; residues interacting with both A⋅T and G⋅C nucleotides are colored in blue. (B) Schematic summary of the DNA contacts in the MEF2A (left) and SRF (right) complexes. The invariant base pairs in the consensus core elements are shown in green, and the variant A⋅T base pairs in the consensus core elements are shown in red. Only contacts between one subunit of the MEF2A and SRF dimers and the DNA are displayed. The black continuous and blue dashed lines indicate interactions in the major and minor grooves, respectively. The numbering scheme for both the protein and DNA components of the SRF–DNA complex has been converted for ease of comparison to that of the MEF2A–DNA complex. The filled circle between bp –1 and 1 indicates the location of the 2-fold symmetry axis. (C) A ribbon representation illustrating a detailed view of the contacts between MEF2A (one subunit) and DNA. The protein backbone and side chains are shown in lilac and dark blue, respectively; A⋅T and G⋅C base pairs are displayed in yellow and green, respectively, and the phosphate backbone is shown as a light blue ribbon.
Two-fold symmetry has a number of important implications with regard to the dipolar couplings and the permissible orientation of the alignment tensor. Specifically, for any body with a 2-fold symmetry axis, one principal axis (in this case the x-axis) of the alignment tensor must be parallel to the 2-fold symmetry axis with the two other axes (in this case y and z) of the alignment tensor orthogonal to it. If this were not the case, then a 180° rotation about the 2-fold symmetry axis would change the orientation of the alignment principal axes with respect to the body; this, however, is not possible since a 180° rotation leaves the body invariant (i.e. because of symmetry a 180° rotation is equivalent to a 360° rotation). As a result, the dipolar couplings will define the relative orientation of the two symmetrical halves of the complex. This includes the relative orientations of the two subunits of the MEF2A dimer, the two halves of the DNA 20mer, and the protein and DNA. Despite the use of dipolar couplings, it should be borne in mind that the intermolecular NOEs still remain the principal determinant of the DNA conformation (including bending).
The interactions with the DNA are confined to the MADS-box (residues 1–58), while dimer contacts involve both the MADS-box and the MEF2S domain (residues 59–73). [Note that from a structural perspective the MADS-box is actually two residues longer and the structured MEF2 domain (MEF2S) is 12 residues shorter than the MEF2 domain designated on the basis of sequence comparisons.] The principal features of the MADS-box dimer comprise an N-terminal extension (residues 1–12), followed by two α-helices (α1, residues 13–37 from each subunit) in a left-handed coiled-coil antiparallel arrangement (with the long axes of the helices at an angle of ∼160°), on top of which lies a four-stranded antiparallel β-sheet (β2–β1–β1′–β2′) with two strands (β1, residues 41–48; β2, residues 52–58) from each subunit. The long axes of the α1 helices are oriented at ∼10° to the long axis of the β-sheet. The α1 helix is itself kinked at Gly26 and the angle between the two halves of the helix (residues 13–26 and residues 26–37) is ∼17°. The predominant feature of the MEF2S domain dimer consists of two short α-helices (α2, residues 61–71), one from each subunit, in an antiparallel coiled-coil arrangement (with the long axes of the two helices oriented at an angle of ∼145°). The α2 helix is connected to the MADS-box by a short turn (residues 59–60) and sits directly upon the β-sheet of the MADS-box dimer. The two helices of the MEF2S domain dimer, which are separated by ∼13 Å, are oriented at ∼90° to the long axis of the underlying MADS-box β-sheet and are directed upwards from the β-sheet at an angle of ∼20°. The C-terminal 12 residues (residues 74–85) are disordered in solution as judged by negative values of the 1H-{15N} NOEs from residue 75 onwards, and by the absence of any non-sequential inter-residue 1H-1H NOEs.
Three discontinuous surfaces form the dimer interface (Figure 2D). The first segment (residues 5–10) is located in the N-terminal extension of the MADS-box, the second segment (residues 16–47) comprises helix α1 and strand β1 of the MADS-box, and the third segment (residues 53–71) comprises strand β2 of the MADS-box and the MEF2S domain. The majority of intersubunit contacts occur between the N-terminal extension of one subunit and the C-terminal part of helix α1 of the other, between helices α1 of each subunit, between strands β1 of each subunit, between strand β1 and β2 of one subunit and the MEF2S domain of the other, and between residues of the MEF2S domain. The total accessible surface area (ASA) buried upon dimerization is ∼1600 Å2 per monomer. The intersubunit interactions between the MADS-boxes and between the MEF2 domains bury ∼1140 and ∼140 Å2 of ASA, respectively, per monomer. In addition, the interaction of the MADS-box of one subunit and the MEF2S domain of the other subunit buries ∼215 and ∼200 Å2 of ASA, respectively. The latter are comparable to the intrasubunit interactions between the MADS-box and the MEF2S domain, which bury ∼200 and ∼250 Å2 of ASA, respectively. Virtually all the intersubunit interactions are hydrophobic in nature. There is, however, a salt bridge between Arg9 of one subunit and Asp39′ of the other, and a hydrogen bond between the side chain of Gln55 and the hydroxyl of Tyr68′.
It is interesting to note that DNA-binding assays using different C-terminally truncated MEF2A derivatives indicates that the minimal domain capable of binding DNA with high affinity and specificity encompasses residues 1–73 (Figure 4). Indeed, the DNA-binding properties of the 1–73 construct (Figure 4, lane 4) are indistinguishable from those of the 1–85, 1–80 and 1–76 constructs (Figure 4, lanes 1–3). Further truncations (compare the 1–70 construct in Figure 4, lane 5) result in a large decrease in DNA-binding affinity. This is in complete agreement with the structural findings since residues 74–85 are found to be disordered in solution. Thus, one can conclude that residues 60–73 of the MEF2 domain (i.e. the MEF2S domain) are sufficient to stabilize and maintain the solubility of the MADS-box dimer, by ensuring that hydrophobic side chains protruding upwards from the β-sheet of the MADS-box (in the view shown in Figure 2B and C) are shielded from solvent. Since both Asn72 and Glu73 are solvent exposed and do not participate in any side chain–side chain interactions, Tyr71, which forms an intersubunit hydrophobic cluster with Phe47′ and Leu53′, must play an important role in stabilizing the structure.
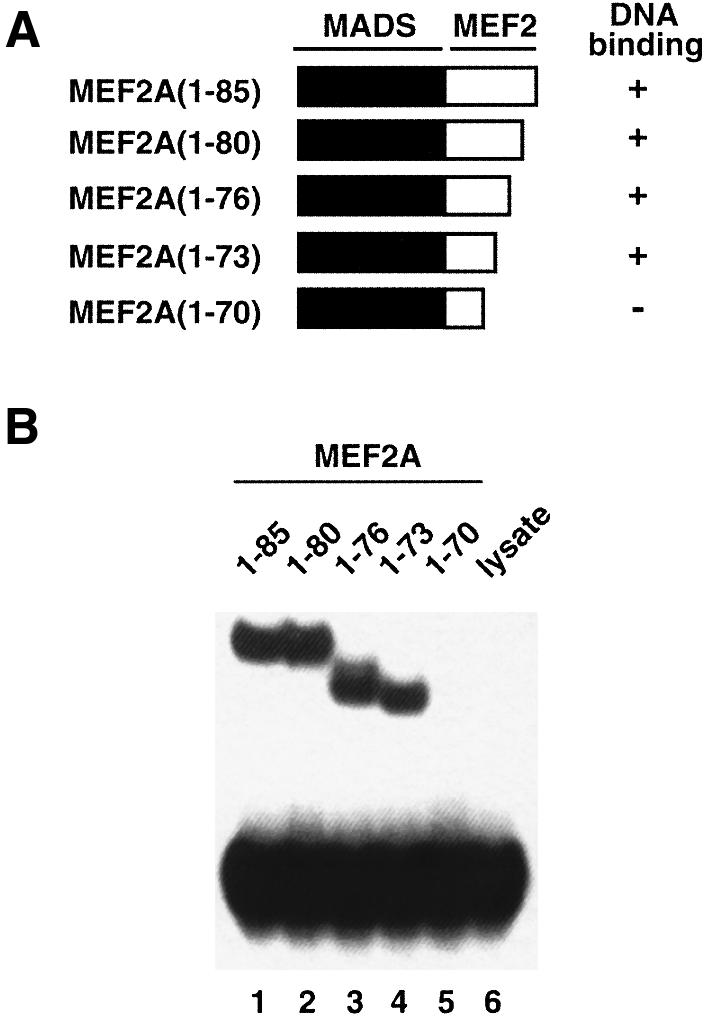
Fig. 4. Mapping the minimal MEF2A DNA-binding domain. (A) Schematic illustration of the truncated MEF2A constructs. (B) Gel retardation analysis of equimolar concentrations of in vitro translated MEF2A proteins bound to the N10 site (central motif CTATTTATAG; Sharrocks et al., 1993a). Approximately equal concentrations (∼50 nM) of protein and DNA were used. Lane 6 is a control containing unprogrammed rabbit reticulocyte lysate.
The DNA in the complex is B-type with average values for the helical rise, helical twist, tilt and roll of ∼3.5 Å, ∼36°, ∼0° and approximately –2°, respectively, and the majority of sugars are in a C2′-endo conformation. (The numbering scheme for the DNA is given in Figure 5B, lower left hand panel.) The propeller twist ranges from ∼1–2° for bp 10/–10 (C⋅G) and 4/–4 (T⋅A) to approximately –20° for bp 1/–1 (T⋅A). The propeller twist for the remaining base pairs ranges from –6 to –17°. The relatively large propeller twist angles (ranging from –14 to –20°) for the central stretch of six A⋅T base pairs (from bp –3 to +3) is correlated with compression and deepening of the minor groove, a concomitant expansion of the major groove, and a higher than average helical twist (40.1 ± 1.5°). Similar correlations have been observed previously in the SRF–DNA complex (Pellegrini et al., 1995). There is a small overall bend of ∼15° in the DNA centered about the 2-fold symmetry axis such that the face of the DNA presented to the protein is slightly concave (Figures 2C and 3A). This bend is accompanied by a relatively large roll angle of approximately –11° for the 1 to –1 base pair step at the 2-fold axis (Figure 2C). In addition, there are two ∼20° bends in opposing directions centered around bp 5/–5, which are directed out of and into the plane of the paper, respectively, in the view shown in Figure 2C. These two bends are most readily seen when viewing the complex from the underside of the DNA (Figure 3C), and are associated with a large positive roll angle (∼10°) for base pair steps 6/–6 to 5/–5, and a large negative roll angle (approximately –17°) for base pair steps 5/–5 to 4/–4. Since these two bends are in opposing directions, they do not alter the overall direction of the DNA (Figure 3C).
All protein–DNA contacts are confined to the N-terminal extension and helix α1 of the MADS-box (Figure 5). The interactions involving the N-terminal extension occur exclusively in the minor groove of the 5 bp on either side of the 2-fold symmetry axis. The majority of contacts involve the sugar–phosphate backbone, with the exception of the N-terminal amino group of Gly1, which is hydrogen bonded to the O2 atom of T3, the guanidino group of Arg2, which is hydrogen bonded to the O2 atom of T4′ (as well as the O4′ atom of the sugar ring of T3′), and the aliphatic portion of the side chain of Arg2, which is in hydrophobic contact with the base of A4. (Note the contacts given relate to a single subunit; the other subunit interacts with the symmetrically related bases of the other half of the DNA palindrome.) The backbone of Gly1 and the side chain of Arg2 lie deep within the minor groove and are oriented parallel to the DNA backbone, whereas the side chains of Lys3 and Lys4 span the minor groove, are oriented orthogonal to the DNA backbone, and contact the phosphates of G5 and A1′, respectively (Figure 5C). Hydrophobic contacts with the DNA are provided by the methyl groups of Ile5 and Ile10, the methylene groups of Arg2, Lys3 and Lys4, and the backbone of Gly1. In contrast to the N-terminal extension, the residues in helix α1 make exclusive contact with the major groove, again principally with the sugar–phosphate backbone. The N-terminal end of helix α1 (residues 13–26) lies in the major groove, oriented at an angle of ∼10° to the phosphate backbone, while residues 27–30 of helix α1 run along the phosphate backbone. Helix α1 spans bp 1–7. There are only four hydrogen bonding interactions (per MEF2A subunit) with the DNA bases in the major groove that are apparent in the present structure: namely, from the guanidino group of Arg14 to the N7 and O6 atoms of G7′, and from the NζH3+ of Lys22 to the N7 and O6 atoms of A4 and G5, respectively. (Note that these hydrogen bonds are inferred from the ensemble of structures and the positions of these side chains are determined from the NOE data.) The methylene groups of the Lys22 side chain also provide hydrophobic contacts with the base of A4. Electrostatic contacts between Arg14(Nε) and C8′(P), Asn15(Nδ) and G5(P), Thr19(Oγ) and A4(P), Arg23(guanidino) and T3(P) and/or A4(P), Gly26(NH) and T3(P), K29(Nζ) and A2(P), and K30(Nζ) to A2(P) and/or T3(P) further stabilize the complex.
Relationship to other MADS-box family proteins
X-ray structures of two protein–DNA complexes of the MADS-box family have been solved previously: a binary complex of the human SRF core domain and a DNA 19mer (Pellegrini et al., 1995), and a ternary complex of the yeast MCM1 core domain, the homeodomain protein MATα2 and a DNA 26mer (Tan and Richmond et al., 1998). The SRF and MCM1 core sequences in the crystal structures span residues 132–223 and 1–100, respectively, with the MADS-box (equivalent to residues 1–58 of MEF2A) comprising residues 142–199 and 17–74, respectively. The structures of SRF and MCM1 are very similar and both proteins employ similar modes of recognition and effect similar degrees of bending (∼70–75°). The mechanisms for generating the central part of the DNA bend appear virtually identical in the two complexes, although a small additional bend in the MCM1–MATα2–DNA complex is observed around base pair 8/–8 (in our numbering scheme). For the sake of simplicity, we have limited the following discussion to a comparison of the SRF–DNA and MEF2A–DNA complexes. For the purposes of clarity we refer to residues and bases in the SRF–DNA complex by their equivalent numbering scheme in the MEF2A–DNA complex. A comparison of the two structures and the DNA contacts is given in Figures 3 and 5.
The structures of the MADS-boxes in MEF2A and SRF are very similar. The backbone Cα r.m.s. difference between the two MADS-boxes is 1.3 Å for the dimer (residues 2–58, Figure 3E), which is of the magnitude expected for two domains with 39% sequence identity (Figure 5A). The structure of the region C-terminal to the MADS-box, however, is quite different in the two proteins (Figure 3E–G). Whereas SRF, AG and MCM1 have a 22 residue SAM domain (residues 59–80), the MEF2 family has a shorter 15 residue, structurally ordered MEF2S domain (residues 59–73). [The complete MEF2 domain extends for another 12 residues, but residues 74–85 are disordered in solution and not required for high affinity DNA binding (Figures 4 and 5A).] The percentage sequence identity between the MEF2S and SAM domains is <5%. In the MEF2S domain there is a helix (α2) from residues 61–71 followed by a two residue coil (Figure 3F). In contrast, in the SAM domain, residues 60–67 form a coil, followed by an α-helix from residues 69–79 (Figure 3G). The presence of a proline at position 67 in the SAM domain (Figure 5A) presumably hinders the formation of an α-helix at the location found in the MEF2S domain. In addition, the orientation of the α2 helix with regard to the underlying sheet of the MADS-box differs by ∼50° in the MEF2S and SAM domains and, more importantly, the α2 helices of the corresponding subunits are located on opposite ends of the underlying β-sheet (Figure 3E–G). In terms of dimer contacts, the α2 helices from each subunit of the MEF2S domain form an antiparallel coiled-coil separated by ∼13 Å (Figures 2C and 3F). The helices of the SAM domain are also oriented approximately antiparallel, but only interact via their N-terminal residues, while the C-terminal end of the helix interacts with the coil of the other subunit (Figure 3G).
The relative contributions of the MEF2S and SAM domains in stabilizing the dimeric structure of the MEF2A and SRF MADS-boxes, respectively, appear to be different. It has been shown that chimeras comprising a MEF2A MADS-box and a SAM domain (MEF:SRF) or an SRF MADS-box and a MEF2 domain (SRF:MEF) retain the DNA-binding specificities of their respective MADS-boxes (West et al., 1997). However, the affinity of the SRF:MEF chimera is severely reduced, whereas that of the MEF:SRF chimera is essentially unchanged relative to MEF2A(1–85) (West et al., 1997). This implies that the MADS-box of MEF2A dimerizes more efficiently than that of SRF and requires fewer stabilizing interactions afforded by the C-terminal residues comprising either a MEF2 or SAM domain. This is consistent with the present findings that the structured MEF2S domain is significantly shorter and has a smaller dimer interface than the SAM domain. The ability of the SAM domain to replace the MEF2 domain in MEF2A can be accounted for by two observations: first, although there is no sequence identity for residues 60–67 between the SAM and MEF2S domains, the distribution of hydrophobic and hydrophilic residues is similar; secondly, the sequence identity for the residues pointing upwards from the β-sheets of the MADS-box in MEF2A and SRF is very high (Figure 5A). Thus, it is not surprising that the SAM domain is capable of interacting with the underlying MADS-box of MEF2A.
The MEF2 and SAM domains are thought to contain sites of interaction for other proteins. Indeed, in the case of SRF, the binding surface for the transcription factor Elk-1 has been mapped to a hydrophobic patch comprising Val53, Thr55, Thr58 and Ile65 formed by strand β2 of the MADS-box and the N-terminal coil of the SAM domain (Ling et al., 1998). This surface is located on the front left side of the views shown in Figure 3B and E. Interestingly, Elk-1 and other transcription factors known to bind to SRF do not bind to MEF2A (Shore and Sharrocks, 1994; Ling et al., 1998). Likewise, the binding partners of MEF2A, such as the recently described protein MITR (Sparrow et al., 1999), do not bind to SRF. This is hardly surprising since the molecular surfaces presented by the MEF2S and SAM domains are different in both shape and charge distribution (compare Figure 3A and B). The surface on MEF2A that corresponds to the Elk-1-binding site on SRF is concave and comprises Phe47, Asn51 and Leu53 of the MADS-box of one subunit and Lys67′ and Tyr71′ of the MEF2S domain of the other (compare Figure 3A and E). An alternative potential site of interaction for MEF2-specific interacting proteins is the deep groove located between the two α2 helices of the MEF2S domain dimer (compare top of Figure 3A). Relevant residues in this location are Leu65, Leu66, Tyr68, Thr69 and Asn72.
Modulation of sequence-specific recognition by MEF2A and SRF
There are a number of critical differences in the protein–DNA interactions observed in the MEF2A and SRF complexes: in particular, the N-terminal extension and its contacts in the minor groove, the interactions in the major groove outside the central 10 bp involving helix α1 and the β-turn that connects stands β1 and β2, and the degree of DNA bending (Figures 3A, B and 5B). On the other hand, the contacts in the major groove between residues of helix α1 and the central 8 bp, as well as the conformation of the central 8 bp (from −4 to 4) of DNA, are similar in the two complexes (Figures 3E and 5B).
Although the sequences of the first five residues of the MADS-box of MEF2A and SRF only differ at position 3 (Lys in MEF2A and Val in SRF), the N-terminal extension makes distinctive contacts at the MEF2A- and SRF-binding sites (Figures 3A, B and 5B). The origin for this difference can be attributed to the presence in SRF and absence in MEF2A of residues N-terminal to Gly1. Indeed, a study on the DNA-binding specificity determinants in MADS-box transcription factors has shown that residues immediately N-terminal to the MADS-box of SRF are important determinants of both DNA-binding affinity and specificity, such that deletion of these residues results in an SRF mutant that recognizes MEF2A-binding sites instead of SRF ones (Sharrocks et al., 1993a; Nurrish and Treisman, 1995). The placement of Lys3 (MEF2A)/Val3 (SRF), Lys4 and Ile5 is very similar in the two complexes (Figure 5B and C). In addition, both Gly1 and Arg2 are buried deep within the minor groove. However, the direction of the polypeptide chain for Gly1 and Arg2 is reversed, such that the side chain of Arg2 (which has an extended conformation) and the backbone of Gly1 in SRF occupy the same position as the backbone of Gly1 and the side chain of Arg2, respectively, in MEF2A. As a consequence, Arg2 in MEF2A contacts the bases of A4 and T4′, and the sugars of G5 and A3′, while the N-terminal amino group of Gly1 is hydrogen bonded to the base of T3 (Figure 5B and C). Note that the position of Gly1 is determined unambiguously by NOEs from the Hα protons of Gly1 to sugar protons of A3′ and T2′ (Figure 1A and B), as well as to the H2 protons of A2 and A4 (not shown). In SRF, on the other hand, Arg2 is hydrogen bonded to the bases of T2, A2′ and A1′, while the backbone of Gly1 is in contact with the sugar of T3′ (Figure 5B). The direction of the main chain adopted by residues 1 and 2 in MEF2A is precluded in SRF by the presence of residues N-terminal to the MADS-box. Indeed, Arg(–1) and Thr(–2) in SRF both make contacts with the DNA (in particular the sugars of G5 and C4′, respectively; compare Figure 5B). However, even if these residues did not contact the DNA, the conformation of the N-terminal extension observed in MEF2A would still be excluded owing to steric clash. The conformation of the N-terminal extension observed in the MCM1 complex is the same as that seen for SRF. In the case of MCM1, not only are there residues N-terminal to the MADS-box, but Gly1 is replaced by Glu. Hence, even if the MCM1 sequence started at residue 1, the backbone of residues 1 and 2 of the N-terminal extension would still adopt the conformation observed in the SRF and MCM1 complex since the one observed in MEF2A would be highly unfavorable owing to steric clash and deleterious electrostatic interactions generated by the side chain of Glu1.
The above observations suggest that the interactions between Gly1 and the DNA and between the side chain of Arg2 and the DNA are key determinants in specificity and are modulated by the presence or absence of residues N-terminal to the MADS-box. The key difference between the MEF2A and SRF/MCM1 DNA sites lies in the nature of bp 4/–4 and 3/–3 of the palindromic 10 bp consensus sequence (Treisman, 1990; Pollock and Treisman, 1991). In MEF2A sites, positions 4/–4 and 3/–3 are occupied by invariant T⋅A and A⋅T base pairs, respectively; in SRF (and MCM1) sites, on the other hand, position 4/–4 is an invariant C⋅G base pair, while position 3/–3 is occupied by either an A⋅T or T⋅A base pair (Figure 5B). In our structure, which lacks an N-terminal Met, Gly1 and Arg2 of MEF2A are responsible for recognizing the T bases in the 3/–3 (A⋅T) and 4/–4 (T⋅A) positions of the consensus sequence, respectively (Figure 5B and C); in SRF and MCM1, on the other hand, Gly1 does not make any base-specific contacts, and the side chain of Arg2 is hydrogen bonded to the variant A⋅T base pairs at positions 2/–2 and 1/–1 of the consensus sequence (Figure 5B).
It has been suggested previously, on the basis of the SRF–DNA structure, that the N-terminal methionine of MEF2A might specify sequence preference of this protein for the A⋅T base pair at the 4/–4 position (Pellegrini et al., 1995). Thus, although recombinant MEF2A produced in Escherichia coli lacks an N-terminal methionine, the question remains as to whether the N-terminal methionine is actually absent in MEF2A produced in vivo in mammalian cells. Several additional lines of evidence suggest that this is indeed the case. First, MEF2A produced in E.coli still discriminates between MEF2 and SRF DNA-binding sites and behaves in an analogous manner to MEF2A protein that has been either produced by in vitro translation in rabbit reticulocyte lysates (A.D.Sharrocks, unpublished results; West et al., 1997) or immunoprecipitated from mammalian cell extracts (Dodou et al., 1995). Secondly, MEF2A is inactive in DNA binding when expressed as a C-terminal fusion with glutathione S-transferase or when cleaved from this fusion protein to leave the additional residues Gly-Ser-Met at the N-terminus of MEF2A (Sharrocks, 1994). This suggests that the inclusion of an N-terminal methionine may in fact inhibit DNA binding of MEF2A as predicted from our structure, rather than acting to determine its unique DNA-binding specificity.
In addition to sequences N-terminal to the MADS-box, amino acids within the MADS-box have been implicated in determining the different DNA-binding specificities of MEF2A and SRF (Sharrocks et al., 1993a; Nurrish and Treisman, 1995). While removing the N-terminal region of SRF switches its specificity towards that of MEF2A, inclusion of the additional mutation of Lys13→Glu is required to complete the specificity switch. The inclusion of another mutation, Val3→Lys, further enhances the affinity of the mutated SRF protein. In MEF2A, Glu13 does not make direct DNA contacts but rather appears to act to repulse the DNA away from the sides of the protein (in the view shown in Figure 3A). In contrast, in SRF, Lys13 binds to the DNA and stabilizes its bent conformation (see discussion below and Pellegrini et al., 1995). Hence, Glu13 indirectly affects DNA-binding specificity, probably due to the change in DNA conformation, which subtly alters the specific protein–DNA interactions observed. Lys3 in MEF2A makes more extensive contacts with the DNA than Val3 in SRF (Figure 5B), suggesting that this might contribute to the relative enhancement in binding affinity seen. This residue, however, might also be indirectly involved in stabilizing the interactions of Arg2 with the DNA. Finally, it is also interesting to note that although Arg14 makes two hydrogen bonds to G7′ (Figure 5B and C), no strong preference for a particular base pair at this position is observed in site-selection studies (Pollock and Treisman, 1991), indicating that despite contributing one-third of the base-specific hydrogen bonds, this interaction does not play a major role in specificity determination.
It is noteworthy that the Arg16→Ala and Arg16→Lys mutations in MEF2A do not affect DNA binding, while the analogous mutation Arg16→Lys in SRF results in a protein with severely reduced DNA-binding efficiency (West et al., 1997). In MEF2A, Arg16 and Gln17 form potential hydrogen bonds with Ser49 and Ser50, respectively. Similarly, in SRF, Arg16 and Tyr17 form hydrogen bonds with Glu49 and Thr50, respectively. Thus, these interactions are involved in orienting the β-loop relative to the underlying α1 helix. One might therefore suppose that the Arg16→Lys mutation in SRF would be rather an innocuous one. The conformation and orientation of the β-loop, however, are critical to SRF’s ability to bend DNA since Thr50 and His52 interact with the phosphate backbone. In contrast, the DNA in MEF2A is barely distorted and the β-loop is not involved in any interactions with the DNA. Consequently, any mutation that perturbs the orientation of the β-loop relative to the underlying helix would be expected to affect DNA binding of SRF but have little or no effect in the case of MEF2A.
Modulation of DNA bending by MEF2A and SRF
Previous biochemical studies have shown that SRF bends the DNA significantly while MEF2A does not (West et al., 1997). These results are entirely consistent, both qualitatively and quantitatively, with the NMR (this paper) and X-ray (Pellegrini et al., 1995) structural studies, which show an overall DNA bend of ∼15 and ∼70–75°, respectively, for the MEF2A–DNA and SRF–DNA complexes. In addition, biochemical work has also suggested an important role for residues 11–14 as determinants of specificity in MEF2A (Sharrocks et al., 1993a) and a critical role in DNA bending played by the N-terminal residue of helix α1, namely Glu13 and Lys13 in MEF2A and SRF, respectively (West et al., 1997). Indeed, the introduction of the mutation Lys13→Glu into SRF severely disrupts its ability to mediate DNA bending. The overall bending of the DNA in SRF is achieved by three individual bends that co-add: one bend of ∼15° at the dyad axis of the DNA, and two bends of ∼30° on either side of the central 8 bp (Figure 3B, D and E). The latter two bends direct the path of the DNA along the side of SRF such that Lys13, Thr18 and Ser21, and Lys24 of helix α1 contact the phosphate of C10′, the base of T8′ and the phosphate of T8′, respectively, while Thr50 and His52 of the β-loop contact the phosphate of A9′ (Figures 3B, E and 5B). In contrast, in the MEF2A complex the bends outside the central 8 bp do not co-add (Figure 3C), and the overall bend is contributed by only a single bend of ∼15° at the dyad axis of the DNA (Figure 3A and E). Associated with this is the absence of any contacts between the β-loop of MEF2A and the DNA (Figures 3A, E and 5). Indeed, the only contacts outside the central 10 bp in the MEF2A complex involve the side chain of Arg14 and the bases of C8′ and G7′ (including two hydrogen bonds from the guanidino group of Arg 14 to G7′) and the sugar–phosphate of C8′ (including a hydrogen bond from the Nε atom of Arg14 to the phosphate of C8′) (Figures 3A, 5B and C). Hydrogen bonding interactions involving residue 14 are precluded in SRF since this position is occupied by a Leu. The electrostatic surfaces of MEF2A and SRF depicted in Figure 3A and B, respectively, highlight the role of the N-terminal residues of helix α1: in MEF2A the presence of a negatively charged Glu at position 13 precludes an upward path of the DNA along the side of the protein owing to unfavorable electrostatic interactions with the phosphate backbone of the DNA, while the presence of the positively charged Arg14 directs the DNA along an essentially linear path. In SRF, on the other hand, the positively charged Lys13 can readily interact with the phosphate backbone, thereby bringing the DNA into close proximity with residues in the β-loop. These interactions are entirely consistent with the observation that the introduction of negatively charged residues into the end of helix α1 and the β-loop of SRF and other MADS-box proteins severely disrupts protein-induced DNA bending (West et al., 1997; West and Sharrocks, 1999; A.G.West and A.D.Sharrocks, unpublished results).
Concluding remarks
We have solved the structure of a specific MEF2A–DNA complex by multidimensional NMR. Specificity is achieved by interactions in the minor groove involving the N-terminal extension and interactions in the major groove involving helix α1. A comparison of the structures of the MEF2A, SRF and MCM1–DNA complexes reveals a key role played by residues N-terminal to the MADS-box in determining specificity and mode of DNA binding, and in the sequence of the N-terminal residues of helix α1 in modulating DNA bending. Moreover, a molecular explanation of links observed between DNA bending and DNA specificity determination is provided. Recently, it has become apparent that a large number of co-regulatory proteins interact with the core DNA-binding domain of the MEF2 proteins, including myogenic bHLH proteins (Molkentin and Olson, 1996), the protein kinase Erk5/BMK (Yang et al., 1998), the co-repressor protein MITR (Sparrow et al., 1999) and the calcium-dependent inhibitory protein Cabin1 (Youn et al., 1999). The availability of the structure of the complex of the DNA-binding domain of MEF2A bound to its specific target site DNA is particularly important in enabling the molecular basis of these interactions to be probed in a rational manner.
Materials and methods
Expression, purification and preparation of the MEF2A protein and the MEF2A–DNA complex
The human MEF2A protein spanning the region from Gly1 to Glu85 was expressed using the pET11a vector and E.coli BL21 (DE3) (Novagen Inc., WI). To avoid cysteine thiol oxidation, residues Cys38 and Cys40 were substituted by Ala using the Quick-Change mutagenesis protocol (Strategene, CA). The nucleotide sequence of the cloned DNA was confirmed by sequencing. It was also confirmed by mass spectroscopy that the N-terminus of the purified MEF2A protein starts with a Gly residue. 1H-15N correlation spectroscopy and gel retardation assays of the wild-type and C38A,C40A mutant MEF2A–DNA complexes indicated that the two Cys to Ala substitutions had no effect on either the structure of MEF2A or its DNA-binding affinity.
Cells were grown at 37°C either in Luria–Bertani medium or in a modified minimal medium for uniform (>95%) 15N and/or 13C labeling with 15NH4Cl and/or [13C6]glucose as the sole nitrogen and carbon sources, respectively. Cells were suspended in 20 vols of buffer A (50 mM Tris–HCl pH 8.2, 10 mM EDTA) and lysed by sonication at 4°C in the presence of 100 µg/ml lysozyme. The insoluble recombinant protein (inclusion bodies) was washed by resuspension in buffer B (buffer A containing 2 M urea and 1% Triton X-100) and then in buffer A. In both cases, the inclusions were pelleted by centrifugation at 20 000 g for 30 min at 4°C. The final pellet was solubilized in 50 mM Tris–HCl pH 8.0, 7.5 M guanidine–HCl, 5 mM EDTA, and applied to a Superdex-75 column (HiLoad 2.6 × 60 cm; Amersham Pharmacia Biotech, NJ) equilibrated in 50 mM Tris–HCl pH 8.0, 4 M guanidine–HCl, 5 mM EDTA, and eluted at a flow rate of 3 ml/min at ambient temperature. MEF2A protein was folded at 4°C by gradually dispensing the protein (7 ml/h) into buffer C (20 mM sodium phosphate pH 6.6, 0.02 mM EDTA, 0.5 M NaCl) to attain a final concentration of ∼0.1 mg/ml protein and 0.1 M guanidine–HCl. The protein was mixed with a 1.1-fold excess of double-stranded oligonucleotide 5′d-CTCGGCTATTAATAGCCGAG (Midland Certified Reagent Company, TX) in buffer C but without NaCl. The complex was concentrated and purified on a Superdex-200 column (HiLoad 2.6 × 60 cm). Peak fractions corresponding to the complex were pooled and concentrated. Samples for NMR contained ∼1 mM MEF2A–DNA complex in 20 mM sodium phosphate pH 6.6, 0.02 mM EDTA and 0.02% NaN3.
Preparation of uniformly 13C/15N-labeled double-stranded MEF oligonucleotide
The MEF2A oligonucleotide 5′d-CTCGGCTATTAATAGCCGAG was synthesized as a tandem repeat 5′d-CCTCGGCTATTAATAGCCGAGGCCTCGGCTATTAATAGCCGAGG (2× MEF2A oligonucleotide) separated by an HaeIII restriction site. Uniformly 15N/13C-labeled double-stranded MEF2A oligonucleotide was synthesized and purified using the Endonuclease Sensitive Repeat Amplification (ESRA) procedure (Louis et al., 1998). An initial amplification reaction of 9.6 ml (120 ng of 2× MEF2A oligonucleotide, 0.08 mM 15N/13C-labeled dNTPs and 2 U of Vent polymerase per 100 µl of reaction mixture) was cycled 4× at 95°C for 1 min and 85°C for 30 min. This amplified mixture was used to further amplify 192 ml of reaction mixture using the same concentration of labeled dNTPs and polymerase. The latter PCR was cycled 20×: 95°C for 1 min, 55°C for 2 min and 72°C for 3 min. After digestion of the PCR products with HaeIII, the double-stranded MEF2A oligonucleotide was purified by anion-exchange chromatography, followed by size-exclusion chromatography on a Superdex-200 column. [15N]MEF2A protein complexed to [15N/13C]doubled-stranded MEF2A oligonucleotide was prepared and purified in the same way as described above for the isotopically labeled MEF2A-unlabeled double-stranded oligonucleotide complex.
DNA-binding assays
DNA-binding assays were carried out as described previously (Sharrocks et al., 1993b) with in vitro translated MEF2A derivatives derived from linear PCR products (Dalgleish and Sharrocks, 2000). The following primer pairs were used: For/Rev [MEF2A(1–85)], ADS587/Rev [MEF2A(1–80)], ADS588/Rev [MEF2A(1–76)], ADS589/Rev [MEF2A(1–73)], ADS590/Rev [MEF2A(1–70)] on the template pAS68.
NMR spectroscopy
Multidimensional NMR experiments were carried out at 35°C on DMX500, DMX600, DMX750 spectrometers equipped with x,y,z-shielded gradient triple resonance probes. Spectra were processed with the NMRPipe package (Delaglio et al., 1995) and analyzed using the programs PIPP, CAPP and STAPP (Garrett et al., 1991). Sequential assignment of 1H, 15N and 13C protein chemical shifts was achieved by means of through-bond heteronuclear scalar correlations along the backbone and side chains (Clore and Gronenborn, 1991, 1998a; Bax and Grzsiek, 1993) using 3D HNCACB, CBCACONH, C(CCO)NH, H(CCO)NH, HCCH-COSY, HCCH-TOCSY and CCH-COSY experiments. Assignment of 1H, 15N and 13C DNA chemical shifts was obtained from analysis of 2D 12C-filtered NOE and HOHAHA experiments recorded on a 1:1 complex of [15N/13C]MEF2A and unlabeled DNA, and from 3D 15N-separated and 13C-separated NOE spectra recorded on a 1:1 complex of [15N]MEF2A and [15N/13C]DNA, using conventional sequential assignment methodology for nucleic acids (Clore and Gronenborn, 1989). 3JHNα, 3JNCγ (aromatic, methyl and methylene), 3JC′Cγ (aromatic, methyl and methylene), JC′C′ and 3JCαCδ couplings were measured by quantitative J correlation spectroscopy (Bax et al., 1994). Backbone φ and ψ torsion angle restraints were derived from 3JHNα and 3JC′C′ coupling constants, in combination with a database search procedure based on 15N, NH, 13Cα, 13Cβ, 13C′ and Hα secondary shifts using the program TALOS (Cornilescu et al., 1999). Side chain torsion angle restraints were derived from analysis of the NOE/ROE and three-bond heteronuclear scalar coupling data. Residual 1DNH and 1DCH dipolar couplings were determined from the difference in 1JNH and 1JCH couplings for a 1:1 complex of [15N]MEF2A and [15N/13C]DNA measured at 35°C in a liquid crystalline medium of 5% 3:1 1,2-ditridecanoyl-sn-glycerol-3-phosphocholine:1,2-di-O-hexyl-sn-glycerol-3-phosphocholine (Ottiger and Bax, 1999) and in isotropic medium using 2D IPAP 1H-coupled {15N, 1H}-HSQC (Ottiger et al., 1998) and 1H-coupled 2D 1H-13C HSQC experiments. The magnitudes of the axial (DaNH) and rhombic (R) components of the alignment tensor DNH were obtained by examining the distribution of normalized residual dipolar couplings (Clore et al., 1998). The values of DaNH and R are –20.0 Hz and 0.39, respectively, and the normalized (to the N–H) dipolar couplings span a range of values from –29.4 to +31.9 Hz. Interproton distance restraints within the protein were derived from a 3D 15N-separated NOE spectrum recorded on a 1:1 complex of [15N]MEF2A and unlabeled DNA; and from 3D 13C-separated NOE, 4D 13C/13C-separated NOE and 4D 13C/15N-separated NOE spectra recorded on a 1:1 complex of [15N/13C]MEF2A and unlabeled DNA. Interproton distance restraints within the DNA were derived from 2D 12C-filtered NOE spectra recorded on a 1:1 complex of [15N/13C]MEF2A and unlabeled DNA, and 3D 15N-separated and 13C-separated NOE spectra recorded on a 1:1 complex of [15N]MEF2A and [15N/13C]DNA. Intermolecular interproton distance restraints were derived from 3D 13C-separated/12C-filtered NOE spectra recorded on a 1:1 complex of [15N/13C]MEF2A and unlabeled DNA and a 1:1 complex of [15N]MEF2A and [15N/13C]DNA, and from a 3D 15N-separated NOE spectrum recorded on a 1:1 complex of [15N]MEF2A and unlabeled DNA.
Structure calculation
Approximate interproton distance and torsion angle restraints were derived from the NOE and coupling constant data as described by Omichinski et al. (1997). Broad torsion angle restraints for the DNA backbone, covering the ranges characteristic of both A- and B-DNA, were employed to prevent problems associated with local mirror images: α = –70 ± 50, β = 180 ± 50, γ = 60 ± 35, ε = 180 ± 50 and ζ = –85 ± 50° (Omichinski et al., 1997). These restraints are justified since both the pattern of NOEs and the 31P spectrum (Gorenstein, 1994) for the DNA in the complex are typical of B-DNA. In addition, the δ angle was restrained to 145 ± 25° in those cases (18 out of 20) where the NOE data indicated that the sugar pucker was unambiguously C2′-endo. Approximate interproton distance restraints for the protein were grouped into four distance ranges, 1.8–2.7 Å (1.8–2.9 Å for NOEs involving NH protons), 1.8–3.3 Å (1.8–3.5 Å for NOEs involving NH protons), 1.8–5.0 Å and 1.8–6.0 Å, corresponding to strong, medium, weak and very weak NOEs, respectively. Approximate interproton distance restraints for the DNA were classified into five ranges, 1.8–2.5, 1.8–3.0, 1.8–3.5, 2.3–5.0 and 3.5–6.0 Å, corresponding to strong, medium-strong, medium, weak and very weak NOEs, respectively. Distances involving ambiguous NOEs, non-stereospecifically assigned methylene protons, methyl groups and Hδ and Hε protons of Tyr and Phe, were represented as a (Σr–6)–1/6 sum (Nilges et al., 1993). A qualitative interpretation of the NOE data indicated unambiguously that the two subunits of the MEF2A dimer were oriented antiparallel to each other. Consequently, as described by Clore et al. (1990) and Lodi et al., (1994), it was relatively straightforward to distinguish intra- from inter-subunit NOEs since the latter were inconsistent with the structure of the monomer (i.e. they corresponded to distances >>5 Å in the monomer). Initially, any NOEs that could potentially arise from both intra- and inter-subunit contacts were treated as ambiguous NOE restraints in the form of a (Σr–6)–1/6 sum; subsequently, all the intersubunit NOEs were resolved during the course of iterative refinement. The structures were calculated by simulated annealing (Nilges et al., 1988) with the protocol described by Omichinski et al. (1997) using the program XPLOR (Brünger, 1993) modified to incorporate pseudopotentials for 3JHNα coupling constants, secondary 13Cα and 13Cβ chemical shifts, residual dipolar couplings and a conformational database potential for proteins and nucleic acids (Clore and Gronenborn, 1998b). Non-bonded contacts were represented by a quartic van der Waals repulsion term (Nilges et al., 1988), and no hydrogen bonding, electrostatic or 6–12 Lennard–Jones empirical potential energy terms are present in the target function used for simulated annealing or restrained minimization. The final force constants for the various terms in the target function used for simulated anneal ing are as follows: 1000 kcal/mol Å2 for bond lengths, 500 kcal/mol rad2 for angles and improper torsions (which serve to maintain planarity and chirality), 4 kcal/mol Å4 for the quartic van der Waals repulsion term (with the van der Waals radii set to 0.8 times their values used in the CHARMM PARAM19/20 protein and PARNAH1ER1 nucleic acid parameters), 100 kcal/mol Å2 for the non-crystallographic symmmetry restraint (which ensures that the two halves of the complex are symmetric), 30 kcal/mol Å2 for the experimental distance restraints (interproton distances and hydrogen bonds), 200 kcal/mol rad2 for torsion angle restraints, 1 kcal/mol Hz2 for coupling constant restraints, 0.5 kcal/mol p.p.m.2 for the secondary 13C chemical shift restraints, 1 kcal/mol Hz2 for the dipolar coupling restraints, and 1.0 for the conformational database potential. The distance and torsion angle restraints are represented by a square-well potential, while the covalent geometry, chemical shift, coupling constant and dipolar coupling restraints are represented by harmonic potentials.
Structural DNA parameters were analyzed using the program COMPDNA (Gorin et al., 1995). Electrostatic calculations were performed with GRASP (Nicholls et al., 1991). Structure figures were generated using the programs MOLMOL (Koradi et al., 1996) and GRASP (Nicholls et al., 1991).
The coordinates have been deposited in the Brookhaven Protein Data Bank (RCSB accession code 1C7U).
Acknowledgments
Acknowledgements
We thank Dan Garrett and Frank Delagio for software support; Rolf Tschudin for technical support; Lewis Pannell for mass spectrometry; and Carole Bewley, John Kuszewski, Mary Starich and Victor Zhurkin for useful discussions. A.D.S. is a Research Fellow of the Lister Institute of Preventative Medicine. This work was supported by the AIDS Targeted Antiviral Program of the Office of the Director of the National Institutes of Health (to G.M.C.) and by the BBSRC (to A.D.S.).
Note added in proof
Following submission of this paper a crystal structure of the MEF2A core bound to DNA was published [Santelli,E. and Richmond,T.J. (2000) Crystal structure of MEF2A core bound to DNA at 1.5 Å resolution. J. Mol. Biol., 297, 437–449]. The NMR and crystal structures are in agreement.
References
- Bax A. and Grzesiek,S. (1993) Methodological advances in protein NMR. Acc. Chem. Res., 26, 131–138. [Google Scholar]
- Bax A., Vuister,G.W., Grzesiek,S., Delaglio,F., Wang,A.C., Tschduin,R. and Zhu,G. (1994) Measurement of homo- and heteronuclear J couplings from quantitative J correlation. Methods Enzymol., 239, 79–106. [DOI] [PubMed] [Google Scholar]
- Black B. and Olson,E.N. (1998) Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu. Rev. Cell Dev. Biol., 14, 167–196. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. (1993) XPLOR: A System for X-ray Crystallography and NMR. Yale University Press, New Haven, CT. [Google Scholar]
- Clore G.M. and Garrett,D.S. (1999) R-factor, free R and complete cross-validation for dipolar coupling refinement of NMR structures. J. Am. Chem. Soc., 121, 9008–9012. [Google Scholar]
- Clore G.M. and Gronenborn,A.M. (1989) Determination of three-dimensional structures of proteins and nucleic acids in solution by nuclear magnetic resonance spectroscopy. CRC Crit. Rev. Biochem. Mol. Biol., 24, 479–564. [DOI] [PubMed] [Google Scholar]
- Clore G.M. and Gronenborn,A.M. (1991) Structures of larger proteins in solution: three- and four-dimensional heteronuclear NMR spectroscopy. Science, 252, 1390–1399. [DOI] [PubMed] [Google Scholar]
- Clore G.M. and Gronenborn,A.M. (1998a) Determining structures of larger proteins and protein complexes by NMR. Trends Biotech., 16, 22–34. [DOI] [PubMed] [Google Scholar]
- Clore G.M. and Gronenborn,A.M. (1998b) New methods of structure refinement for macromolecular structure determination by NMR. Proc. Natl Acad. Sci. USA, 95, 5891–5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore G.M., Appella,E., Yamada,M., Matsushima,K. and Gronenborn,A.M. (1990) Three-dimensional structure of interleukin-8 in solution. Biochemistry, 29, 1689–1696. [DOI] [PubMed] [Google Scholar]
- Clore G.M., Gronenborn,A.M. and Bax,A. (1998) A robust method for determining the magnitude of the fully asymmetric alignment tensor of oriented macromolecules in the absence of structural information. J. Magn. Reson., 133, 216–222. [DOI] [PubMed] [Google Scholar]
- Cornilescu G., Delaglio,F. and Bax,A. (1999) Protein backbone angle restraints from searching a database for protein chemical shift and sequence homology. J. Biomol. NMR, 13, 289–302. [DOI] [PubMed] [Google Scholar]
- Dalgleish P. and Sharrocks,A.D. (2000) The mechanism of complex formation between Fli-1 and SRF transcription factors. Nucleic Acids Res., 28, 560–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F., Grzesiek,S., Vuister,G.W., Zhu,G., Pfeifer,J. and Bax,A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX PIPES. J. Biomol. NMR, 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Dodou E., Sparrow,D.B., Mohun,T. and Treisman,R. (1995) MEF2 proteins, including MEF2A, are expressed in both muscle and non-muscle cells. Nucleic Acids Res., 23, 4267–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett D.S., Powers,R., Gronenborn,A.M. and Clore,G.M. (1991) A common sense approach to peak picking in two-, three- and four-dimensional spectra using automatic computer analysis of contour diagrams. J. Magn. Reson., 94, 214–220. [DOI] [PubMed] [Google Scholar]
- Gorenstein D.G. (1994) Conformation and dynamics of DNA and protein–DNA complexes by 31P NMR. Chem. Rev., 94, 1315–1338. [Google Scholar]
- Gorin A.A., Zhurkin,V.B. and Olson,W.K. (1995) B-DNA twisting correlates with base-pair morphology. J. Mol. Biol., 247, 34–48. [DOI] [PubMed] [Google Scholar]
- Gossett L.A., Kelvin,D.J., Sternberg,E.A. and Olson,E.N. (1989) A new myocyte-specific enhancer-binding factor that recognizes a conserved element associated with multiple muscle-specific genes. Mol. Cell. Biol., 9, 5022–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson T.A., Taylor,A. and Kedes,L. (1989) DNA bending is induced by a transcription factor that interacts with the human c-FOS and α-actin promoters. Proc. Natl Acad. Sci. USA, 86, 2162–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J., Jiang,Y., Li,Z., Kravchenko,V.V. and Ulevitch,R.J. (1997) Activation of the transcription factor MEF2C by the MAP kinase p38 in inflammation. Nature, 386, 296–299. [DOI] [PubMed] [Google Scholar]
- Hirel P.H., Schmitter,M.J., Dessen,P., Fayat,G. and Blanquet,S. (1989) Extent of N-terminal methionine excision from Escherichia coli proteins is governed by side-chain length of the penultimate amino acid. Proc. Natl Acad. Sci. USA, 86, 8247–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y., Kravchenko,V.V., Taping,R.I., Han,J., Ulevitch,R.J. and Lee,J.-D. (1997) BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J., 16, 7054–7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koradi R., Billeter,M. and Wüthrich,K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph., 14, 51–55. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur,M.W., Moss,D.S. and Thornton,J.M. (1993) PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283–291. [Google Scholar]
- Ling Y., West,A.G., Roberts,E.C., Lakey,J.H. and Sharrocks,A.D. (1998) Interaction of transcription factors with serum response factor: identification of the Elk-1 binding surface. J. Biol. Chem., 273, 10506–10514. [DOI] [PubMed] [Google Scholar]
- Lodi P.J., Garrett,D.S., Kuszewski,J., Tsang,M.L.-S., Weatherbee,J.A., Leonard,W.J., Gronenborn,A.M. and Clore,G.M. (1994) High-resolution solution structure of the β-chemokine hMIP-1β by multidimensional NMR. Science, 263, 1762–1767. [DOI] [PubMed] [Google Scholar]
- Louis J.M., Martin,R.G., Clore,G.M. and Gronenborn,A.M. (1998) Preparation of uniformly isotope-labeled DNA oligonucleotides for NMR spectroscopy. J. Biol. Chem., 273, 2374–2378. [DOI] [PubMed] [Google Scholar]
- Mao Z., Bonni,A., Xia,F., Nadal-Vicens,M. and Greenberg,M.E. (1999) Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science, 286, 785–790. [DOI] [PubMed] [Google Scholar]
- Molkentin J.D. and Olson,E.N. (1996) Combinatorial control of muscle development by basic helix–loop–helix and MADS-box transcription factors. Proc. Natl Acad. Sci. USA, 93, 9366–9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls A., Sharp,K.A. and Honig,B. (1991) Protein folding and association: insights from interfacial and thermodynamic properties of hydrocarbons. Proteins, 11, 281–296. [DOI] [PubMed] [Google Scholar]
- Nilges M. (1993) A calculational strategy for the structure determination of symmetric dimers by 1H NMR. Proteins, 17, 297–309. [DOI] [PubMed] [Google Scholar]
- Nilges M., Gronenborn,A.M., Brünger,A.T. and Clore,G.M. (1988) Determination of three-dimensional structures of proteins by simulated annealing with interproton distance restraints: application to crambin, potato carboxypeptidase inhibitor and barley serine proteinase inhibitor 2. Protein Eng., 2, 27–38. [DOI] [PubMed] [Google Scholar]
- Nurrish S.J. and Treisman,R. (1995) DNA binding specificity determinants in MADS-box transcription factors. Mol. Cell. Biol., 15, 4076–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omichinski J.G., Pedone,P.V., Felsenfeld,G., Gronenborn,A.M. and Clore,G.M. (1997) The solution structure of a specific GAGA factor–DNA complex reveals a modular binding mode. Nature Struct. Biol., 4, 122–132. [DOI] [PubMed] [Google Scholar]
- Ornatsky O.I., Cox,D.M., Tangirala,P., Andreucci,J.J., Qinn,Z.A., Wrana,J.L., Prywes,R., Yu,Y.T. and McDermott,J.C. (1999) Post-translational control of the MEF2A transcriptional regulatory protein. Nucleic Acids Res., 27, 2646–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottiger G. and Bax,A. (1999) Bicelle-based liquid crystals for NMR-measurement of dipolar couplings at acidic and basic pH values. J. Biomol. NMR, 13, 187–191. [DOI] [PubMed] [Google Scholar]
- Ottiger G., Delaglio,F. and Bax,A. (1998) Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J. Magn. Reson., 131, 173–178. [DOI] [PubMed] [Google Scholar]
- Pellegrini L., Tan,S. and Richmond,T.J. (1995) Structure of serum response factor core bound to DNA. Nature, 376, 490–498. [DOI] [PubMed] [Google Scholar]
- Pollock R. and Treisman,R. (1991) Human SRF-related proteins: DNA-binding properties and potential regulatory targets. Genes Dev., 5, 2327–2341. [DOI] [PubMed] [Google Scholar]
- Schwarz-Sommer Z., Huijser,P., Nacken,W., Saedler,H. and Sommer,H. (1990) Genetic control of flower development by homeotic genes in Antirrhinum majus. Science, 250, 931–936. [DOI] [PubMed] [Google Scholar]
- Sharrocks A.D. (1994) A T7 expression vector for producing N- and C-terminal fusion proteins with glutathione S-transferase. Gene, 138, 105–108. [DOI] [PubMed] [Google Scholar]
- Sharrocks A.D., Von Hesler,F. and Shaw,P.E. (1993a) The identification of elements determining the different DNA binding specificities of the MADS-box proteins p67SRF and RSRFC4. Nucleic Acids Res., 21, 215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharrocks A.D., Gille,H. and Shaw,P.E. (1993b) Identification of amino acids essential for DNA binding and dimerization in p67SRF: implications for a novel DNA binding motif. Mol. Cell. Biol., 13, 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore P. and Sharrocks,A.D. (1994) The transcription factors Elk-1 and SRF interact by direct protein–protein contacts mediated by a short region of Elk-1. Mol. Cell. Biol., 14, 3283–3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore P. and Sharrocks,A.D. (1995) The MADS-box family of transcription factors. Eur. J. Biochem., 229, 1–13. [DOI] [PubMed] [Google Scholar]
- Smith D.L., Desai,A.B. and Johnson,A.D. (1995) DNA bending by the a1 and α2 homeodomain proteins from yeast. Nucleic Acids Res., 23, 1239–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow D.B., Miska,E.A., Langley,E., Reynaud-Deonauth,S., Kotecha,S., Towers,N., Spohr,G., Kouzarides,T. and Mohun,J.J. (1999) MEF-2 function is modified by a novel co-repressor, MITR. EMBO J., 18, 5085–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S. and Richmond,T.J. (1998) Crystal structure of the yeast MATα2/MCM1/DNA ternary complex. Nature, 391, 660–666. [DOI] [PubMed] [Google Scholar]
- Treisman R. (1990) The SRE: a growth factor responsive transcriptional regulator. Semin. Cancer Biol., 1, 47–58. [PubMed] [Google Scholar]
- West A.G. and Sharrocks,A.D. (1999) MADS-box transcription factors adopt alternative mechanisms for bending DNA. J. Mol. Biol., 286, 1311–1323. [DOI] [PubMed] [Google Scholar]
- West A.G., Shore,P. and Sharrocks,A.D. (1997) DNA binding by MADS-box transcription factors: a molecular mechanism for differential DNA bending. Mol. Cell. Biol., 17, 2876–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.C., Ornatsky,O.I., McDermott,J.C., Cruz,T.F. and Prody,C.A. (1998) Interaction of myocyte enhancer factor 2 (MEF2) with a mitogen-activated kinase, ERK5/BMK1. Nucleic Acids Res., 26, 4771–4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn H.D., Sun,L., Prywes,R. and Liu,J.O. (1999) Apoptosis of T cells mediated by Ca2+-induced release of the transcription factor MEF2. Science, 286, 790–793. [DOI] [PubMed] [Google Scholar]
- Zhao M., New,L., Kravchenko,V., Kato,Y., Gram,H., Di Padova,F., Olson,E., Ulevitch,R.J. and Han,J. (1999) Regulation of the MEF2 family of transciption factors by p38. Mol. Cell. Biol., 19, 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]