

Abstract
The bacterial LmrA protein and the mammalian multidrug resistance P-glycoprotein are closely related ATP-binding cassette (ABC) transporters that confer multidrug resistance on cells by mediating the extrusion of drugs at the expense of ATP hydrolysis. The mechanisms by which transport is mediated, and by which ATP hydrolysis is coupled to drug transport, are not known. Based on equilibrium binding experiments, photoaffinity labeling and drug transport assays, we conclude that homodimeric LmrA mediates drug transport by an alternating two-site transport (two-cylinder engine) mechanism. The transporter possesses two drug-binding sites: a transport-competent site on the inner membrane surface and a drug-release site on the outer membrane surface. The interconversion of these two sites, driven by the hydrolysis of ATP, occurs via a catalytic transition state intermediate in which the drug transport site is occluded. The mechanism proposed for LmrA may also be relevant for P-glycoprotein and other ABC transporters.
Keywords: drug efflux/LmrA/membrane protein/multidrug resistance/transport mechanism
Introduction
Multidrug resistance in pro- and eukaryotic cells is often associated with the enhanced expression of multidrug transport proteins (van Veen and Konings, 1997). A protein in Lactococcus lactis, LmrA, mediates drug and antibiotic resistance by extruding amphiphilic compounds from the inner leaflet of the cytoplasmic membrane (Bolhuis et al., 1996; van Veen et al., 1996). LmrA is a structural and functional homolog of the mammalian multidrug resistance P-glycoprotein, overexpression of which is one of the major causes of resistance of cancers to chemotherapy (Gottesman et al., 1995). Bacterial LmrA can substitute for P-glycoprotein in human lung fibroblast cells, and both proteins have a very similar drug and modulator specificity (van Veen et al., 1998).
LmrA and P-glycoprotein are both members of the ATP-binding cassette (ABC) superfamily of proteins (Higgins, 1992). ABC transporters form one of the largest of all protein families with substrate specificities ranging from the uptake of sugars, amino acids and organic ions to the export of large protein toxins. In eukaryotic cells, ABC transporters serve a variety of physiological roles, many of which are associated with disease phenotypes, including the TAP peptide transporter required for Class I antigen presentation, the transporters associated with adrenoleukodystrophy and Zellweger’s syndrome, the multidrug resistance-associated (MRP) proteins, sulfonylurea receptors (SUR), the bile salt transporter (BSEP) in hepatocytes, and the ABC1 transporter associated with Tangier disease and familial high-density lipoprotein deficiency (Klein et al., 1999). Most ABC transporters have a common domain organization with four core domains (Higgins, 1992). Two of these domains, the transmembrane domains, are hydrophobic and span the membrane multiple times. There is now compelling evidence that each transmembrane domain of P-glycoprotein consists of six transmembrane segments (putative α-helices), giving a total of 12 transmembrane segments per P-glycoprotein molecule (Kast et al., 1996; Loo and Clarke, 1999a). These domains are thought to form the pathway through which the solute crosses the membrane and confer substrate specificity through one or more substrate-binding sites. The other two domains bind and hydrolyze ATP and couple the energy of ATP hydrolysis to the transport process. These ABC domains, in particular, exhibit significant sequence identity between ABC transporters irrespective of their phylogenetic origin or substrate specificity.
P-glycoprotein, like many eukaryotic ABC transporters, has all four domains fused into one single polypeptide, and there is evidence that this monomeric, four-domain protein is the functional unit (Higgins et al., 1997; Rosenberg et al., 1997; Loo and Clarke, 1999a). The protein appears to have arisen by a gene duplication event, fusing two related half-molecules each consisting of one ABC domain and one transmembrane domain. In contrast, LmrA appears to be a half-transporter consisting of an N-terminal transmembrane domain with six membrane-spanning segments, fused to an ABC domain. It has been predicted that LmrA functions as a homodimer to form a full transporter with four core domains (van Veen et al., 1996); we show in this paper that this is indeed the case.
P-glycoprotein and LmrA utilize the free energy of ATP to pump drugs out of the cell. The two proteins exhibit a basal rate of ATP hydrolysis that is stimulated 3- to 4-fold in vitro by transport substrates, and have a similar apparent affinity for Mg-ATP (Senior et al., 1995; Martin et al., 1997; van Veen et al., 1998). For P-glycoprotein, both the ABC domains have been shown to bind and hydrolyze ATP (Hrycyna et al., 1998), and several observations provide strong support to an alternating catalytic sites model in which the ABC domains of P-glycoprotein act alternately to hydrolyze ATP: (i) mutations (Urbatsch et al., 1998) or covalent modifications (Senior and Bhagat, 1998; Loo and Clarke, 1999a) that inactivate ATP hydrolysis by one of the ABC domains abolish all drug-stimulated ATPase activity, demonstrating catalytic cooperativity between the two ABC domains of P-glycoprotein; (ii) o-vanadate, a potent inhibitor of P-glycoprotein and LmrA-associated ATPase and drug transport activities, permits only a single turnover of P-glycoprotein by trapping either the first or the second ABC domain in a catalytic transition state conformation, thus preventing the other ABC domain from doing so (Urbatsch et al., 1995); and (iii) photoaffinity labeling experiments with azido-nucleotides suggest that ATP binding to one ABC domain stimulates ATP hydrolysis at the other ABC domain (Ueda et al., 1999), and that 1 mol of Mg-8-azido-ADP is bound per mol of P-glycoprotein after hydrolysis of Mg-8-azido-ATP (Senior et al., 1995).
However, the mechanism by which P-glycoprotein and LmrA couple the hydrolysis of ATP to the movement of drugs across the membrane is not known. Indeed, the number and nature of the drug-binding site(s) are also ill-defined. There is strong evidence that the transmembrane domains of P-glycoprotein form the binding site(s) for transported drugs (Ueda et al., 1997; Loo and Clarke, 1999b). Genetic analysis has revealed that replacement of a large number of individual amino acids can affect drug specificity (Gottesman et al., 1995), but since these amino acids are scattered throughout the transmembrane segments and intervening loops, genetic analysis has failed to define a drug-binding pocket. Photoaffinity labeling studies with drug analogs have indicated two regions within the transmembrane domains of P-glycoprotein encompassing transmembrane segments 5 and 6 in the N-terminal half, and transmembrane segments 11 and 12 in the C-terminal half (Greenberger, 1993; Morris et al., 1994), which may form part of one large drug interaction domain, or which may represent two or more distinct drug-binding sites (Dey et al., 1997). Owing to the lack of specificity of the cross-linking reaction, photoaffinity labeling has been unable to identify the amino acid residues involved in drug binding. The notion that P-glycoprotein contains at least two drug-binding sites is supported by studies employing continuous fluorescence measurements of P-glycoprotein-mediated drug transport in plasma membrane vesicles (Shapiro and Ling, 1997). In addition to binding sites for transported drugs, detailed pharmacological analysis shows at least one binding site in P-glycoprotein and LmrA for allosteric modulators such as the indolizin sulfone SR33557 (Martin et al., 1997) and the 1,4-dihydropyridine nicardipine (Ferry et al., 1992; van Veen et al., 1998) that are bound to, but not transported by the protein.
In this study we have carried out a detailed analysis of the drug-binding sites of LmrA by transport measurements, vinblastine equilibrium binding determinations, and photoaffinity labeling using N-(4′,4′-azo-n-pentyl)-21-deoxy-[3H]ajmalinium (APDA) (Müller et al., 1994; van Veen et al., 1996). The results demonstrate the presence of two vinblastine-binding sites: a low-affinity binding site exposed at the outside (extracellular) surface of the membrane, which is allosterically coupled to a high-affinity binding site exposed at the inside (intracellular) surface of the membrane. Only the low-affinity, outside-facing vinblastine-binding site is accessible in the vanadate-trapped transporter. The data suggest an alternating two-site mechanism in which the transport protein oscillates between two configurations, each containing a high-affinity, inside-facing, transport-competent drug-binding site, and a low-affinity, outside-facing drug-release site. The ATP-dependent interconversion of one configuration into the other proceeds via a catalytic transition state conformation in which the transport-competent drug-binding site is occluded. This model is analogous to a two-cylinder engine, and has implications for the mechanism of action of P-glycoprotein and other ABC transporters.
Results
LmrA and P-glycoprotein have an identical domain organization
From its primary amino acid sequence, LmrA appears to be a ‘half’ ABC transporter consisting of one transmembrane domain and one ATP-binding domain. To assess whether two LmrA molecules cooperate to form a single transporter, the functional properties of defined covalently linked dimers of LmrA were studied. Two copies of the lmrA coding sequence were fused, in frame, connected by a linking sequence identical to the ‘linker’ region that connects the two halves of P-glycoprotein. The coding sequence for this LmrA dimer was cloned into the lactococcal expression vector pNZ8048 under the control of the nisin-inducible nisA promoter, which has previously been used for the expression of LmrA (Margolles et al., 1999). In addition to the wild-type dimeric LmrA protein (designated KK), single mutant dimeric LmrA proteins were generated in the same vector by introducing lysine to methionine substitutions (van Veen et al., 1998) in the Walker A region of either the N- or the C-terminal ABC domain of the dimer (designated MK and KM, respectively). A double mutant was also generated with a lysine to methionine substitution in both ABC domains (designated MM). His6 tags were placed at the N-termini of each of the proteins.
Upon the addition of 10 ng/ml nisin to L.lactis cells containing plasmids encoding the ‘dimeric’ lmrA genes, equivalent amounts of a 132 kDa polypeptide could be detected using an anti-His6 tag monoclonal antibody (Figure 1A). Wild-type LmrA is able to reduce the accumulation of ethidium bromide in the cell by mediating its efflux (van Veen et al., 1996). Measurements of ethidium bromide accumulation in non-expressing control cells, cells expressing wild-type LmrA and cells expressing the KK form of dimeric LmrA demonstrated that the KK form (having wild-type nucleotide-binding domains) was transport active (Figure 1B). The difference in ethidium accumulation between cells containing monomeric LmrA or dimeric (KK) LmrA was consistent with a difference in the level of expression of the two proteins [∼10% of total membrane protein for LmrA compared with 2% of total membrane protein for dimeric (KK) LmrA; see the later section ‘Direct determination of LmrA:vinblastine binding stoichiometry’ for experimental details]. In contrast, the MK, KM and MM mutant forms of dimeric LmrA had all lost the ability to transport ethidium from the cell. Cells expressing these mutant forms displayed a similar ethidium accumulation level to that observed in non-expressing control cells, which lack significant LmrA activity.
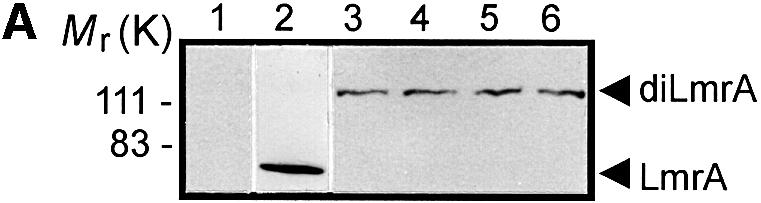


Fig. 1. Expression and activity of covalently linked dimers of LmrA. (A) Western blot of total membrane protein (20 µg) from inside-out membrane vesicles lacking LmrA (lane 1), or containing LmrA (lane 2), the wild-type (KK) form of dimeric LmrA (lane 3), or the dimeric form of LmrA with a K to M substitution in the Walker A region of the first ABC domain (MK, lane 4), the second ABC domain (KM, lane 5), or both ABC domains (MM, lane 6). The western blot was probed with an anti-His6 tag antibody. The migration of molecular mass markers is indicated. An arrowhead indicates the position of monomeric LmrA (66 kDa) and dimeric LmrA (132 kDa). (B) Accumulation of ethidium bromide in cells expressing LmrA, or the KK, MK, KM or MM forms of dimeric LmrA. Cells were pre-energized for 3.5 min in the presence of 20 mM glucose, after which 2 µM ethidium bromide was added. Ethidium accumulation in the cells was measured by fluorimetry. The uptake of ethidium in control cells not expressing LmrA was similar to that in cells expressing the KM, MK or MM form of dimeric LmrA (not shown). (C) Vanadate-sensitive ATPase activity in inside-out membrane vesicles lacking LmrA (control), or containing LmrA, or the KK, MK, KM or MM form of dimeric LmrA. The ATPase activity was measured at a Mg-ATP concentration of 1 mM in the presence of 20 µM verapamil.
LmrA displays a basal ATPase activity, which is stimulated by verapamil and inhibited by o-vanadate (van Veen et al., 1998). Analysis of the verapamil-stimulated, LmrA-associated ATPase activity in inside-out membrane vesicles of L.lactis demonstrated that the KK form of dimeric LmrA had essentially wild-type ATPase activity, whereas the KM, MK and MM mutant forms had lost all LmrA-associated ATPase activity (Figure 1C). Thus, KM, MK and MM mutants of dimeric LmrA have similar properties to the KM, MK and MM mutants of P-glycoprotein, which lack all ATPase and transport activity (Müller et al., 1996; Loo and Clarke, 1999a). These data are consistent with the notion that LmrA functions as a homodimer, analogous to monomeric P-glycoprotein, with both halves of the dimer required for transport.
The dimeric nature of the LmrA transporter was also studied by the co-reconstitution into proteoliposomes of two different LmrA proteins: the cysteine-less wild-type LmrA and a mutant form of LmrA in which the N-ethylmaleimide (NEM)-reactive glycine to cysteine (G386C) substitution in the Walker A region (Loo and Clarke, 1999a) was introduced by site-directed mutagenesis. The wild-type and mutant forms were purified by Ni2+-nitrilotriacetic acid (NTA) affinity chromatography, mixed at molar ratios of 6:0, 3:3, 2:4 and 0:6, respectively, and reconstituted into n-dodecyl-β-d-maltoside-destabilized liposomes. After reconstitution, the LmrA-associated transport activity in proteoliposomes was measured using fluorescent 1-myristoyl-2-[6-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]caproyl]-sn-glycero-3-phosphoethanolamine (C6-NBD-PE) (Margolles et al., 1999). In the absence of NEM treatment after the reconstitution procedure, equal rates were observed for the LmrA-mediated transbilayer movement of NBD-PE in proteoliposomes containing the wild-type and mutant proteins at the various ratios (Figure 2). Hence, the single-cysteine mutant form of LmrA had retained its transport activity compared with that of wild-type LmrA. When the proteoliposomes were incubated for 30 min in the presence of 1 µM NEM, allowing NEM to react with the cysteine residue in the G386C mutant form of LmrA, the LmrA-mediated transport of C6-NBD-PE in proteoliposomes containing wild-type LmrA only (wild-type:mutant ratio of 6:0) was not affected; the transport rate after NEM treatment was 98 ± 5% (n = 3) of the transport rate before NEM treatment. In contrast, the transport rate in NEM-treated proteoliposomes containing wild-type and mutant proteins at a ratio of 3:3, 2:4 and 0:6 was reduced to 22 ± 5, 13 ± 4 and 0 ± 4% (n = 3) of the transport rate in proteoliposomes without NEM treatment, respectively. This inhibition pattern suggested that two wild-type LmrA monomers cooperate to form a functional transporter, as 100, 25, 10 and 0% of the dimers would contain two wild-type monomers at a wild-type:mutant ratio of 6:0, 3:3, 2:4 and 0:6, respectively. On the other hand, the observed inhibition pattern is not consistent with the presence of three or four (or more) wild-type LmrA monomers in the minimal functional unit, as 100, 13, 4 and 0% of the trimers would contain three wild-type monomers, and 100, 6, 1 and 0% of the tetramers would contain four wild-type monomers at a wild-type:mutant ratio of 6:0, 3:3, 2:4 and 0:6, respectively. These data are in agreement with the electron microscopy analysis of purified and functionally reconstituted LmrA, which revealed small, uniform particles with a diameter of 8.5 nm by 5 nm (S.Scheuring, A.Margolles, H.W.van Veen, W.N.Konings and A.Engel, unpublished data), similar to that previously observed for the monomeric P-glycoprotein (Rosenberg et al., 1997). Taken together, these data demonstrate that the dimeric state of LmrA is a prerequisite for function, analogous to the monomeric P-glycoprotein, and that functional cross-talk between the two LmrA monomers is essential for transport.

Fig. 2. Negative dominance of an NEM-inactivated single-cysteine LmrA mutant in proteoliposomes containing co-reconstituted wild-type and mutant LmrA proteins. The transport activity in proteoliposomes was measured using fluorescent C6-NBD-PE as a substrate. Inset: typical NBD fluorescence traces obtained for proteoliposomes containing wild-type LmrA in assay mixtures supplemented with ATP or non-hydrolyzable AMP-PNP demonstrate the experimental and analytical methods used. At the onset of transport measurement, C6-NBD-PE was present in donor liposomes in which the NBD fluorescence was low due to the presence of N-Rh-PE quencher. Addition of proteoliposomes at the arrow resulted in the movement of C6-NBD-PE, but not of the N-Rh-PE quencher, from donor liposomes to proteoliposomes with a concomitant increase in NBD fluorescence. The increase in NBD fluorescence observed was deconvoluted by a double exponential fit of the data (see Materials and methods), which is shown displaced from the fluorescence traces for clarity, yielding a rate constant k1 for the interbilayer movement of C6-NBD-PE between donor liposomes and proteoliposomes, and a rate constant k2 for the transbilayer movement of the probe in the proteoliposomes. Main figure: NBD-PE transport was measured in the presence of ATP or AMP-PNP, using proteoliposomes containing the cysteine-less wild-type LmrA and the G386C mutant form of LmrA at the molar ratios indicated, before (filled circles) and after (open circles) NEM treatment of the proteoliposomes. In all experiments k1 values were of the order of 0.038–0.041 s–1, and were at least 10 and 70 times the k2 values obtained for incubations containing ATP or AMP-PNP, respectively. Hence, the transbilayer movement of C6-NBD-PE could be explicitly characterized, separate from the interbilayer movement. The relative transbilayer transport rate, as depicted in the graph, was calculated from the estimated k2 values using the equation: vrel = vATP/vAMP-PNP = k2ATP/k2AMP-PNP. The k2 values obtained for proteoliposomes in incubations supplemented with AMP-PNP were relatively constant, and of the order of 0.00052–0.00058 s–1.
Vinblastine and Hoechst 33342 affect the transport of each other via LmrA
LmrA and P-glycoprotein are functionally interchangeable proteins with very similar drug and modulator specificities (van Veen et al., 1998). Previously, we have used 2-[2-(4-ethoxyphenyl)-6-benzimidazolyl]-6-(1-methyl)-(4-piperazil)-benzimidazole (Hoechst 33342) and vinblastine to monitor drug transport by LmrA expressed in human lung fibroblast cells (van Veen et al., 1998). Hoechst 33342 is fluorescent when it resides in the lipid environment of the membrane, and is essentially non-fluorescent in the aqueous phase. Transport of Hoechst 33342 from inside-out membrane vesicles of L.lactis by LmrA resulted in a decrease in Hoechst 33342 fluorescence (Figure 3A). Interestingly, the rate of LmrA-mediated Hoechst 33342 transport in inside-out membrane vesicles was enhanced in the presence of low concentrations of vinblastine. In a similar fashion, Hoechst 33342 at low concentrations also stimulated the ATP-dependent accumulation of vinblastine in inside-out membrane vesicles of LmrA-overexpressing L.lactis (Figure 3B). Both drugs inhibited the transport of each other when applied at 10-fold higher concentrations (Figure 3). Under the experimental conditions, vinblastine did not affect the intrinsic fluorescence of Hoechst 33342, and vinblastine and Hoechst 33342 did not affect the partitioning of each other in the phospholipid bilayer (data not shown; Putman et al., 1999). The observation that vinblastine and Hoechst 33342 are able to stimulate the transport of each other by LmrA at low drug concentrations is not readily explained if the LmrA transporter contains only a single drug-binding site. Instead, these data and the reciprocal inhibition of transport at high drug concentrations suggest at least two binding sites with overlapping drug specificities, which are functionally coupled.


Fig. 3. LmrA-mediated transport of Hoechst 33342 and vinblastine. (A) Effect of vinblastine on the transport of Hoechst 33342 was measured by fluorimetry. After 30 s of pre-incubation of inside-out membrane vesicles containing LmrA in transport buffer, 2 µM Hoechst 33342 was added and its binding to the membrane vesicles followed in time until a steady state was reached. Hoechst 33342 transport was initiated by the addition of 2 mM Mg-ATP. Incubations with Mg-ATPγS served as a control. Vinblastine (2 µM; VBL), added to the membrane vesicles prior to the addition of Hoechst 33342, did not affect Hoechst 33342 fluorescence in experiments with ATPγS (not shown), but significantly stimulated the ATP-dependent transport of Hoechst 33342. In the presence of 20 µM vinblastine, ATP-dependent Hoechst 33342 transport was completely inhibited. The fluorescence trace obtained under these conditions was indistinguishable from that of the depicted control experiment and is therefore not shown. (B) Effect of Hoechst 33342 on the LmrA-mediated transport of [3H]vinblastine. The uptake of vinblastine in inside-out membrane vesicles in buffer supplemented with 40 nM [3H]vinblastine and 2 mM Mg-ATPγS (open squares, open circles, open triangles) or 2 mM Mg-ATP (filled squares, filled circles, filled triangles) was measured in the absence (open and filled circles) or presence of 2 µM Hoechst 33342 (open and filled squares) or 20 µM Hoechst 33342 (open and filled triangles).
LmrA contains two non-identical, allosterically linked vinblastine-binding sites
The drug-binding sites involved in LmrA-mediated vinblastine transport were studied in more detail by equilibrium binding using [3H]vinblastine. The specific binding of vinblastine to LmrA in inside-out membrane vesicles was plotted as a function of the free vinblastine concentration in the water phase, which could be determined more accurately than the free vinblastine concentration in the phospholipid bilayer, and which increased linearly with the amount of vinblastine associated with the membrane at total vinblastine concentrations up to 1 µM in accordance with a membrane/water partition coefficient of 278 (data not shown). Figure 4A shows that the specific binding of vinblastine to LmrA increased as a function of the free vinblastine concentration in a saturable fashion. No specific binding of vinblastine was observed in inside-out membrane vesicles in the absence of LmrA. The binding data could not readily be fitted to a hyperbolic curve, as shown by the residuals between the experimental data and the best-fit hyperbola as a function of the free vinblastine concentration in the assays (Figure 4B). The hyperbola overestimates the amount of drug binding at vinblastine concentrations below 50 nM, and underestimates binding at vinblastine concentrations between 50 and 160 nM. Similar results were obtained for the KK, KM, MK and MM forms of dimeric LmrA expressed in inside-out membrane vesicles of L.lactis (data not shown). Thus, experimental measurements of vinblastine binding to LmrA are not consistent with a single drug-binding site. Instead, the experimental vinblastine binding data fit a cooperative, two-site drug-binding model:




Fig. 4. Vinblastine equilibrium binding to LmrA. (A) Specific binding of [3H]vinblastine to inside-out membrane vesicles without LmrA (control) or with LmrA, as a function of the free vinblastine concentration. Superimposed on the data are the best fit curves obtained for a single-site binding model (hyperbolic, dotted curve), the cooperative two-site binding model (sigmoidal, solid curve) and the Hill equation (indistinguishable from the sigmoidal, solid curve). (B) Residual variance between the data and the best fitting hyperbola and sigmoidal curves from (A), plotted as a function of the free vinblastine concentration. Note the systematic deviation of the residual variance of the data for the hyperbola, indicating that the single-site model provides an inadequate fit to the data. The residual variance between the data and the sigmoidal curve shows only random scatter, suggesting that the data are adequately fitted by the cooperative two-site binding model. (C) Heterologous displacement of vinblastine from LmrA by CP100-356. LmrA was saturated with [3H]vinblastine at a concentration of 192 nM, and vinblastine displacement from LmrA by CP100-356 measured at increasing concentrations of CP100-356. The data were fitted by the cooperative two-site drug-binding model in which direct competition was introduced between vinblastine and CP100-356 for binding to the two drug-binding sites in the LmrA transporter. A relative binding value of 1 represents 1.42 nmol vinblastine/mg of membrane protein. (D) Residual variance between the experimental data and the best fitting curve from (C), showing random scatter of the data around the predicted curve.

where T is the transport protein and S is a drug molecule. In this model, an initial vinblastine-binding event with low affinity (Kd1) initiates a second vinblastine-binding event with high affinity (Kd2). Figure 4B shows that, for such a cooperative two-site drug-binding model, the residual variance between the experimental data and the predicted sigmoidal curves randomly scatters around the best fit. Based on the model, Kd1 and Kd2 for LmrA can be determined to be 150.2 ± 16.7 nM vinblastine (n = 4) and 29.8 ± 5.5 nM vinblastine (n = 4), respectively, with a maximal binding (Bmax) of 1.49 ± 0.06 nmol vinblastine/mg of membrane protein (n = 4). The binding data were also well fitted by the Hill equation. The best fit obtained using the Hill equation is indistinguishable from that obtained using the cooperative two-site binding model (Figure 4A), with a Hill number (nHill) of 1.96 ± 0.06 (n = 4), a Bmax of 1.43 ± 0.07 nmol vinblastine/mg of membrane protein (n = 4) and an average binding constant KHill of 0.015 ± 0.002 nM vinblastine–1 (n = 4). For the two-site model, KHill equals 1/(Kd1 × Kd2)1/n. Using the previously determined Kd1 and Kd2, KHill can be calculated to be 0.014 ± 0.004 nM vinblastine–1, close to that obtained by direct curve fitting. Thus, the vinblastine equilibrium binding measurements are best fitted by a model in which there are two strongly cooperative vinblastine-binding sites in the LmrA transporter.
Displacement studies also indicate two vinblastine-binding sites in LmrA
The calcium-channel blocker CP100-356 is a competitive inhibitor of vinblastine binding to LmrA (van Veen et al., 1998). The affinities of the two vinblastine-binding sites for CP100-356 were estimated from its ability to displace vinblastine. In this type of experiment the drug-binding sites of the LmrA transporter were initially saturated with radiolabeled vinblastine. Increasing amounts of non-radiolabeled CP100-356 displaced the radiolabeled vinblastine from LmrA in a biphasic fashion (Figure 4C). The displacement data fitted the cooperative, two-site drug-binding model, assuming direct competition by CP100-356 for binding to each of the two vinblastine-binding sites (Figure 4D). The data revealed vinblastine dissociation constants that were very similar to those determined by the direct binding data above [Kd1 =157.0 ± 18.8 nM vinblastine (n = 3); Kd2 = 35.1 ±8.2 nM vinblastine (n = 3)]. The inhibitor constants Ki1 and Ki2 for the displacement of vinblastine by CP100-356 were 2.0 ± 0.9 nM CP100-356 (n = 3) and 596.4 ±78.1 nM CP100-356 (n = 3), respectively, suggesting that CP100-356 binds with low affinity to the low-affinity vinblastine-binding site of the LmrA transporter and with high affinity to the high-affinity vinblastine-binding site of the LmrA transporter.
Direct determination of LmrA:vinblastine binding stoichiometry
If, as indicated by the above data, there are two vinblastine-binding sites on the LmrA transporter, then two molecules of vinblastine should be bound to a single LmrA homodimer under saturating binding conditions. To measure the number of vinblastine-binding sites on the LmrA transporter directly, inside-out membrane vesicles containing wild-type monomeric or dimeric (KK) LmrA were used. For LmrA, the number of LmrA transporter molecules in membrane vesicles was determined as 755 ± 102 pmol/mg of membrane protein (n = 4) by quantitative immunoblotting, and as 806 ± 182 pmol/mg of membrane protein (n = 3) by densitometric analysis of total membrane protein on a Coomassie Blue-stained SDS–PAGE gel (see Materials and methods). Thus, similar values were obtained by two different methods. For the KK form of dimeric LmrA, the number of transporter molecules in membrane vesicles was estimated by quantitative immunoblotting as 151 ± 27 pmol/mg of membrane protein (n = 3). To measure the number of vinblastine molecules bound to LmrA in these same membranes, the vinblastine-binding sites in the LmrA transporter were completely saturated by decreasing the number of transporter molecules in a binding reaction from ∼76 to 2 pmol/ml at a fixed concentration of 160 nM vinblastine. As the total number of vinblastine molecules available per transporter was increased from 0.1 up to 80, the average number of vinblastine molecules bound per transporter increased to a value close to two (Figure 5). Similar data were obtained for the KK form of dimeric LmrA (data not shown). Thus, each homodimer of LmrA binds two vinblastine molecules.

Fig. 5. Direct determination of the vinblastine/LmrA transporter stoichiometry. The average number of vinblastine molecules bound per LmrA homodimer is plotted as a function of the total number of vinblastine molecules available per transporter. Specific binding of vinblastine to inside-out membrane vesicles containing LmrA was measured in equilibrium binding experiments in which the total amount of membrane protein was varied, at a fixed concentration of 160 nM vinblastine. The number of LmrA molecules per milligram of membrane protein in inside-out membrane vesicles was determined by quantitative immunoblotting and densitometry. The data show that each homodimer of LmrA binds two vinblastine molecules under conditions that saturate the vinblastine-binding sites in the protein.
Only the low-affinity drug-binding site is accessible in the ADP/vanadate-trapped LmrA transporter
ABC-type multidrug transporters can be trapped with o-vanadate in a form that is thought to mimic the physiological state following ATP hydrolysis in which ADP and phosphate are bound and from which they are normally released (Urbatsch et al., 1995). The efficiency of photoaffinity labeling of LmrA in the presence of 130 nM APDA, a drug that can be transported by the protein (van Veen et al., 1996), was significantly reduced in the presence of vinblastine, suggesting competition between vinblastine and APDA for binding to LmrA (Figure 6A). In addition, photoaffinity labeling was significantly inhibited by vanadate trapping of LmrA. Thus, the vanadate-trapped protein has reduced affinity or accessibility for the drug-binding sites, presumably due to conformational coupling between the nucleotide-binding domains and the drug-binding sites on the transmembrane domains. Heterologous displacement by CP100-356 of vinblastine from the vanadate-trapped LmrA transporter showed significant differences from that observed for the non-trapped transporter, and suggested the presence of only a single population of vinblastine-binding sites with drug-binding properties similar to those of the low-affinity vinblastine-binding site in the non-trapped transporter [Kd = 161.3 ± 21.9 nM vinblastine (n = 3), Ki = 620.5 ± 62.9 nM CP100-356 (n = 3)] (Figure 6B and C). Furthermore, direct determination of the vinblastine/transporter stoichiometry in the vanadate-trapped transporter revealed a vinblastine/transporter stoichiometry of close to one (Figure 6D), in contrast to the value of two determined for the non-trapped transporter (Figure 5). Although similar results were obtained for vinblastine binding to the vanadate-trapped KK form of dimeric LmrA, vinblastine binding to the KM, MK and MM forms of dimeric LmrA was not affected by the presence of vanadate (data not shown). These data are consistent with previous observations suggesting that the KM, MK and MM forms of P-glycoprotein do not exhibit vanadate-induced nucleotide trapping (Ueda et al., 1999). Taken together, the results demonstrate that of the two vinblastine-binding sites accessible in the LmrA transporter, only the low-affinity vinblastine-binding site is accessible in the vanadate-trapped protein.
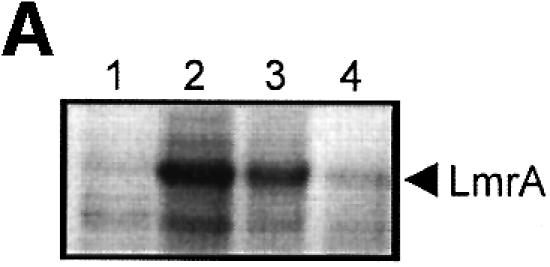



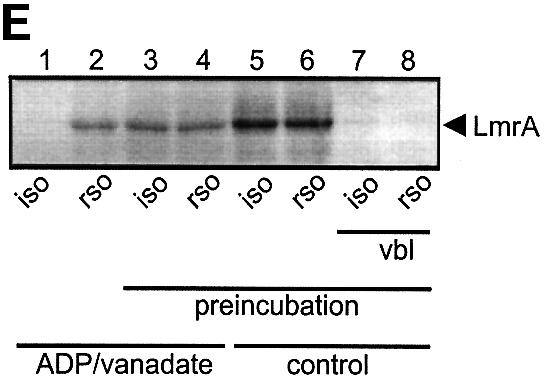
Fig. 6. Vanadate-trapped LmrA contains a single, low-affinity drug-binding site, which is exposed on the outside surface of the membrane. (A) Vanadate-trapped LmrA exhibits reduced affinity for [3H]APDA in photoaffinity labeling experiments. Inside-out membrane vesicles lacking LmrA (lane 1) or containing LmrA (lane 2), vanadate-trapped LmrA (lane 3), or LmrA in buffer supplemented with 3 µM vinblastine (lane 4), were incubated for 2 h at room temperature with 130 nM [3H]APDA. Subsequently, the four samples were UV irradiated, and analyzed by SDS–PAGE and autoradiography. Each lane contained 25 µg of protein. The arrow indicates photolabeled LmrA. (B) Heterologous displacement of [3H]vinblastine by CP100-356 from vanadate-trapped LmrA. Vinblastine displacement was performed as described in the legend to Figure 4C, in the presence of 2 mM Mg-ADP and 2 mM o-vanadate. A relative binding value of 1 represents 1.42 nmol vinblastine/mg of membrane protein. The data suggest the presence of a single vinblastine-binding site in vanadate-trapped LmrA with binding characteristics similar to those of the low-affinity site in the non-trapped protein. (C) Residual variance between the experimental data and the best fitting curve from (B). (D) Direct determination of the vinblastine/LmrA transporter stoichiometry suggests the presence of a single vinblastine-binding site in vanadate-trapped LmrA. Experimental details are similar to those described in the legend to Figure 5, with 2 mM Mg-ADP plus 2 mM o-vanadate included in the vinblastine binding assays. (E) Orientation of the low-affinity drug-binding site in vanadate-trapped LmrA. The accessibility of the low-affinity drug-binding site in vanadate-trapped LmrA from the inside or outside surface of the membrane was determined by photoaffinity labeling with [3H]APDA. LmrA-containing inside-out (ISO) membrane vesicles (lanes 1 and 3) in buffer supplemented with 2 mM Mg-ADP and 2 mM o-vanadate, and LmrA-containing right-side-out (RSO) membrane vesicles loaded with 2 mM Mg-ADP and 2 mM o-vanadate (lanes 2 and 4), were pre-incubated for 2 h at room temperature in the absence (lanes 1 and 2) or presence (lanes 3 and 4) of 130 nM [3H]APDA, and cooled on ice–water. Subsequently, 130 nM [3H]APDA was added to the incubations of lane 1 and 2, and the four reaction mixtures were immediately UV irradiated for 5 min. Non-vanadate-trapped LmrA in inside-out and right-side-out membrane vesicles in the absence (lane 5 and 6, respectively) or presence (lane 7 and 8, respectively) of 5 µM vinblastine were pre-incubated with APDA for 2 h at room temperature prior to UV irradiation. Samples containing 15 µg of membrane protein were analyzed by SDS–PAGE and autoradiography. The arrow indicates photolabeled LmrA.
Low- and high-affinity drug-binding sites in LmrA are present on opposite membrane surfaces
The vanadate-trapped conformation of LmrA, with a single low-affinity drug-binding site, is consistent with the notion that an ATP hydrolysis-induced conformational change moves a high-affinity drug-binding site from the inside of the membrane to the outside with a concomitant change to low affinity. To determine whether the low-affinity vinblastine-binding site is exposed at the inside or outside of the membrane, the relatively hydrophilic drug APDA was used for photoaffinity labeling of the vanadate-trapped LmrA transporter in inside-out and right-side-out oriented membrane vesicles. Methods are available to prepare membrane vesicles from bacteria with a well-defined inside-out or right-side-out orientation (Rosen and Tsuchiya, 1979; Otto et al., 1982; van der Does et al., 1996). In the absence of pre-incubation and equilibration of membrane vesicles with APDA, specific photoaffinity labeling of vanadate-trapped LmrA was obtained in right-side-out membrane vesicles, but not in inside-out membrane vesicles (Figure 6E). In contrast, when the membrane vesicles were incubated for 2 h at 22°C prior to photoaffinity labeling, allowing APDA to reach the interior of the membrane vesicles, labeling of vanadate-trapped LmrA in inside-out and right-side-out membrane vesicles was identical. Thus, the low-affinity vinblastine-binding site present in vanadate-trapped LmrA is exposed to the outside of the cell membrane. Furthermore, the amount of photoaffinity labeling of non-trapped LmrA in inside-out and right-side-out membrane vesicles pre-incubated with APDA was increased 2- to 3-fold compared with the labeling obtained for vanadate-trapped LmrA. This result is consistent with the vinblastine displacement studies and vinblastine/transporter stoichiometry determinations, and suggests that the second (high-affinity) vinblastine-binding site in non-trapped LmrA is inaccessible in the vanadate-trapped transporter.
Discussion
In order to mediate the active extrusion of drugs, LmrA and P-glycoprotein have to couple two distinct activities: (i) conformational changes in the nucleotide-binding domains induced by the binding and/or hydrolysis of ATP; and (ii) the reorientation of drug-binding sites in the transmembrane domains with alternate exposure to each face of the membrane, and alternate changes in the drug-binding affinity to facilitate drug binding to and release from the transporter. Conformational changes in P-glycoprotein upon binding and/or hydrolysis of ATP, and upon binding of drugs, have been detected by Fourier transform IR spectroscopy (Sonveaux et al., 1996), cross-linking of cysteine residues introduced in the protein (Loo and Clarke, 1999a), immunoreactivity of the protein with the monoclonal antibody UIC2 (Mechetner et al., 1997), partial trypsin digestion (Wang et al., 1998), intrinsic tryptophan fluorescence measurements (Sonveaux et al., 1996, 1999), and fluorescence labeling of the ABC domains (Sharom et al., 1999), indicating the existence of different P-glycoprotein conformations associated with different steps in the transport process.
The data obtained in this study strongly suggest the presence of two cooperative drug-binding sites on the LmrA transporter. First, a stoichiometry of two molecules of vinblastine bound per transporter molecule of LmrA was determined. Secondly, vinblastine equilibrium binding experiments demonstrate the presence of a low-affinity vinblastine-binding site in the LmrA transporter on the outside surface of the membrane, which is allosterically coupled to a high-affinity vinblastine-binding site in the protein on the inside surface of the membrane. Only the low-affinity, outside-facing vinblastine-binding site is accessible in the ADP/vanadate-trapped LmrA transporter, providing evidence for the coupling between ATP hydrolysis and the outwardly directed transbilayer movement of the high-affinity vinblastine-binding site. Thirdly, competitive inhibitors of vinblastine binding also showed behavior consistent with two alternating binding sites. Finally, these alternating sites were shown to be directly related to drug transport by the observation of reciprocal stimulation of LmrA-mediated vinblastine and Hoechst 33342 transport at low drug concentrations, and reciprocal inhibition at high drug concentrations. These two transport sites may exist in addition to regulatory sites to which allosteric inhibitors of LmrA and P-glycoprotein bind but are not transported (Ferry et al., 1992; Martin et al., 1997; van Veen et al., 1998).
In this alternating two-site transport model (Figure 7), the hydrolysis of ATP by the ABC domain of one half of the transporter is coupled to drug efflux through the movement of the inside-facing, high-affinity drug-binding site (which binds a drug molecule in the inner leaflet of the membrane) to the outside of the membrane with a concomitant change to low affinity (resulting in the release of the drug molecule into the extracellular medium) (arrows 1 and 2 in Figure 7). In the ADP/vanadate-trapped transporter, the high-affinity drug-binding site is occluded. In addition, ATP binding and/or hydrolysis must facilitate the return of an unliganded, outside-facing low-affinity site to an inside-facing high-affinity site. Presumably, ATP binding by the ABC domain of the other half-molecule is also induced. The alternating two-site model for drug binding and transport is consistent with the alternating catalytic sites mechanism proposed for ATP hydrolysis by P-glycoprotein (Senior and Gadsby, 1997), and now provides a model that links the drug-binding and catalytic cycles. The vinblastine equilibrium binding experiments suggest that the partial reactions in the ATP hydrolysis model are reversible and that, in the absence of ADP/vanadate, vinblastine binding results in the formation of a ternary drug–transporter complex that possesses a low-affinity vinblastine-binding site and high-affinity vinblastine-binding site (arrows a and b in Figure 7). This ternary drug–transporter complex is formed via an intermediate binary drug–transporter complex in which the high-affinity vinblastine-binding site is not accessible, and which is trapped by ADP/vanadate.

Fig. 7. Alternating two-site (two-cylinder engine) transport model. Rectangles represent the transmembrane domains of LmrA. Circles, squares and hexagons represent different conformations of the nucleotide-binding domains. The ATP-bound (circle) state is associated with a high-affinity drug-binding site on the inside of the transporter. The ADP-bound (square) state is associated with a low-affinity drug-binding site on the outside of the transporter. The ADP-Pi (hexagonal) state is associated with an occluded drug-binding site, and represents the ADP/vanadate-trapped form of the ABC domain. In and out refer to the inside and outside of the phospholipid bilayer, respectively. The upper and lower horizontal branches (as indicated by arrows a and b) summarize the events that occur during equilibrium drug binding to the LmrA transporter. Arrow a: an initial drug-binding event with low affinity to one LmrA molecule in the dimer yields a binary drug–protein intermediate, which can be stabilized through the binding of Mg-ADP plus o-vanadate. Arrow b: a second drug-binding event with high affinity to the other LmrA molecule in the dimer yields a ternary drug–protein complex. In the presence of ATP, the transport cycle turns counterclockwise (as indicated by arrows 1 and 2). Arrow 1: ATP hydrolysis at the second ABC domain in the LmrA dimer is coupled to: (i) drug efflux at the second membrane domain through the movement of a liganded inside-facing high-affinity site to the outside of the membrane with the concomitant change to low affinity, via a catalytic transition intermediate in which the transport site at the second membrane domain is inaccessible; (ii) the reorientation of an empty outside-facing low-affinity site at the first membrane domain to an inside-facing high-affinity site; and (iii) ATP binding at the first ABC domain. Arrow 2: the first and second LmrA molecules in the dimer have reversed their relationship, and the next ATP hydrolysis step will occur at the first ABC domain. Thus, in a complete drug transport cycle, the LmrA dimer exposes four drug-binding sites in two pairs of two sites via an alternating two-site mechanism. It is important to be aware that although binding sites are presented in separate transmembrane domains in this scheme (for reasons of clarity), they may equally well be present at the interface between transmembrane domains.
LmrA is functionally indistinguishable from mammalian P-glycoprotein (van Veen et al., 1998). Published data for P-glycoprotein, including the biphasic fluorescence quenching by transport substrates of 2-[4′-maleimidylanilino]naphthalene 6-sulfonic acid-labeled P-glyco protein (Sharom et al., 1999), the stimulation of photoaffinity labeling by transport substrates (Safa et al., 1994), the reduced efficiency of photoaffinity labeling with drug analogs after vanadate trapping of P-glycoprotein (Dey et al., 1997; Ramachandra et al., 1998), and the stimulatory effects of rhodamine 123 and Hoechst 33342 (Shapiro and Ling, 1997), and of colchicine and synthetic hydrophobic peptides (Sharom et al., 1996) on the transport of each other, although indirect, are consistent with the two-site model for LmrA.
There is now a substantial body of knowledge about the physiological roles of ABC transporters and their fundamental importance in many cellular processes. However, the molecular basis of transport has remained elusive. The data presented here suggest that the LmrA transporter may function in a fashion analogous to a two-cylinder engine. Insight into the molecular basis of drug transport and recognition by multidrug transporters is vital for our ability to treat multidrug-resistant tumors, and infectious diseases caused by multidrug-resistant microorganisms. The alternating two-site model (two-cylinder engine) presented here provides a basis for further understanding the nature of drug-binding sites, the mechanism of drug translocation and other mechanistic questions relating to ABC-type multidrug transporters. As ABC transporters share sequence identity and a common domain organization, the alternating two-site mechanism may also apply to other members of the ABC superfamily.
Materials and methods
Genetic manipulations
To express monomeric LmrA in L.lactis, the lmrA gene together with upstream regions coding for an N-terminal His6 tag and an enterokinase cleavage site were PCR amplified from pCHLmrA (van Veen et al., 1998) and cloned into the nisA promoter-containing expression vector pNZ804, giving pNHLmrA (Margolles et al., 1999). To express a covalently linked dimer of LmrA, the lmrA gene was PCR amplified from pCHLmrA. The forward primer 5′-TCTAGACCACCATGGGGCATCACCATCACCATCACGATGACGATGACAAAGCCGAAAGAGG-3′ and the reverse primer 5′-CAGGTCGACGGATCCTTGACCAACAGTC-3′ were used to generate an lmrA gene with: (i) a His6 tag, an enterokinase cleavage site and an NcoI site upstream of the His tag; and (ii) a BamHI site and a deleted translational stop codon at the 3′-end of the lmrA gene. A second copy of the lmrA gene, without the His6 tag and enterokinase cleavage site, and with an XbaI site at the 5′-end and a SacI site at the 3′-end, was generated using the forward primer 5′-GATGACAAAGCCTCTAGAGAAAGAGGTCC-3′ and the reverse primer 5′-ATGGCCGACGAGCTCTTATTATTGACCAACAGTC-3′. The human multidrug resistance P-glycoprotein linker region (E634-S671) was amplified from the MDR1 gene from pKSMDR1 (D.Gill, unpublished data), using the forward primer 5′-CAGACAGCAGGATCCGAAGTTG-3′ to introduce a BamHI site at the 5′-end and the reverse primer 5′-GGCTTGTGATCCTCTAGAACTCCTACG-3′ to introduce an XbaI site at the 3′-end. The PCR products were cloned, separately, as NcoI–BamHI, XbaI–SacI and BamHI–XbaI fragments, respectively, in pNZ8048. Subsequently, the BamHI–XbaI fragment and the XbaI–SacI fragment were subcloned in two consecutive steps into pNZ8048 containing the NcoI–BamHI fragment, giving pNZdiLmrA(KK). To express covalently linked dimers of LmrA with the K388M mutation in the first and/or the second ABC domain, the lmrA gene was amplified from pCHLmrAK388M (van Veen et al., 1998) and subcloned as described above, giving pNZdiLmrA(KM), pNZdiLmrA(MK) and pNZdiLmrA(MM). The single-cysteine (G386C) mutant form of lmrA was generated by PCR amplification of the lmrA sequence from pNHLmrA using the mutagenic oligonucleotide 5′-GGTCCTTCTGGTTGTGGTAAATCAACC-3′ and forward and reverse primers as described (van Veen et al., 1998). The G386C mutant lmrA sequence was cloned as an NcoI–XbaI fragment in pNZ8048, yielding pNHLmrAG386C. All PCR amplified DNA fragments were sequenced to ensure that only the intended changes had been introduced.
Cells, membrane vesicles and proteoliposomes
Lactococcus lactis MG1363 was grown in M17 medium (Difco) at 30°C, to an OD660 of ∼1. Nisin was added to a final concentration of 40 pg/ml to induce transcription from the nisA promoter in pNZ8048-derived plasmids, after which the cell suspension was incubated for 1 h at 30°C. The cells were then split into two portions to prepare inside-out and right-side-out membrane vesicles as described previously (Otto et al., 1982; Putman et al., 1999). Membrane vesicles were resuspended in 10 mM Tris–HCl pH 7.4 containing 20% (v/v) glycerol to a final protein concentration of ∼10 mg/ml, and frozen in small aliquots in liquid nitrogen. The sideness of inside-out and right-side-out membrane vesicle preparations generated by these methods has been confirmed by: (i) the accessibility of substrates to the active site of enzymes; (ii) the accessibility of antigenic determinants; (iii) the morphology of freeze-etched membrane vesicles; (iv) the direction of F0F1-H+-ATPase-mediated proton translocation; and (v) the direction of solute transport (Rosen and Tsuchiya, 1979; Otto et al., 1982; van der Does et al., 1996). For the preparation of LmrA-containing proteoliposomes, purified LmrA was reconstituted in preformed dodecylmaltoside-destabilized liposomes containing L.lactis lipids, as described previously (Margolles et al., 1999).
Immunochemistry
Protein was analyzed by SDS–PAGE (10%) followed by western blot analysis using the anti-His6 tag monoclonal antibody DIA900 (Dianova). For immunochemical quantification of the total amount of His-tagged LmrA or dimeric LmrA in inside-out membrane vesicles, membrane vesicles were solubilized in 1× SDS–PAGE sample buffer at a concentration of 10 mg of total membrane protein/ml, and further diluted in 1× SDS–PAGE buffer. A standard solution of purified His-tagged LmrA with known protein concentration [determined from the OD280 and the calculated molar extinction coefficient of the protein (ε280 = 45 090)], was also diluted in 1× SDS–PAGE buffer. Samples and standards (10 µl), containing 10–100 µg of membrane protein, were blotted on a PVDF membrane, stained immunochemically using anti-His tag antibody, and detected by enhanced chemiluminescence (ECL). The number of His tags in a protein sample was estimated by comparison of the density of the ECL signal obtained for this sample with that obtained for the standard samples in which the number of His tags was known.
Transport assays
Ethidium transport. Nisin-induced cells were washed in 50 mM (K)HEPES pH 7.4 containing 2 mM MgSO4, resuspended to an OD660 of 0.5, and kept on ice until use. Ethidium accumulation in cells was measured by fluorimetry as described (van Veen et al., 1996), with excitation and emission wavelengths of 500 and 580 nm, respectively, and slit widths of 2 nm each.
Hoechst 33342 transport. Inside-out membrane vesicles were diluted to a concentration of 1 mg/ml in 2 ml of 50 mM (K)HEPES pH 7.4, supplemented with 2 mM MgSO4, 0.1 mg/ml creatine kinase and 5 mM phosphocreatine. After 1 min of incubation at 30°C, 2 µM Hoechst 33342 was added and the binding of the dye to the membrane vesicles was followed by fluorimetry (excitation and emission wavelength of 355 and 457 nm, respectively, and slit widths of 2 nm) until a steady state was reached. Subsequently, 2 mM Mg-ATP or Mg-ATPγS [both in 50 mM (K)HEPES buffer pH 7.4] was added, and Hoechst 33342 fluorescence followed as a function of time.
Vinblastine transport. Inside-out membrane vesicles (0.1 mg of membrane protein) were diluted as described above in a final volume of 100 µl. After 1 min of incubation at 30°C in the presence of 2 mM Mg-ATP or Mg-ATPγS, 40 nM [G-3H]vinblastine was added. At time points indicated in the legend to Figure 3B, 3 ml of ice-cold 20 mM Tris–HCl pH 7.4 containing 2 mM MgSO4 were added to the reaction mixture. Samples were filtered through a Whatman GF/F glass fiber filter that had been pre-equilibrated overnight at 22°C in wash buffer supplemented with 10% bovine serum albumin (BSA). Filter-retained radioactivity was measured by liquid scintillation counting.
NBD-PE transport. Donor liposomes containing 3 mol% C6-NBD-PE, 6 mol% l-α-phosphatidylethanolamine-N-(lissamine rhodamine B sulfonyl) (egg) (N-Rh-PE) and 91 mol% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine were prepared as described previously (Margolles et al., 1999). NBD-phospholipid transport was measured in 50 mM KPi buffer pH 7.0 containing 2 mM MgSO4, 0.1 mg/ml of creatine kinase, 5 mM phosphocreatine, 1.25 mM Mg-ATP or Mg-AMP-PNP, and 58 µg of donor liposomes. For NEM treatment, proteoliposomes were incubated in the presence of 1 µM NEM for 30 min at 20°C, and washed three times with 50 mM KPi buffer pH 7.0 containing 2 mM MgSO4. Transport was initiated after 2 min by the addition of proteoliposomes to a final protein concentration of 0.7 µg/ml and a donor liposome/proteoliposome ratio of 4. NBD fluorescence [excitation 475 nm (15 nm slit width), emission 530 nm (4 nm slit width)] was monitored at 25°C. The NBD fluorescence increase was fit to:
![]()
where F(t) is the fluorescence as a function of time t, F0 is the fluorescence of NBD-PE at t = 0 s, k1 is the rate constant for the interbilayer movement of NBD-PE between donor liposomes and proteoliposomes, k2 is the rate constant for the transbilayer movement of NBD-PE in proteoliposomes, and A1 and A2 are the amplitudes of the fluorescence development associated with the interbilayer and transbilayer movement of NBD-PE, respectively.
ATPase assay
The ATPase activity of inside-out membrane vesicles was based on a colorimetric ascorbic acid/ammonium molybdate assay in the presence of 20 µM verapamil to measure the liberation of inorganic phosphate from ATP (van Veen et al., 1998). To obtain the LmrA-associated ATPase activity, the ATPase activity in the presence of 1 mM o-vanadate was subtracted from the total ATPase activity.
Photoaffinity labeling
Lactococcus lactis membrane vesicles (50 µg of membrane protein) were diluted in 10 mM Tris–HCl pH 7.4 containing 10 mM MgSO4, 130 nM [3H]APDA (1 Ci/mmol) (Müller et al., 1994), and other additions as indicated in the legend to Figure 6. Unless indicated otherwise, the membrane vesicles were incubated for 2 h at 22°C in the dark in the presence of APDA prior to the photolabeling. To study the ADP/vanadate-induced inhibition of APDA binding to LmrA, 2 mM o-vanadate and 2 mM Mg-ADP were added to the reaction mixtures prior to the addition of APDA, and the reaction mixtures pre-incubated for 15 min at 22°C. Photolabeling of the samples was performed in 1.5 ml microfuge tubes at 4°C by irradiation for 5 min at 350 nm in a Rayonet Photochemical Chamber Reactor Model RPR-100 (The Southern New England Ultraviolet Company, Branford, CT) with 16 UV lamps of 24 W each, and a 10′′ reactor barrel. Samples were solubilized in 5× SDS–PAGE sample buffer at 22°C, and analyzed by SDS–PAGE and autoradiography.
Drug-binding and displacement assays
For equilibrium binding of vinblastine to LmrA, inside-out membrane vesicles (4 µg of membrane protein, or as indicated otherwise) were incubated for 2 h with [G-3H]vinblastine at the indicated concentrations at 22°C in the dark, in a final volume of 100 µl of 50 mM Tris–HCl pH 7.4. The incubation period of 2 h was sufficient to obtain steady-state binding of vinblastine to LmrA and membranes at the indicated vinblastine concentrations. After the incubation period, LmrA was stable for at least two additional hours, as judged from SDS–PAGE analysis, and the steady-state level of vinblastine binding to LmrA. After incubation, 3 ml of ice-cold 20 mM Tris–HCl pH 7.4 containing 2 mM MgSO4 were added. Samples were filtered through Whatman GF/F glass fiber filters that had been pre-equilibrated overnight at 22°C in 20 mM Tris–HCl pH 7.4 containing 2 mM MgSO4 and 10% BSA. Filter-retained radioactivity was measured by liquid scintillation counting. Non-specific binding (usually <30% total) was defined as the amount of [G-3H]vinblastine bound in the presence of 20 µM unlabeled vinblastine and was subtracted from all values. For vinblastine displacement studies, CP100-356 was included in the binding assays at concentrations as indicated in Figures 4 and 6. To study ADP/vanadate-induced inhibition of vinblastine binding to LmrA, 2 mM o-vanadate and 2 mM Mg-ADP were added to the reaction mixture prior to the addition of radiolabeled vinblastine and CP100-356, if required, and the reaction mixture was pre-incubated for 15 min at 22°C.
Modeling of drug binding
For the single-site drug-binding model, the vinblastine binding data were fitted to the hyperbolic equation:

in which vinblastine binding is represented by B, the maximal vinblastine binding by Bmax, the free drug concentration by [S] and the dissociation constant by Kd.
The data were also fitted by the Hill equation:

were KHill is the average binding constant and nHill is the Hill number.
For the cooperative two-site drug-binding model, the fractional saturation Y of the vinblastine-binding sites at steady state equals the ratio of the number of liganded drug-binding sites over the total number of drug-binding sites:

in which T, TS and TS2 refer to unliganded transporter, and to the binary and ternary complex, respectively. Using the equations

equation (1) can be rearranged into:

where B represents the binding of vinblastine at a variable free vinblastine concentration [S], Bmax is the maximal vinblastine binding, and Kd1 and Kd2 are the dissociation constants for the binary complex and ternary complex, respectively. Equation (2) was used for curve fitting of the vinblastine equilibrium binding data.
Heterologous vinblastine displacement by CP100-356 of vinblastine bound to the ternary drug–transporter–drug (TS2) complex was modeled by the multistage equilibrium:
![]()
and was fitted by:

where Ki1 and Ki2 are the dissociation constants for the ternary complex containing one or two CP100-356 molecules, respectively, and Kd2 and Kd1 are the dissociation constants for the ternary complex containing two or one vinblastine molecule, respectively.
Data analysis
Kinetic data were routinely analyzed using the multi-variable analysis programe Sigma Plot (Jandel Scientific). All statistical analyses were performed using Student’s t-test with a 95% confidence interval for the sample mean.
Acknowledgments
Acknowledgements
We would like to thank Piet Borst, Christine Hrycyna, Richard Callaghan and Catherine Martin for discussions, and Simon Scheuring and Andreas Engel for sharing data prior to publication. This research was funded by the EU program on Structural Biology, the Dutch Cancer Society, the Medical Research Council and Cancer Research Campaign. A.M. is a recipient of a TMR fellowship of the EU, H.W.v.V. is a fellow of the Royal Netherlands Academy of Arts and Sciences and C.F.H. is a Howard Hughes International Research Scholar.
References
- Bolhuis H., van Veen,H.W., Molenaar,D., Poolman,B., Driessen,A.J.M. and Konings,W.N. (1996) Multidrug resistance in Lactococcus lactis: evidence for ATP-dependent drug extrusion from the inner leaflet of the cytoplasmic membrane. EMBO J., 15, 4239–4245. [PMC free article] [PubMed] [Google Scholar]
- Dey S., Ramachandra,M., Pastan,I., Gottesman,M.M. and Ambudkar,S.V. (1997) Evidence for two non-identical drug-interaction sites in the human P-glycoprotein. Proc. Natl Acad. Sci. USA, 94, 10594–10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferry D.R., Russell,M.A. and Cullen,M.H. (1992) P-glycoprotein possesses a 1,4-dihydropyridine-selective drug acceptor site which is allosterically coupled to a vinca-alkaloid-selective binding site. Biochem. Biophys. Res. Commun., 188, 440–445. [DOI] [PubMed] [Google Scholar]
- Gottesman M.M., Hrycyna,C.A., Schoenlein,P.V., Germann,U.A. and Pastan,I. (1995) Genetic analysis of the multidrug transporter. Annu. Rev. Genet., 29, 607–649. [DOI] [PubMed] [Google Scholar]
- Greenberger L.M. (1993) Major photoaffinity drug labeling sites for iodoarylazidoprazosin in P-glycoprotein are within, or immediately C-terminal to, transmembrane domains 6 and 12. J. Biol. Chem., 268, 11417–11425. [PubMed] [Google Scholar]
- Higgins C.F. (1992) ABC transporters: from microorganisms to man. Annu. Rev. Cell Biol., 8, 67–113. [DOI] [PubMed] [Google Scholar]
- Higgins C.F., Callaghan,R., Linton,K.J., Rosenberg,M.F. and Ford,R.C. (1997) Structure of the multidrug resistance P-glycoprotein. Semin. Cancer Biol., 8, 135–142. [DOI] [PubMed] [Google Scholar]
- Hrycyna C.A., Ramachandra,M., Ambudkar,S.V., Ko,Y.H., Pedersen,P.L., Pastan,I. and Gottesman,M.M. (1998) Mechanism of action of human P-glycoprotein ATPase activity. Photochemical cleavage during a catalytic transition state using orthovanadate reveals cross-talk between the two ATP sites. J. Biol. Chem., 273, 16631–16634. [DOI] [PubMed] [Google Scholar]
- Kast C., Canfield,V., Levenson,R. and Gros,P. (1996) Transmembrane organisation of mouse P-glycoprotein determined by epitope insertion and immunofluorescence. J. Biol. Chem., 271, 9240–9248. [DOI] [PubMed] [Google Scholar]
- Klein I., Sarkadi,B. and Varadi,A. (1999) An inventory of the human ABC proteins. Biochim. Biophys. Acta, 1461, 237–262. [DOI] [PubMed] [Google Scholar]
- Loo T.W. and Clarke,D.M. (1999a) Determining the structure and mechanism of the human multidrug resistance P-glycoprotein using cysteine-scanning mutagenesis and thiol-modification techniques. Biochim. Biophys. Acta, 1461, 315–325. [DOI] [PubMed] [Google Scholar]
- Loo T.W. and Clarke,D.M. (1999b) The transmembrane domains of the human multidrug resistance P-glycoprotein are sufficient to mediate drug binding and trafficking to the cell surface. J. Biol. Chem., 274, 24759–24765. [DOI] [PubMed] [Google Scholar]
- Margolles A., Putman,M., van Veen,H.W. and Konings,W.N. (1999) The purified and functionally reconstituted multidrug transporter LmrA of Lactococcus lactis mediates the transbilayer movement of specific fluorescent phospholipids. Biochemistry, 38, 16298–16306. [DOI] [PubMed] [Google Scholar]
- Martin C., Berridge,G., Higgins,C.F. and Callaghan,R. (1997) The multidrug resistance reversal agents SR33557 modulates vinca alkaloid binding to P-glycoprotein by an allosteric interaction. Br. J. Pharmacol., 122, 765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechetner E.B., Schott,B., Morse,B.S., Stein,W.D., Druley,T., Davis,K.A., Tsuruo,T. and Roninson,I.B. (1997) P-glycoprotein function involves conformational transitions detectable by differential immunoreactivity. Proc. Natl Acad. Sci. USA, 94, 12908–12913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris D.I., Greenberger,L.M., Bruggeman,E.P., Cardarelli,C., Gottesman,M.M., Pastan,I. and Seamon,K.B. (1994) Localization of the forskolin labeling sites to both halves of P-glycoprotein: similarity of the sites labeled by forskolin and prazosin. Mol. Pharmacol., 46, 329–337. [PubMed] [Google Scholar]
- Müller M., Mayer,R., Hero,U. and Keppler,D. (1994) ATP-dependent transport of amphiphilic cations across the hepatocyte canalicular membrane mediated by MDR1 P-glycoprotein. FEBS Lett., 343, 168–172. [DOI] [PubMed] [Google Scholar]
- Müller M., Bakos,E., Welker,E., Varadi,A., Germann,U.A., Gottesman,M.M., Morse,B.S., Roninson,I.B. and Sarkadi,B. (1996) Altered drug-stimulated ATPase activity in mutants of the human multidrug resistance protein. J. Biol. Chem., 271, 1877–1883. [DOI] [PubMed] [Google Scholar]
- Otto R., Lageveen,R.G., Veldkamp,H. and Konings,W.N. (1982) Lactate efflux-induced electrical potential in membrane vesicles of Streptococcus cremoris. J. Bacteriol., 149, 733–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putman M., Koole,L., van Veen,H.W. and Konings,W.N. (1999) The secondary multidrug transporter LmrP contains multiple drug interaction sites. Biochemistry, 38, 13900–13905. [DOI] [PubMed] [Google Scholar]
- Ramachandra M., Ambudkar,S.V., Chen,D., Hrycyna,C.A., Dey,S., Gottesman,M.M. and Pastan,I. (1998) Human P-glycoprotein exhibits reduced affinity for substrates during a catalytic transition state. Biochemistry, 37, 5010–5019. [DOI] [PubMed] [Google Scholar]
- Rosen B.P. and Tsuchiya,T. (1979) Preparation of everted membrane vesicles from Escherichia coli for the measurement of calcium transport. Methods Enzymol., 56, 233–241. [DOI] [PubMed] [Google Scholar]
- Rosenberg M.F., Callaghan,R., Ford,R.C. and Higgins,C.F. (1997) Structure of the multidrug resistance P-glycoprotein to 2.5 nm resolution determined by electron microscopy and image analysis. J. Biol. Chem., 272, 10685–10694. [DOI] [PubMed] [Google Scholar]
- Safa A.R., Agresti,M., Bryk,D. and Tamai,I. (1994) N-(p-azido-3-[125I]iodophenethyl)spiperone binds to specific regions of P-glycoprotein and another multidrug binding protein, spiperophilin, in human neuroblastoma cells. Biochemistry, 33, 256–265. [DOI] [PubMed] [Google Scholar]
- Senior A.E. and Bhagat,S. (1998) P-glycoprotein shows strong catalytic cooperativity between the two nucleotide sites. Biochemistry, 37, 831–836. [DOI] [PubMed] [Google Scholar]
- Senior A.E. and Gadsby,D.C. (1997) ATP hydrolysis and mechanism in P-glycoprotein and CFTR. Semin. Cancer Biol., 8, 143–150. [DOI] [PubMed] [Google Scholar]
- Senior A.E., Al-Shawi,M.K. and Urbatsch,I.L. (1995) The catalytic cycle of P-glycoprotein. FEBS Lett., 377, 285–289. [DOI] [PubMed] [Google Scholar]
- Shapiro A.B. and Ling,V. (1997) Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur. J. Biochem., 250, 130–137. [DOI] [PubMed] [Google Scholar]
- Sharom F.J., Yu,X., Didiodato,G. and Chu,J.W.K. (1996) Synthetic hydrophobic peptides are substrates for P-glycoprotein and stimulate drug transport. Biochem. J., 320, 421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharom F.J., Liu,R., Romsicki,Y. and Lu,P. (1999) Insights into the structure and substrate interactions of the P-glycoprotein multidrug transporter from spectroscopic studies. Biochim. Biophys. Acta, 1461, 327–344. [DOI] [PubMed] [Google Scholar]
- Sonveaux N., Shapiro,A.B., Goormaghtigh,E., Ling,V. and Ruysschaert,J.M. (1996) Secondary and tertiary structure changes of reconstituted P-glycoprotein. J. Biol. Chem., 271, 24617–24624. [DOI] [PubMed] [Google Scholar]
- Sonveaux N., Vigano,C., Shapiro,A.B., Ling,V. and Ruysschaert,J.M. (1999) Ligand-mediated tertiary structure changes of reconstituted P-glycoprotein. J. Biol. Chem., 274, 17649–17654. [DOI] [PubMed] [Google Scholar]
- Ueda K., Taguchi,Y. and Morishima,M. (1997) How does P-glycoprotein recognize its substrate? Semin. Cancer Biol., 8, 151–159. [DOI] [PubMed] [Google Scholar]
- Ueda K., Matsuo,M., Tanabe,K., Morita,K., Kioka,N. and Amachi,T. (1999) Comparative aspects of the function and mechanism of SUR1 and MDR1 proteins. Biochim. Biophys. Acta, 1461, 305–313. [DOI] [PubMed] [Google Scholar]
- Urbatsch I.L., Sankaran,B., Weber,J. and Senior,A.E. (1995) P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J. Biol. Chem., 270, 19383–19390. [DOI] [PubMed] [Google Scholar]
- Urbatsch I.L., Beaudet,L., Carrier,I. and Gros,P. (1998) Mutations in either nucleotide-binding site of P-glycoprotein (Mdr3) prevent vanadate trapping of nucleotide at both sites. Biochemistry, 37, 4592–4602. [DOI] [PubMed] [Google Scholar]
- van der Does C., den Blaauwen,T., de Wit,J.G., Manting,E.H., Groot,N.A., Fekkes,P. and Driessen,A.J.M. (1996) SecA is an intrinsic subunit of the Escherichia coli preprotein translocase and exposes its carboxyl terminus to the periplasm. Mol. Microbiol., 22, 619–629. [DOI] [PubMed] [Google Scholar]
- van Veen H.W. and Konings,W.N. (1997) Multidrug transporters from bacteria to man: similarities in structure and function. Semin. Cancer Biol., 8, 183–191. [DOI] [PubMed] [Google Scholar]
- van Veen H.W., Venema,K., Bolhuis,H., Oussenko,I., Kok,J., Poolman,B., Driessen,A.J.M. and Konings,W.N. (1996) Multidrug resistance mediated by a bacterial homolog of the human multidrug transporter MDR1. Proc. Natl Acad. Sci. USA, 93, 10668–10672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Veen H.W., Callaghan,R., Soceneantu,L., Sardini,A., Konings,W.N. and Higgins,C.F. (1998) A bacterial antibiotic-resistance gene that complements the human multidrug-resistance P-glycoprotein gene. Nature, 391, 291–295. [DOI] [PubMed] [Google Scholar]
- Wang G., Pincheira,R. and Zhang,J.-T. (1998) Dissection of drug-binding-induced conformational changes in P-glycoprotein. Eur. J. Biochem., 255, 383–390. [DOI] [PubMed] [Google Scholar]