

Abstract
Extracellular concentrations of Ca2+ change rapidly and transiently in the brain during excitatory synaptic activity. To test whether such changes in Ca2+ can play a signaling role we examined the effects of rapidly lowering Ca2+ on the excitability of acutely isolated CA1 and cultured hippocampal neurons. Reducing Ca2+ excited and depolarized neurons by activating a previously undescribed nonselective cation channel. This channel had a single-channel conductance of 36 pS, and its frequency of opening was inversely proportional to the concentration of Ca2+. The inhibition of gating of this channel was sensitive to ionic strength but independent of membrane potential. The ability of this channel to sense Ca2+ provides a novel mechanism whereby neurons can respond to alterations in the extracellular concentration of this key signaling ion.
The entry of Ca2+ into neurons provides a major signal for the regulation of Ca2+-dependent channels and enzymes and contributes to the induction of various forms of synaptic plasticity including long-term potentiation (1). In contrast, little attention has been paid to the possibility that changes in extracellular Ca2+ might themselves play a specific signaling role in controlling postsynaptic excitability. Excitatory activity causes a transient decrease in the extracellular concentration of Ca2+ due to activation of both postsynaptic glutamate receptors of the N-methyl-d-aspartate (NMDA) subtype and voltage-dependent Ca2+ channels (2–6). Decreases in extracellular Ca2+ are most pronounced in the pyramidal cell layers of the hippocampus, where it can fall by as much as 0.6 mM during trains of stimulation. These changes in Ca2+ are further enhanced during seizure activity (2–6).
Lowering Ca2+ reduces its charge-shielding effects on groups located at the membrane surface (7, 8). By this mechanism, Ca2+ nonselectively influences the activation of voltage-dependent currents (8). Calcium also alters the gating and permeation properties of ion channels (7). In some cases, the selectivity of a channel can be lost when Ca2+ is reduced. For example, Na+ will readily permeate Ca2+ channels when extracellular Ca2+ is lowered to nanomolar concentrations (9). A loss of selectivity may account for a rapidly inactivated inward current recorded in neurons in response to a step decrease in extracellular Ca2+ (10).
Ca2+ also blocks a variety of nonselective cation channels. For example, Ca2+ suppresses a nonselective cation current in Xenopus oocytes (11). Membrane depolarization or lowering Ca2+ relieves the inhibition and reveals the presence of this current. Other nonselective cation channels blocked by Ca2+ include nucleotide-gated (12, 13), stretch-activated cation (14), hemi-gap junction (15), and Ca2+-selective channels (16). For most of these channels Ca2+ likely binds to a site located within the channel pore, thus accounting for the voltage dependence of the currents (11).
Activation of NMDA receptors results in an influx of Ca2+ into neurons and a decrease in extracellular Ca2+. NMDA responses inactivate with the influx of Ca2+ (17–20), and this may occur through the binding of calmodulin to the C termini of NMDA receptor subunits (21). Inactivation of these currents can be minimized if extracellular Ca2+ is reduced to or below 200 μM during the application of NMDA (17–20). Using similar approaches we have found that decreasing Ca2+ can itself activate an inward current. Therefore, we have investigated the responses of both cultured and acutely isolated CA1 hippocampal neurons to transient decreases in the extracellular concentration of Ca2+.
METHODS
Cultured mouse hippocampal neurons were grown as previously described (22) and were used for recordings 12–20 days after plating. CA1 pyramidal neurons were isolated from neonatal rats according to Wang and MacDonald (23). The extracellular solution contained 140 mM NaCl/5.4 mM KCl/25 mM Hepes/33 mM glucose, with (0.0005–0.001 mM) or without tetrodotoxin, pH 7.4, using NaOH; 320–335 mosmolar. NMDA and extracellular solutions containing various concentrations of Ca2+ were applied using a multibarreled perfusion apparatus.
Patch electrodes contained 140 mM CsCl or CsF/35 mM CsOH/10 mM Hepes/2 mM MgCl2/11 mM EGTA/2 mM TEA/1 mM CaCl2/4 mM MgATP, pH 7.3, using CsOH; 300 mosmolar. In some experiments no CaCl2 was added and EGTA was replaced with 11 mM 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA). The relative Na+ and K+ permeability ratios PNa/PCs and PK/PCs were determined from the reversal potentials in Na+-rich (150 mM NaCl/25 mM Hepes/33 mM glucose) or K+-rich (20 mM KCl/130 mM N-methyl-d-glucamine/25 mM Hepes/33 mM glucose) solutions using the constant field equation in the following form:
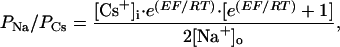 |
1 |
where E is the reversal potential and F, R, and T have their usual meanings.
The single-channel open probability was determined from the ratio of the time spent in the open state to the duration of recording: Po = (t1 + t2 +… + tn)/Nttot, where t is the amount of time that n channels are open and N is the maximum number of levels observed in the patch. Data are expressed as the mean ± SEM, and a paired Student’s t test was employed.
RESULTS
In the presence of physiological concentrations of both Ca2+ and Mg2+ a rapid reduction in Ca2+ from 1.5 to 0.5 mM strongly depolarized and excited neurons (Fig. 1A). This graded depolarization was readily reversible and it was insensitive to tetrodotoxin (Fig. 1B). In such recordings (Fig. 1 C and D) a threshold for excitation was detected with as little as a 100 μM decrease in Ca2+.
Figure 1.

The effect of lowering Ca2+ on the excitability of hippocampal neurons. (A) The membrane potential of a cultured neuron was recorded using the whole-cell current-clamp configuration (KCl instead of CsF in the recording pipette). Decreasing Ca2+ from 1.5 to 0.5 mM for a period of 2 s depolarized and strongly excited the cell. (B) In the presence of 0.5 μM tetrodotoxin and 1.5 mM Mg2+, reductions of Ca2+ from 1.3 to 1.2, 1.0, 0.5, 0.2, and 0 mM respectively, evoked graded depolarizations. (C) In a series of six recordings, the amplitude of the depolarization was recorded and values plotted versus the concentration of Ca2+. The control solution contained 1.3 mM Ca2+, and all of the solutions contained 1.5 mM Mg2+. (D) The normalized dose–response curve for the depolarization induced by lowering Ca2+. The depolarization was normalized to that induced by the solution containing no added Ca2+. The nominal concentration of Ca2+ in this solution was estimated to be about 20 μM (24). The logistic equation in the form R = Rmax/[1 + (D/EC50)nH] was used to fit the points. R represents the normalized depolarization at any given Ca2+ concentration, Rmax is the response with no added Ca2+, the EC50 value is the concentration of Ca2+ that produced 50% of the maximal response, and nH is the estimated Hill coefficient (EC50 = 0.39 ± 0.11 mM; nH = 1.4 ± 0.3; n = 6).
To examine the mechanism of this excitation, cells were bathed in a solution lacking Mg2+ and voltage clamped to a holding potential of −60 mV. Reducing Ca2+ from 2 to 0 mM (nominally free) induced a slowly activating (τ = 992 ± 85 ms, n = 28) sustained inward current that rapidly recovered (Fig. 2 A and B). The amplitude of this current ranged from 150 to 1,500 pA (mean 803 ± 120 pA, n = 8, acutely isolated CA1 neurons; 745 ± 30 pA, n = 76, cultured hippocampal neurons). The reversal potential for the response to low Ca2+ was approximately 0 mV (Fig. 2B).
Figure 2.

Lowering Ca2+ induces a nonspecific cation current in cultured and isolated hippocampal neurons. (A) A step reduction in the concentration of Ca2+ evoked a substantial inward current in a cultured hippocampal neuron. This inward current was associated with an increase in membrane conductance as assessed by repeating a depolarizing voltage step. (B) In a group of 15 neurons the current–voltage relationships (I-V) were determined for the response to a change from 2 to 0 mM Ca2+ while holding at values from −60 to +20 mM. The currents demonstrated a near-linear I-V curve and reversed at +0.3 ± 1.8 mV. (C) Superimposed responses to NMDA (100 μM, 3 μM glycine) with and without a simultaneous decrease in Ca2+ (to 200 μM) are shown for two cultured neurons (a and b). The smallest current in each case is the superimposed response to a decrease in Ca2+ alone. A response of a HEK293 cell to a decrease in Ca2+ (2 mM to 200 μM) alone (NMDA was not applied) is shown before and following a partial block with Gd3+ (2 μM) (c). These currents ranged from 50 to 1500 pA in different HEK cells.
In some neurons we examined NMDA-induced currents as well as responses to lowered concentrations of Ca2+. NMDA currents demonstrated various degrees of inactivation (Fig. 2C, a and b). When the concentration of Ca2+ was simultaneously reduced to 200 μM, inactivation of the currents was reduced (Fig. 2C). However, lowering Ca2+ itself generated a current of a magnitude, that, with appropriate kinetics, was sufficient to account for the apparent loss of inactivation in some cases (Fig. 2C, a) and contribute to the apparent reduction of inactivation in others (Fig. 2C, b). The responses to low Ca2+ were insensitive to competitive antagonists of NMDA (AP5, 100 μM) and AMPA receptors (CNQX, 100 μM), demonstrating that these responses were not the consequence of activation of glutamate receptors. We also examined the response of human epithelial kidney (HEK293) cells to step reductions in Ca2+ because recombinant NMDA receptors are most often expressed in these cells. Reductions of Ca2+ in the absence of NMDA also evoked large inward currents in some of these cells (Fig. 2C, c).
The current evoked by lowering Ca2+ was dependent on the presence of either extracellular Na+ or K+ (Fig. 3A). Reducing the concentrations of both NaCl and KCl in the extracellular solution by half shifted the reversal potential toward more negative values (a 15.3 ± 2.8 mV shift, n = 4; Fig. 3 B and C) as anticipated for a nonselective cation current. A more detailed analysis showed the following rank order of relative permeability ratios: Na+ (1.3) ≥ K+ (1.28) > Cs+ (1). In contrast, no anion permeability was detected and there was no shift in the reversal potential when all extracellular Cl− was substituted with gluconate (Fig. 3 D and E). In addition, no currents were detected in response to a decrease of Ca2+ from 2.0 to 0.5 mM when all monovalent cations were replaced with the nonpermeant ion N-methyl-d-glucamine (not shown), suggesting that the Ca2+ permeability of these channels is unlikely to be substantial. Suction or pressure applied to the patch electrode did not alter the responses to lowered Ca2+.
Figure 3.

The inward current evoked by decreasing Ca2+ is carried by Na+ and K+ but not by Cl−. (A) Example traces showing the responses of cultured hippocampal neurons to a decrease in Ca2+. Ca2+ was reduced from 2.0 to 0 mM in four different extracellular solutions: (i) No Na+ or K+, replacement with 150 mM NMDG; (ii) in the presence of 140 NaCl and 5.4 mM KCl; (iii) in 20 mM NaCl; and (iv) in 20 mM KCl. In the absence of monovalent cations, step reductions of Ca2+ evoked little or no inward current, demonstrating that the response was not due to some kind of nonspecific breakdown of the membrane. However, when the usual extracellular solution (140 NaCl, 5.4 KCl) was used, an inward current with an amplitude of about 500 pA was recorded from the same cell. A smaller current was recorded when the extracellular solution contained only 20 mM Na+ or only K+. The holding potential was −60 mV. (B) A step reduction in the concentration of Ca2+ was used to evoke responses in the presence of a solution containing 150 mM NaCl and 5.4 mM KCl or a second solution with half the concentration of Na+ (75 mM) and K+ (2.7 mM). In each case the holding potential of the cell was varied between −60 and +20 mV. (C) Such data (n = 3) was then used to construct I-V curves. When the concentration of monovalent ions was reduced by one-half, the reversal potential shifted toward more hyperpolarized values (see text). (D) Substitution of all of the extracellular Cl− with gluconate had little influence on the responses to lowered Ca2+ and did not alter the I-V curve (n = 3) for this response (E).
A number of drugs, reported to block nonselective cation channels, including flufenamic acid (25), spermine (26, 27), and amiloride (28), reversibly reduced the amplitude of this inward current (−60 mV, 100 μM flufenamic acid, 39.7 ± 5.2%, n = 6, P < 0.01; 1 mM amiloride, 63.7 ± 8.4%, n = 7, P < 0.01; 1 mM spermine, 49.0 ± 10.4%, n = 3, P < 0.05). On the other hand, the inward current evoked by lowering Ca2+ was unaltered by the inclusion of nucleotides (e.g., GTP[γS], 1.5 mM; GDP[βS], 0.5 mM) in the recording pipette, suggesting that it was not mediated by a G protein-coupled channel or by a nucleotide-gated cation channel (29), nor was it eliminated by the addition of high concentrations of either EGTA or BAPTA (Ca2+ chelators) to the recording pipette.
Hablitz et al. (10) reported that reductions in Ca2+ evoked a rapidly inactivating inward current in avian dorsal root ganglia neurons that was blocked by verapamil and Cd2+. They attributed this current to voltage-dependent Ca2+ channels that had lost their ionic selectivity. In contrast, the current we observed in central neurons was noninactivating and it was not blocked (not shown) by the Ca2+ channel antagonists verapamil (50 μM) or nitrendipine (50 μM).
We next examined the relative sensitivity of this current to various divalent cations (Fig. 4). A change in Ca2+ from 2 to 0 mM was used to activate the current, and differing concentrations of test cations were then added to construct dose-inhibition relationships. The concentration of Ca2+ that inhibited 50% of the current (IC50) was 0.15 mM (±0.04 mM; n = 7) (Fig. 4B). Magnesium was less potent with an IC50 of 0.34 mM (± 0.10 mM, n = 5) as were Ba2+ (IC50 = 0.32 ± 0.10 mM, n = 5) and Cd2+ (IC50 = 0.37 ± 0.05 mM, n = 4).
Figure 4.

Divalent and polyvalent cations block the inward currents induced by lowering Ca2+. (A) Dose-dependent block of inward currents. The inward current was induced by a reduction of Ca2+ from 2 to 0 mM at a holding potential of −60 mV. Different concentrations of cations were added to the low Ca2+ solution, and the normalized inhibition of this current was plotted against the concentration of a given cation (B). The dose-inhibition curves were fitted using the logistic equation. The doses that produced 50% block of the current were: Gd3+ (◊), 0.0014 ± 0.00049 mM (n = 5); neomycin (⧫), 0.0043 ± 0.0011 mM (n = 6); Ca2+ (○), 0.15 ± 0.04 mM (n = 7); Ba2+ (•), 0.32 ± 0.10 mM (n = 4); Mg2+ (▵), 0.34 ± 0.14 mM (n = 5); and Cd2+ (▴), 0.37 ± 0.05 mM (n = 4). Higher concentrations of neomycin activated an additional inward current, thus apparently limiting the inhibition of the nonspecific cation current. (C) The apparent affinity of divalent cations was increased when the ionic strength in the extracellular solution was reduced. The open symbols represent Ca2+ (○) and Mg2+ (▵) dose-inhibition curves in the presence of solutions of normal ionic strength (140 mM NaCl, 5.4 mM KCl), and the filled symbols represent Ca2+ (•) and Mg2+ (▴) in the presence of solution of reduced ionic strength (35 mM NaCl, 1.4 mM KCl, supplemented with sucrose). The recording pipettes contained either CsCl (or CsF) and 2 mM TEA.
Gadolinium and neomycin are potent activators of a recently cloned calcium-sensing receptor (30). Furthermore, mechanosensitive, nonselective cation channels are highly sensitive to Gd3+ (31). Therefore, we tested the effects of Gd3+ and neomycin on the current generated by reducing Ca2+ (Fig. 4 A and B). Gadolinium was the most potent blocker of this current, with an IC50 of 1.4 μM (±0.49, n = 5), followed by neomycin (4.3 ± 1.1 μM, n = 6).
Calcium functions to stabilize membrane potential by shielding negatively charged groups on the plasma membrane. By this means, Ca2+ can influence the gating properties of a number of ion channels (7, 8). To further explore the possibility that this mechanism accounted for the activation of the nonselective cation current, we reduced the ionic strength of the extracellular solution by 25%. This would reduce the shielding effects of monovalent cations and thus reduce their potential competition with divalent cations at these sites. Consistent with this hypothesis, reducing the ionic strength increased the apparent affinity for Ca2+ and Mg2+ by a factor of 3 to 4 (Fig. 4C).
Many of the previously described nonselective cation channels are voltage-dependent or at least demonstrate a voltage-dependent blockade by Ca2+. We therefore examined whether or not the nonselective cation current in neurons showed any voltage dependency. A series of voltage steps was made in the presence and absence of Ca2+ (Fig. 5A). No voltage relaxations were observed during these steps (Fig. 5A, c) and the current-voltage relationships were linear, demonstrating that this current is not voltage-dependent. In another series of cells we activated this current by decreasing Ca2+ to one of two different values (0 or 0.5 mM) while the holding potential was set at values between −60 and +20 mV (Fig. 5B). The steady-state relationships between holding potential and current amplitude were about linear for both Ca2+ concentrations.
Figure 5.

The nonselective cation current demonstrates little or no voltage dependence. (A) An example recording from a cultured hippocampal neuron (holding, −40 mV). In (a) the response to a series of voltage steps from −120 to +80 mV in the presence of Ca2+ (2 mM) is shown. The same series was then repeated following a decrease of Ca2+ to nominally free values (b), and the difference responses (b-a) were calculated (c). The dotted line marks the initial holding current. No current relaxations were observed, and the I-V curve (d) was linear. Identical results were observed in another four cells. The reversal potential was slightly more positive than 0 mV because of the pipette solution that had been diluted (120 mM CsF). (B) Steady-state currents in response to two different changes in Ca2+ concentration (0 and 0.5 mM) were also examined. The I-V curves (n = 3) for each were approximately linear over the range tested, and both reversed at 0 mV, demonstrating that Ca2+ did not cause an obvious voltage-dependent block.
Using outside-out patches we then examined whether or not we could detect single-channel events in response to decreases in Ca2+. Reducing Ca2+ consistently induced inward current channel openings (Fig. 6A). The relationship between holding potential and the amplitude of single-channel currents was near linear (Fig. 6B), yielding a single-channel conductance of 36 pS. In another series of patches, the open probability (Po) of these channels was substantially reduced by 2 μM Gd3+ (Fig. 6C) without any change in the single-channel amplitude (control Po = 0.21 ± 0.13; Gd3+ Po = 0.10 ± 0.07, P < 0.05, n = 5). Increasing Ca2+ to 0.5 mM also decreased the single-channel open probability (0 mM Ca2+, Po = 0.24 ± 0.09; 0.5 mM Ca2+, Po = 0.09 ± 0.05, P < 0.01, n = 4) without affecting either single-channel amplitude or open times. This evidence demonstrates that both Gd3+ and Ca2+ primarily act to inhibit the gating of this nonselective cation channel.
Figure 6.

Single-channel recordings in outside-out patches taken from cultured hippocampal neurons. (A) An example record showing the activation of single-channel openings in response to a step reduction in the concentration (2 to 0 mM) of Ca2+. The holding potential was −60 mV. (B) I-V relationship for single-channel currents. Representative trances show the activation of these currents at holding potentials ranging from −60 to 0 mV. The I-V curve was constructed from the results of recordings from eight such patches. The channel showed a linear I-V relationship with a slope conductance of 36 ± 2.6 pS (n = 8). (C) Effect of Gd3+ on single-channel currents. Gd3+ (0.002 mM) significantly decreased the channel-open probability (control Po = 0.21 ± 0.13; Gd3+ Po = 0.10 ± 0.07, n = 5, P < 0.05) without having any effect on single-channel amplitude.
DISCUSSION
Our results demonstrate that hippocampal neurons possess a unique nonselective cation channel whose gating is sensitive to changes in the concentration of extracellular Ca2+. Even in the presence of Mg2+, which can itself inhibit gating of this channel, decreases in Ca2+ were sufficient to activate inward currents and excite neurons. Furthermore, during intense synaptic excitation, decreases in extracellular Ca2+ are often associated with simultaneous decreases in extracellular Mg2+ (2–6), which might further accentuate activation in these channels.
The current we have described resembles in some aspects that observed in Xenopus oocytes. Both currents are activated by lowering Ca2+, both are slowly activating and show little inactivation, and both are mediated by channels that demonstrate little selectivity between Na+ and K+ but have little Ca2+ permeability and exclude anions (11). Also, like stretch-activated channels, both oocyte and hippocampal currents are highly sensitive to Gd3+ (11, 14, 31). The hippocampal channel is also sensitive to flufenamic acid and high concentrations of amiloride, as are stretch-activated cation channels. However, the nonselective cation channel observed in hippocampal neurons demonstrates no intrinsic voltage dependency, and the block by Ca2+ and Gd3+ is not voltage-dependent in contrast with most other nonselective cation channels (12, 13). Some hemi-gap channels are also voltage-dependent but they are permeable to anions, which differentiates them from the hippocampal channel (32).
The sensitivity of the hippocampal nonselective cation channel to various cations and polyvalent cations is remarkably similar to that reported for the calcium-sensing receptor (30). At least one form of this receptor has been cloned from the rat brain, and it is expressed in cultured hippocampal neurons (33). This receptor is linked to G protein activation and it is most homologous to metabotropic glutamate receptors. However, it is highly unlikely that this receptor itself forms an ion channel (30). Furthermore, Ca2+ and polyvalent cations activate this receptor while they inhibit the gating of the nonselective cation channel expressed in hippocampal neurons. The lack of dependence of this nonselective cation channel upon the presence of intracellular GTP also suggests that there is no direct link between this receptor and the nonselective cation channel described in this paper. It seems more likely that both activation of calcium-sensing receptor and gating of the nonselective cation channel are controlled by a similar mechanism, that is, a shielding of negative surface charges.
The high selectivity of Ca2+ channels is compromised by lowering Ca2+ to concentrations below the micromolar range (16, 34). However, the nonselective cation channels described here could be activated by decreases in Ca2+ at a far higher range of concentration (e.g., from 2.0 to 0.5 mM). Furthermore, they demonstrated no voltage dependency, and they were insensitive to at least some Ca2+ channel antagonists (e.g., verapamil and nitrendipine). Similarly, the selectivity of K+ channels (35, 36) may be lost when Ca2+ is lowered. These modified K+ channels were insensitive to Mg2+ and were blocked by extracellular K+. This strongly contrasts with the blockade of hippocampal nonselective cation channels by Mg2+ and their high relative permeability to K+.
Our results demonstrate that the practice of simultaneously lowering Ca2+ when applying NMDA may be inappropriate. The relatively slow onset of the nonselective current, together with its lack of inactivation, will tend to contaminate and obscure NMDA-induced inactivation. However, this contamination clearly does not account for all of the effects of lowered Ca2+ on inactivation of NMDA currents (Fig. 2C, b). The nonselective cation channel also shares a number of other properties with NMDA channels (e.g., both are blocked by divalent cations, spermine, and flufenamic acid). Its presence may have complicated the interpretation of some of these studies (25–27).
Tetanic stimulation of hippocampal afferents depolarizes CA1 neurons sufficiently to relieve the voltage-dependent blockade of NMDA receptors by Mg2+. The resulting sustained influx of Ca2+ through these receptors then contributes to the induction of long-term potentiation (1). However, the resulting decrease in extracellular Ca2+ could also potentially activate nonselective cation channels, leading to a further depolarization and amplification of the excitatory signal. Such a mechanism could also serve to synchronize and amplify paroxysmal bursting of neurons, which is characteristic of seizure activity. In this respect, the nonselective cation channels might be anticipated to play a role in the epileptogenic-like bursting of neurons that is observed when hippocampal slices are superfused with solutions containing either low concentrations of Ca2+ (37) or Mg2+ (38).
Acknowledgments
We thank Ms. L. Brandes and E. Czerwinska for technical assistance and Drs. P. Ascher, M. W. Salter, and B. A. Orser for comments on the manuscript. This work was supported by the Medical Research Council of Canada and by the National Centres of Excellence. Z.X. is a joint Medical Research Council and National Centres of Excellence fellow, and W.L. is a National Centres of Excellence fellow.
ABBREVIATION
- NMDA
N-methyl-d-aspartate
References
- 1.Bliss T V P, Collingridge G L. Nature (London) 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 2.Heinemann U, Lux H D, Gutnick M J. Exp Brain Res. 1977;27:237–243. doi: 10.1007/BF00235500. [DOI] [PubMed] [Google Scholar]
- 3.Heinemann U, Stabel J, Rausche G. Prog Brain Res. 1990;83:197–214. doi: 10.1016/s0079-6123(08)61250-9. [DOI] [PubMed] [Google Scholar]
- 4.Heinemann U, Hamon B. Exp Brain Res. 1986;65:1–10. doi: 10.1007/BF00243826. [DOI] [PubMed] [Google Scholar]
- 5.Kohr G, Heinemann U. Epilepsy Res. 1989;4:187–200. doi: 10.1016/0920-1211(89)90003-x. [DOI] [PubMed] [Google Scholar]
- 6.Krnjevic K, Morris M E, Reiffenstein R J. Can J Physiol Pharmacol. 1982;60:1643–1657. doi: 10.1139/y82-243. [DOI] [PubMed] [Google Scholar]
- 7.Hille B. Ionic Channels of Excitable Membranes. Sunderland, MA: Sinauer; 1992. [Google Scholar]
- 8.Zhou W, Jones S W. J Gen Physiol. 1995;105:441–462. doi: 10.1085/jgp.105.4.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hess P, Tsien R W. Nature (London) 1984;309:453–456. doi: 10.1038/309453a0. [DOI] [PubMed] [Google Scholar]
- 10.Hablitz J J, Heinemann U, Lux H D. Biophys J. 1986;50:753–757. doi: 10.1016/S0006-3495(86)83515-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arellano R O, Woodward R M, Miledi R. J Physiol (London) 1995;484:593–604. doi: 10.1113/jphysiol.1995.sp020689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Root M J, MacKinnon R. Neuron. 1993;11:459–466. doi: 10.1016/0896-6273(93)90150-p. [DOI] [PubMed] [Google Scholar]
- 13.Frings S, Seifert R, Godde M, Kaupp U B. Neuron. 1995;15:169–179. doi: 10.1016/0896-6273(95)90074-8. [DOI] [PubMed] [Google Scholar]
- 14.Yang X, Sachs F. Science. 1989;243:1068–1071. doi: 10.1126/science.2466333. [DOI] [PubMed] [Google Scholar]
- 15.Malchow R P, Qian H, Ripps H. J Neurosci Res. 1993;35:237–245. doi: 10.1002/jnr.490350303. [DOI] [PubMed] [Google Scholar]
- 16.Almers W, McCleskey E W. J Physiol (London) 1984;353:585–608. doi: 10.1113/jphysiol.1984.sp015352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clark G D, Clifford D B, Zorumski C F. Neuroscience. 1990;39:787–797. doi: 10.1016/0306-4522(90)90261-2. [DOI] [PubMed] [Google Scholar]
- 18.Vyklicky L., Jr J Physiol (London) 1993;470:575–600. doi: 10.1113/jphysiol.1993.sp019876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Legendre P, Rosenmund C, Westbrook G L. J Neurosci. 1993;13:674–684. doi: 10.1523/JNEUROSCI.13-02-00674.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenmund C, Westbrook G L. J Physiol (London) 1993;470:705–729. doi: 10.1113/jphysiol.1993.sp019884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ehlers M D, Zhang S, Bernhardt J P, Huganir R L. Cell. 1996;84:745–755. doi: 10.1016/s0092-8674(00)81052-1. [DOI] [PubMed] [Google Scholar]
- 22.MacDonald J F, Mody I, Salter M W. J Physiol (London) 1989;414:17–34. doi: 10.1113/jphysiol.1989.sp017674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L-Y, MacDonald J F. J Physiol. 1995;486.1:83–95. doi: 10.1113/jphysiol.1995.sp020792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoth M. Pflügers Arch. 1995;430:315–322. doi: 10.1007/BF00373905. [DOI] [PubMed] [Google Scholar]
- 25.Lerma J, Del Río R M. Mol Pharmacol. 1992;41:217–222. [PubMed] [Google Scholar]
- 26.Lerma J. Neuron. 1992;8:343–352. doi: 10.1016/0896-6273(92)90300-3. [DOI] [PubMed] [Google Scholar]
- 27.Benveniste M, Mayer M L. J Physiol (London) 1993;464:131–163. doi: 10.1113/jphysiol.1993.sp019627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lane J W, McBride D W, Jr, Hamill O P. Br J Pharmacol. 1992;106:283–286. doi: 10.1111/j.1476-5381.1992.tb14329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eismann E, Bonigk W, Kaupp U B. Cell Physiol Biochem. 1993;3:332–351. [Google Scholar]
- 30.Brown E M, Gamba G, Riccardi D, Lombardl M, Butters R, Klfor O, Sun A, Hedlger M A, Lytton J, Hebert S C. Nature (London) 1993;366:575–580. doi: 10.1038/366575a0. [DOI] [PubMed] [Google Scholar]
- 31.Sackin H. Annu Rev Physiol. 1995;57:333–353. doi: 10.1146/annurev.ph.57.030195.002001. [DOI] [PubMed] [Google Scholar]
- 32.De Vries S W, Schwartz E A. J Physiol. 1992;445:201–230. doi: 10.1113/jphysiol.1992.sp018920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruat M, Molliver M E, Snowman A M, Snyder S H. Proc Natl Acad Sci USA. 1995;92:3161–3165. doi: 10.1073/pnas.92.8.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellinor P T, Yang J, Sather W A, Zhang J F, Tsien R W. Neuron. 1995;15:1121–1132. doi: 10.1016/0896-6273(95)90100-0. [DOI] [PubMed] [Google Scholar]
- 35.Armstrong C M, Lopez-Barneo J. Science. 1987;236:712–714. doi: 10.1126/science.2437654. [DOI] [PubMed] [Google Scholar]
- 36.Armstrong C M, Miller C. Proc Natl Acad Sci USA. 1990;87:7579–7582. doi: 10.1073/pnas.87.19.7579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jefferys J G R, Haas H L. Nature (London) 1982;300:448–450. doi: 10.1038/300448a0. [DOI] [PubMed] [Google Scholar]
- 38.Mody I, Lambert J D C, Heinemann U. J Neurophysiol. 1987;57:869–888. doi: 10.1152/jn.1987.57.3.869. [DOI] [PubMed] [Google Scholar]