

Abstract
The Saccharomyces cerevisiae protein kinase Rim15 was identified previously as a component of the Ras/cAMP pathway acting immediately downstream of cAMP-dependent protein kinase (cAPK) to control a broad range of adaptations in response to nutrient limitation. Here, we show that the zinc finger protein Gis1 acts as a dosage-dependent suppressor of the rim15Δ defect in nutrient limitation-induced transcriptional derepression of SSA3. Loss of Gis1 results in a defect in transcriptional derepression upon nutrient limitation of various genes that are negatively regulated by the Ras/cAMP pathway (e.g. SSA3, HSP12 and HSP26). Tests of epistasis as well as transcriptional analyses of Gis1-dependent expression indicate that Gis1 acts in this pathway downstream of Rim15 to mediate transcription from the previously identified post-diauxic shift (PDS) element. Accordingly, deletion of GIS1 partially suppresses, and overexpression of GIS1 exacerbates the growth defect of mutant cells that are compromised for cAPK activity. Moreover, PDS element-driven expression, which is negatively regulated by the Ras/cAMP pathway and which is induced upon nutrient limitation, is almost entirely dependent on the presence of Gis1.
Keywords: cAPK/Gis1/nutrient signaling/PDS element/yeast
Introduction
The yeast Saccharomyces cerevisiae copes with diverse environmental conditions by coordinated regulation of growth, cell cycle progression and metabolic activities. The Ras/cAMP pathway is essential for the control and integration of these processes, in particular with respect to the nutritional status. The dramatic reprogramming of the metabolism at the diauxic transition, when glucose becomes limiting, as well as some of the subsequent adaptations during both the post-diauxic phase, when cells grow respiratively on ethanol, and during entry into stationary phase, are negatively controlled by the Ras/cAMP pathway. Thus, cells with elevated cAMP-dependent protein kinase (cAPK) activity fail to accumulate carbohydrate reserves, to undergo a proper diauxic transition and to arrest in G1 upon nutrient limitation. As a result, such cells remain highly sensitive to heat stress and rapidly lose viability in stationary phase. In contrast, cAPK-deficient cells exhibit physiological changes normally associated with nutrient limitation, such as G1 cell cycle arrest, accumulation of storage carbohydrates and increased resistance towards heat and oxidative stress (for reviews and further details on the Ras/cAMP pathway, see Tatchell, 1986; Broach and Deschenes, 1990; Thevelein and de Winde, 1999; for a review on stationary phase, see Werner-Washburne et al., 1993).
The mechanism by which cAPK controls growth is still an issue of conjecture. One mechanism by which cells may control proliferation has recently been suggested to be based on cAPK-mediated regulation of G1 cyclin translation (Hall et al., 1998). In addition, it was found that modulation of Rap1 transcriptional activity by cAPK mediates growth-regulated expression of ribosomal protein genes (Klein and Struhl, 1994). However, positive cAPK-mediated regulation of ribosomal protein synthesis may not be the only reason for the dependence of growth on cAPK (Klein and Struhl, 1994; Neumann-Silberberg et al., 1995) and some of the growth-related effects of cAPK may be due to changes in additional transcriptional control mechanisms. For instance, transcriptional derepression/activation of a large number of yeast genes (e.g. CTT1, DDR1, HSP12 and TPS2) is negatively regulated by cAPK through one or more AG4 stress-responsive elements (STREs), which confer transcriptional activation in response to a wide range of stresses, including heat, oxidative and osmotic shocks, and nutrient limitation (for reviews, see Mager and De Kruijff, 1995; Ruis and Schüller, 1995). Control of STRE-driven gene expression by cAPK is mediated via Msn2 and Msn4, a pair of partially redundant zinc finger transcription factors that recognize and bind to STRE (Estruch and Carlson, 1993; Martínez-Pastor et al., 1996; Schmitt and McEntee, 1996; Görner et al., 1998). Surprisingly, loss of Msn2 and Msn4 renders cells largely independent of cAPK activity, suggesting that an essential function of cAPK is to inhibit expression of growth-inhibitory genes that are under the control of Msn2 and Msn4 (Smith et al., 1998). In agreement with this suggestion, Msn2/Msn4 function is also required for expression of YAK1, a gene whose product was shown previously to antagonize cAPK-dependent growth (Garrett and Broach, 1989; Garrett et al., 1991; Smith et al., 1998). cAPK-mediated control of gene expression is also exerted via additional transcriptional regulators. Accordingly, expression of the glucose-repressible ADH2 gene has been found to be regulated mainly by cAPK-dependent inactivation of the transcriptional activator Adr1 (Cherry et al., 1989), while expression of GAC1 and SSA3 was reported possibly to be regulated by cAPK-dependent activation of the transcriptional repressor Sok2 (Ward et al., 1995). Since overexpression of SOK2 can partially relieve dependence on cAPK function, Sok2 acts as a cAPK-dependent repressor of one or more genes whose products inhibit cell proliferation.
Several observations suggest the existence of additional cAPK-dependent transcriptional control mechanisms. For instance, expression of GSY2 was recently found to be partially repressed by the cAPK pathway through an unknown STRE-independent mechanism (Parrou et al., 1999). Similarly, a large-scale analysis of gene expression in a msn2 msn4 double mutant (Boy-Marcotte et al., 1998), as well as a screen for cAMP-repressible genes (Tadi et al., 1999), revealed a subset of Msn2/Msn4-independent genes whose expression is negatively controlled by cAMP. One cAPK-regulated element that may confer Msn2/Msn4-independent transcriptional control is the T(T/A)AG3AT post-diauxic shift (PDS) element, which mediates transcriptional activation in response to nutritional limitation (Boorstein and Craig, 1990). Unlike the very similar STRE, it does not mediate transcriptional activation in response to other stresses such as heat shock (Boorstein and Craig, 1990), indicating that STRE and PDS elements may be controlled by different cAPK-dependent mechanisms. In agreement with such a model, Msn2 and Msn4 were found to be dispensable for the nutrient limitation-induced transcriptional activation of SSA3, a gene with several PDS elements, but no conserved STRE in its promoter region (Martínez-Pastor et al., 1996). In this context, it is interesting to note that transcriptional derepression/activation of SSA3 at the diauxic transition is almost entirely dependent on the presence of the protein kinase Rim15, which has been identified as a component of the Ras/cAMP pathway acting immediately downstream and under negative control of cAPK to control a broad range of adaptations in response to nutrient limitation (Reinders et al., 1998).
Here we describe the identification of GIS1 as a dosage-dependent suppressor of the rim15Δ defect to derepress/activate SSA3 upon nutrient limitation. GIS1 encodes a zinc finger transcription factor, which has recently been isolated as a damage-responsive repressor of the photolyase-encoding PHR1 gene (Jang et al., 1999), as well as a high-copy suppressor of the Gal– phenotype of a snf1 mig1 srb8 mutant (Balciunas and Ronne, 1999). Loss of Gis1 results in a defective response of mutant cells to nutrient limitation, including a defect in transcriptional derepression/activation of SSA3. Tests of epistasis as well as transcriptional analyses of Gis1-dependent expression suggest that Gis1 acts in the Ras/cAMP pathway downstream of Rim15 specifically to control transcriptional activation through the PDS element. Interestingly, overexpression of GIS1 prevents growth, and loss of Gis1 partially relieves the dependence on cAPK function, indicating that Gis1 activates one or more genes whose products inhibit cell proliferation.
Results
Identification of GIS1 as dosage suppressor of rim15Δ
We have previously reported that loss of Rim15 results in a defective response to nutrient limitation: mutant cells lacking Rim15 fail to accumulate trehalose, to derepress HSP12, HSP26 and SSA3, to induce thermotolerance and to arrest properly in G1 (Reinders et al., 1998). To identify potential downstream effectors of Rim15, we performed a screen for dosage-dependent suppressors of the rim15Δ defect in nutrient limitation-induced transcriptional derepression of SSA3. Accordingly, strain AR2 (rim15Δ/rim15Δ) containing the reporter plasmid pWB204Δ-236 (SSA3-lacZ; Boorstein and Craig, 1990) was transformed with a genomic DNA library under the control of the strong, inducible GAL1 promoter (Ramer et al., 1992). We rescued library plasmids from cells in which the reporter gene showed galactose-dependent transcription after replica-plating and incubation for 2 days on selective X-gal plates (containing 2% galactose and 1% raffinose), and assigned the plasmids to 48 different classes by restriction analysis. To isolate specifically downstream effectors of the Ras/cAMP/Rim15 pathway, we further tested the positive plasmids for their ability to exacerbate the growth defect of a strain with attenuated cAPK activity. To this end, one representative plasmid of each class was assayed for galactose-dependent inhibition of growth at 31.5°C of a tpk2ts strain (SGY446). Partial sequencing of a plasmid from one of the six remaining positive classes showed it to contain the entire GIS1 open reading frame, including 470 bp of the 5′-untranslated region, fused to the GAL1 promoter of the library plasmid. GIS1 encodes a 90 kDa protein, which contains one classical C2H2 zinc finger followed by an alternative C2HC zinc finger at the C-terminal end (Böhm et al., 1997). Comparison of the predicted Gis1 sequence with all other deduced amino acid sequences in yeast revealed a second gene product, Rph1, with particularly high homology to the zinc finger-containing C-terminal region of Gis1 (Balciunas and Ronne, 1999; Jang et al., 1999). Interestingly, while genetic studies suggested different roles for Gis1 and Rph1 (Balciunas and Ronne, 1999), in vivo footprinting, binding competition and transcriptional studies indicated that Gis1 and Rph1 may act as functionally redundant transcriptional repressors of an STRE-like upstream repressing sequence (URSPHR1; Jang et al., 1999).
Gis1 regulates nutrient limitation-induced transcription of several cAPK/Rim15-dependent genes
To determine the consequences of the loss of Gis1, we replaced the complete GIS1 coding region by a PCR-based gene deletion method (Baudin et al., 1993; Wach et al., 1994; see Materials and methods). In agreement with previous studies (Balciunas and Ronne, 1999; Jang et al., 1999), deletion of GIS1 caused no obvious defect in germination of spores, or in exponential growth on YPD at 30°C. Thus, GIS1 is not essential for growth or germination. The effect of gis1Δ on SSA3 transcription was assessed by using the SSA3-lacZ reporter plasmid pWB204Δ-236. Comparison with wild-type cells revealed that gis1Δ cells, similarly to rim15Δ cells, were strongly defective for nutrient limitation-induced derepression of SSA3-lacZ (Table I). Overexpression of GIS1, in contrast, was able not only to suppress fully the SSA3-lacZ derepression defect of rim15Δ cells, but also to increase SSA3-lacZ induction significantly in wild-type, rim15Δ and gis1Δ mutant cells when compared with the control plasmid-containing wild-type strain. Finally, the increase in SSA3-lacZ induction mediated by RIM15 overexpression was found to be almost entirely dependent on the presence of Gis1 (Table I). The simplest interpretation of these results is that Gis1 may act downstream of Rim15 to mediate one or more Rim15-controlled, nutrient limitation-induced responses. Therefore, we also analyzed whether deletion of GIS1 causes additional defects in response to nutrient limitation. As is seen in Figure 1, SSA3, HSP12 and HSP26 were repressed in exponentially growing wild-type cells and induced upon glucose starvation. Under the same conditions, gis1Δ mutant cells, similarly to rim15Δ and gis1Δ rim15Δ double mutant cells, were almost entirely defective for derepression of SSA3 and partially defective for the maintenance of derepression of HSP12 (Figure 1, lane 4). Derepression of HSP26, in contrast, was found to be only slightly defective in gis1Δ mutant cells, while being strongly defective in rim15Δ and rim15Δ gis1Δ double mutant cells (Figure 1, lanes 3 and 4). Notably, glucose limitation-induced TPS2 transcription remained largely unaffected by gis1Δ and/or rim15Δ. As a control for the physiological status of the cells, we also examined the expression pattern of a gene, SSB1, the transcription of which is repressed upon nutrient limitation (Werner-Washburne et al., 1989). Transcript levels of SSB1 were high in exponentially growing cells and very low in glucose-starved cells of all four strains (Figure 1, lanes 1–4). Together, these results show that Gis1 not only is involved in nutrient limitation-induced derepression of SSA3, but also partially regulates expression of additional cAPK-repressible genes such as HSP12 and HSP26 (Engelberg et al., 1994; Varela et al., 1995).
Table I. Epistatic relationship between RIM15 and GIS1.
| Plasmid | Strain |
||
|---|---|---|---|
| Wild type | rim15Δ | gis1Δ | |
| YCpIF2 | 153.0 ± 23.1 | 33.3 ± 9.0 | 22.1 ± 4.0 |
| YCpIF2-RIM15 | 280.9 ± 54.6 | 249.6 ± 58.5 | 34.1 ± 2.9 |
| YCpIF2-GIS1 | 264.5 ± 45.8 | 281.5 ± 54.4 | 258.7 ± 36.3 |
Transformants of wild-type (AR1-1A), rim15Δ (AR1-1C) and gis1Δ (CDV100-10D) cells were grown to early stationary phase (2 days) on SD medium containing 2% galactose and 1% raffinose to induce GAL1-driven transcription of RIM15 and GIS1 in the corresponding YCpIF2-derived plasmids. β-galactosidase activities were measured (as described in Miller, 1972) to monitor the induction of an SSA3-lacZ fusion gene (from plasmid pWB204Δ-236). Notably, all YCpIF2-GIS1 transformants reached much lower cell densities (<30%) than corresponding YCpIF2 and YCpIF2-RIM15 transformants. Values represent means ± SDs of β-galactosidase activities (in Miller units) of three independent experiments.

Fig. 1. Effects of gis1Δ, rim15Δ and gis1Δ rim15Δ on the expression of various genes following glucose starvation. Homozygous wild-type (YEF473), and gis1Δ/gis1Δ (CDV101), rim15Δ/rim15Δ (AR2) and gis1Δ/gis1Δ rim15Δ/rim15Δ (CDV104) mutant cells were harvested by centrifugation and resuspended in S medium containing 0.1% glucose. Total RNAs were extracted immediately after resuspension (1) and following further incubation for 90 (2), 180 (3) and 270 min (4). Equal amounts of RNAs (10 µg) were probed with SSA3, HSP12, HSP26, TPS2 and SSB1 fragments after electrophoresis and blotting. The application and transfer of equal amounts of RNA were verified by ethidium bromide staining.
The strong defect in HSP26 derepression observed in rim15Δ cells is only partially mimicked by loss of Gis1, indicating that Gis1 mediates only a subset of Rim15-controlled responses. Therefore, diploid gis1Δ/gis1Δ cells were also analyzed for a variety of other phenotypic traits that are characteristic of rim15Δ cells (e.g. the defects in trehalose accumulation, in proper G1 arrest, in long-term stationary phase survival and in sporulation; Vidan and Mitchell, 1997; Reinders et al., 1998). When compared with isogenic wild-type cells (YEF473), 4-day-old stationary phase gis1Δ/gis1Δ cells (CDV101) exhibited normal trehalose levels (0.207 ± 0.029 g/g protein versus 0.176 ± 0.008 g/g protein for GIS1/GIS1 cells) and were only slightly defective for proper G1 arrest (3.5 ± 0.4% budded cells versus 0.8 ± 0.1% budded cells for GIS1/GIS1 cells). In addition, gis1Δ/gis1Δ cells exhibited no obvious sporulation defect (data not shown). However, gis1Δ/gis1Δ cells were found to be significantly more sensitive than wild-type cells, and only slightly less sensitive than rim15Δ and rim15Δ gis1Δ cells, to prolonged nutrient starvation (Figure 2). These results suggest that Gis1 controls transcriptional activation of a subset of Rim15-dependent genes, the expression of which is important for long-term survival under conditions of nutrient starvation. In agreement with such a role for Gis1, we found Gis1 protein levels to be very low in exponentially growing cells, as estimated by immune blots of myc13-tagged Gis1 (Figure 3, lane 1), and to increase following glucose exhaustion at the diauxic transition (Figure 3, lanes 2–5). Interestingly, Gis1 protein levels were induced similarly in rim15Δ cells entering the diauxic transition (Figure 3), indicating that potential Rim15-dependent regulation of Gis1 activity is exerted at a post-transcriptional level.

Fig. 2. Effects of gis1Δ, rim15Δ and gis1Δ rim15Δ on stationary phase survival. Homozygous wild-type (YEF473; filled circle) and gis1Δ/gis1Δ (CDV101; filled triangle), rim15Δ/rim15Δ (AR2; open circle) and gis1Δ/gis1Δ rim15Δ/rim15Δ (CDV104; filled square) mutant cells were grown to stationary phase on YPD medium. The percentage of viable cells was determined by the colony-forming efficiency on YPD agar at the times indicated.

Fig. 3. Immunoblot analysis of Gis1-myc13 in wild-type (NB14) and rim15Δ mutant (NB15) cells entering the diauxic transition phase. Samples were taken 2 h before glucose exhaustion (1), at the time of glucose exhaustion (2), and 2 (3), 4 (4) and 6 h (5) following glucose exhaustion. Twenty micrograms of extract protein were loaded in each lane.
Loss of Gis1 partially suppresses cdc25ts, cdc35ts and tpkts
The data presented above suggest that Gis1 may act in the Ras/cAMP pathway downstream of Rim15 to control transcriptional activation upon nutrient limitation. To determine the potential contribution of Gis1 to cAPK-dependent growth, we tested whether loss of Gis1 could suppress growth defects that are associated with attenuated cAPK activity. To this end, temperature-sensitive Ras GTP-exchange factor and adenylate cyclase mutant strains, harboring corresponding cdc25ts and cdc35ts mutations, respectively, as well as a temperature-sensitive cAPK mutant strain with only one functional tpk2ts gene, were transformed with the gis1Δ::kanMX2 cassette. In all three mutant strains, deletion of GIS1 did not restore growth at the non-permissive temperature, indicating that gis1Δ is not able to suppress the total loss of cAPK activity (data not shown). However, when tested at a semi-permissive temperature (i.e. at 34°C), the relatively low growth rates of each of the three temperature-sensitive mutant strains were significantly increased by loss of Gis1. Accordingly, deletion of GIS1 caused growth rates of cdc25ts, cdc35ts and tpk2ts cells to increase from 0.158 ± 0.008 (OL86) to 0.205 ± 0.006 h–1 (NB19), from 0.147 ± 0.007 (PD6517) to 0.181 ± 0.008 h–1 (NB21) and from 0.151 ± 0.005 (SGY446) to 0.191 ± 0.002 h–1 (NB23), respectively. Together, these results show that loss of Gis1 partially relieves dependence on cAPK function.
GIS1 overproduction exacerbates the growth defect of cdc25ts, cdc35ts and tpkts strains
In order to determine whether Gis1, as suggested by the results presented above, may antagonize cAPK-dependent growth, we tested whether GIS1 overexpression could exacerbate the growth defect associated with attenuated cAPK activity. Overexpression of GIS1 from the galactose-inducible GAL1 promoter inhibited growth even in wild-type cells (Figure 4A; see also Table I). In contrast, wild-type cells overexpressing GIS1 from the constitutive ADH1 promoter had only a slight growth defect, which was apparent after growth for 1 day on plates (data not shown), but not when plates were incubated for >2 days (Figure 4A). In accordance with this result, assessment of growth in liquid cultures revealed that the growth rate of ADH1-GIS1-overexpressing wild-type cells was reduced by ∼50% in comparison with control cells (data not shown). When transformed into cdc25ts and cdc35ts mutants, the GIS1 overexpression plasmid YCpADH1-GIS1, but not the corresponding control plasmid YCpADH1, seriously compromised both mutants for growth even at the permissive temperature of 27°C (Figure 4B). Similarly, overexpression of GIS1 from the ADH1 promoter inhibited growth of tpk2ts cells at the permissive temperature of 27°C, while it had little effect on the growth of isogenic TPK2 wild-type cells. Thus, Gis1 overproduction partially inhibits growth in wild-type cells and exacerbates the growth defects of cAPK-compromised mutants.
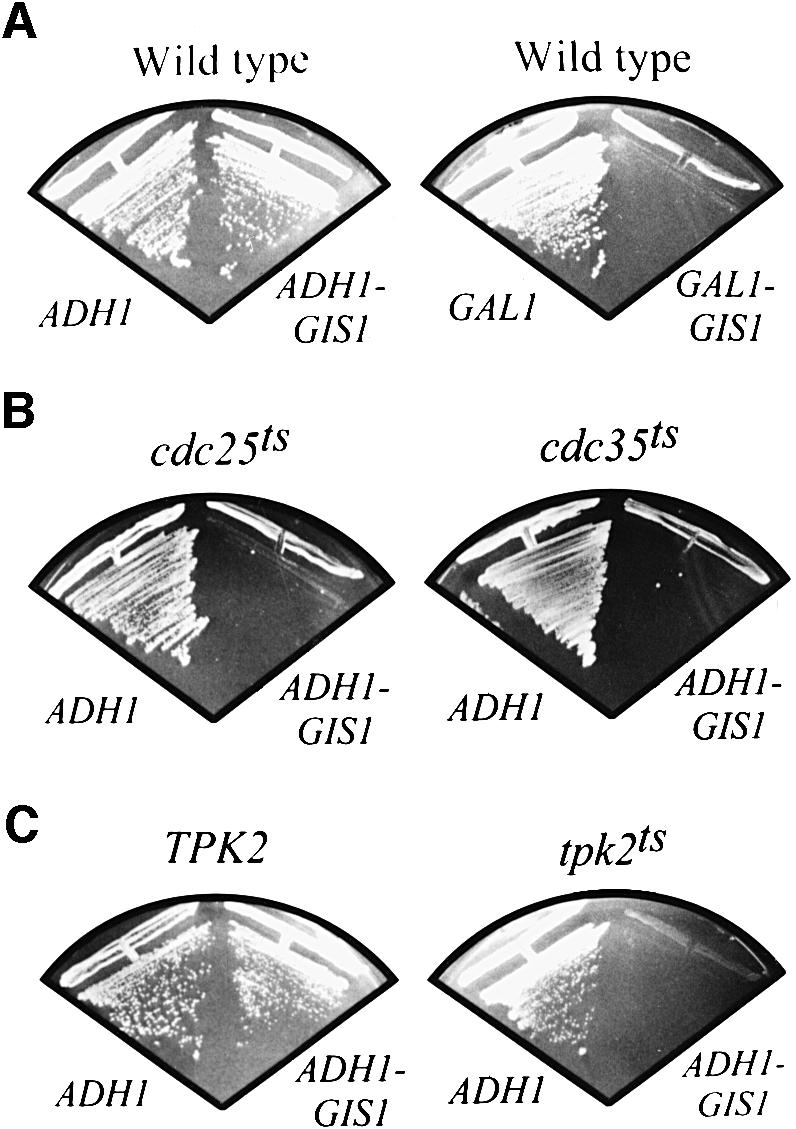
Fig. 4. GIS1 overproduction inhibits growth of wild-type cells and exacerbates the growth defect of cdc25ts, cdc35ts and tpk2ts strains. (A) Wild-type strain YEF473 was transformed with plasmids YCpADH1 (ADH1), YCpADH1-GIS1 (ADH1-GIS1), YCpIF2 (GAL1) and YCpIF2-GIS1 (GAL1-GIS1), streaked on glucose- (ADH1 plasmids) or galactose-containing (GAL1 plasmids) selective plates and incubated for 3 days at 30°C. (B) Strains OL86 (cdc25ts) and PD6517 (cdc35ts) were transformed with plasmids YCpADH1 and YCpADH1-GIS1, streaked on glucose-containing SD plates and incubated for 3 days at 27°C. (C) Strain SGY446 (tpk2ts) and its isogenic wild-type strain ASY18 (TPK2) were transformed with plasmids YCpADH1 and YCpADH1-GIS1, streaked on glucose-containing SD plates and incubated for 3 days at 27°C.
To study further the effects of GIS1 overexpression in wild-type cells, and to compare these effects with overexpression of RIM15 and the GIS1 homolog RPH1, strain YEF473 was transformed with YCpADH1, YCpADH1-RIM15, YCpADH1-GIS1 and YCpADH1-RPH1, and analyzed for transcription of SSA3, HSP12, HSP26, TPS2 and SSB1 in exponentially growing cells. Transcript levels of SSA3, HSP12 and HSP26 were barely detectable in wild-type cells growing exponentially on glucose-containing medium, but were strongly induced following overexpression of GIS1 under the same conditions (Figure 5, lanes 1 and 3). Similarly, albeit to a significantly lower extent, overexpression of RIM15 was found to cause transcriptional induction of HSP12 and HSP26, while transcription of the same genes was not induced by overexpression of RPH1 (Figure 5, lanes 1, 2 and 4). In contrast, the level of TPS2 expression detected in exponentially growing cells remained largely unaffected by GIS1 and RIM15 overexpression, but was significantly reduced by overexpression of RPH1, suggesting that Rph1 may function as a transcriptional repressor of TPS2 under these conditions (Figure 5, lane 4). Together, these results show that overexpression of GIS1 allows derepression/activation of cAPK-repressible genes even under repressing conditions.
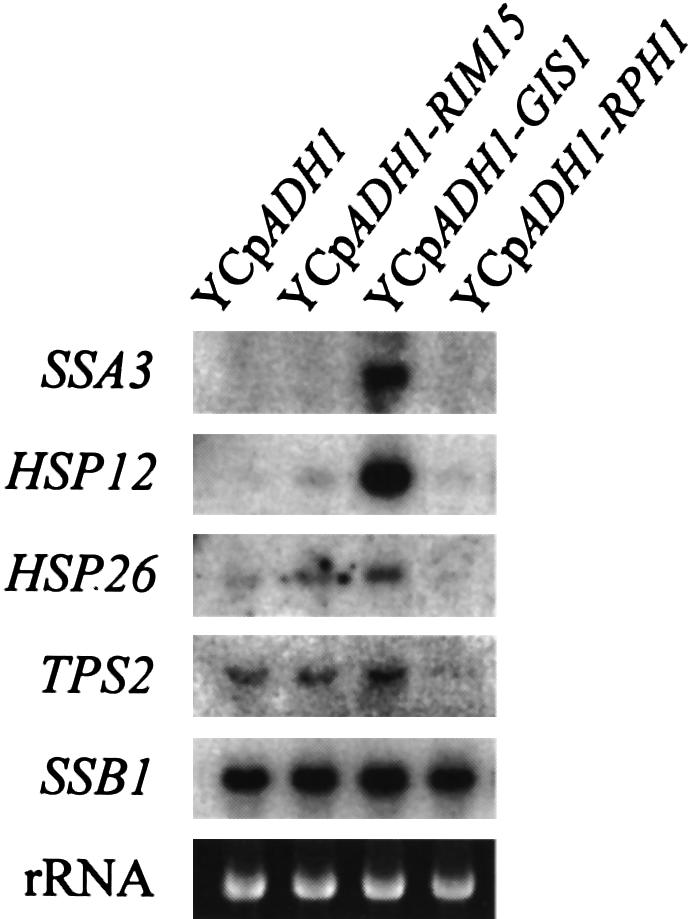
Fig. 5. Overexpression of GIS1, but not of RPH1, induces transcriptional activation of SSA3, HSP12 and HSP26. Wild-type (YEF473) cells transformed with either YCpADH1, YCpADH1-RIM15, YCpADH1-GIS1 or YCpADH1-RPH1 were grown to exponential phase on SD media containing 2% glucose. Total RNAs from exponentially growing transformants were extracted and equal amounts of RNAs (10 µg) were probed with SSA3, HSP12, HSP26, TPS2 and SSB1 fragments after electrophoresis and blotting. The application and transfer of equal amounts of RNA were verified by ethidium bromide staining.
Transcriptional activation of the PDS element, but not of STRE, is dependent on Gis1
The data presented above suggest that Gis1 functions as a transcriptional activator of PDS element- and/or STRE-controlled genes. Therefore, we next determined whether Gis1 acts as an activator of transcription using the one-hybrid technique. To this end, we fused the entire Gis1 to the LexA DNA-binding domain (DBD) in plasmid pEG202 (see Materials and methods). This fusion protein was able to activate transcription strongly from a promoter containing eight lexA-binding sites (Table II). This is in agreement with a previously reported observation that a partial Gis1 protein, lacking the C-terminal zinc finger domain, can activate transcription in a one-hybrid assay (Balciunas and Ronne, 1999). Interestingly, we were not able to detect transcriptional activity of a corresponding LexA–Rph1 fusion protein when assayed under the same conditions. Moreover, transcriptional activity of Gis1, measured by the one-hybrid assay, was equally strong in exponentially growing as in stationary phase wild-type cells, but slightly reduced in corresponding rim15Δ cells (Table II). These results show that Gis1 can act as a transcriptional activator that, under the conditions of the one-hybrid assay, is unresponsive to the nutritional status and only partially dependent on the presence of Rim15. These results may be explained if overexpression of GIS1 from the strong ADH1 promoter in pEG202 results in protein levels that exceed a certain threshold beyond which Gis1 becomes largely independent of activation by upstream regulatory proteins (see Discussion).
Table II. Transcriptional activation by LexA–Gis1.
| Expressed proteins | Wild type |
rim15Δ |
||
|---|---|---|---|---|
| LOG | STAT | LOG | STAT | |
| LexA | 1.1 ± 0.1 | 7.1 ± 2.8 | 5.1 ± 0.1 | 1.6 ± 0.3 |
| LexA–Gis1 | 1268.4 ± 65.6 | 1157.0 ± 167.1 | 981.9 ± 256.7 | 922.3 ± 25.0 |
| LexA–Rph1 | 9.5 ± 2.5 | 11.5 ± 5.0 | 25.2 ± 2.9 | 25.0 ± 5.1 |
Proteins were expressed from plasmids pEG202-GIS1 and pEG202-RPH1 and from vector pEG202 (Zervos et al., 1993). Target plasmid was pSH18-34 containing eight LexA operators (Gyuris et al., 1993). Transformants of wild-type (AR1-1A) and rim15Δ (AR1-1C) cells were grown with selection for both plasmids. β-galactosidase activities were measured (as described in Miller, 1972) to monitor the induction of the LexAops-lacZ fusion gene (from plasmid pSH18-34). Values represent means ± SDs of β-galactosidase activities (in Miller units) of three independent transformants.
Gis1-dependent transcriptional activation may be mediated via STREs and/or PDS elements. To assess quantitatively the role of Gis1 for STRE- and PDS element-driven expression, nutrient limitation-induced expression of heterologous STRE-LEU2-lacZ and PDS-LEU2-lacZ genes was tested in different strain backgrounds. As expected, nutrient limitation significantly induced both STRE- and PDS element-driven expression in wild-type cells (Figure 6A and B). The induction ratio (defined as the ratio of nutrient limitation-induced expression to basal level expression) of the STRE-LEU2-lacZ reporter was reduced slightly in rim15Δ cells (26.8% decrease), remained unchanged in gis1Δ and rph1Δ cells, and was strongly reduced in both msn2 msn4 (89.8% decrease) and msn2 msn4 gis1Δ (94.1% decrease) cells when compared with the induction ratio in wild-type cells (Figure 6A). These results show that STRE-driven expression, while being strongly dependent on the presence of Msn2 and Msn4, is completely independent of Gis1 and Rph1, and only partially dependent on Rim15. The induction ratio of the PDS-LEU2-lacZ reporter, in contrast, was strongly reduced in rim15Δ (85.1% decrease), gis1Δ (96.5% decrease) and msn2 msn4 gis1Δ (95.0% decrease) cells, while it remained virtually unchanged in msn2 msn4 and rph1Δ cells when compared with the induction ratio in wild-type cells (Figure 6B). Thus, PDS element-driven expression is completely independent of Msn2, Msn4 and Rph1, strongly dependent on Rim15, and almost entirely dependent on the transcriptional activator Gis1.

Fig. 6. Effects of rim15Δ, gis1Δ, msn2 msn4, msn2 msn4 gis1Δ and rph1Δ mutations on the basal level expression and nutrient limitation-induced expression of STRE- and PDS element-driven LEU2-lacZ reporter genes. Wild-type, rim15Δ, gis1Δ, msn2 msn4, msn2 msn4 gis1Δ and rph1Δ cells carrying a single chromosomally integrated STRE-LEU2-lacZ reporter gene (strains CDV120, CDV121, CDV122, CDV123, CDV124 and IP17, respectively) (A) or a corresponding PDS-LEU2-lacZ reporter gene (strains CDV125, CDV126, CDV127, CDV128, CDV129 and IP13, respectively) (B) were grown to exponential (open bars) and to early stationary phase (2 days; cross-hatched bars) on YPD medium. The chromosomal genotypes are indicated below the ordinate; the induction ratios following nutrient limitation are indicated above the chromosomal genotypes. β-galacto sidase activities were measured (as described in Miller, 1972) to monitor the induction of the STRE-LEU2-lacZ and PDS-LEU2-lacZ fusion genes. Values represent means ± SDs of β-galactosidase activities (in Miller units) of three independent experiments.
Finally, STRE-driven basal level expression has previously been reported to be enhanced when cells were growing on rich medium containing ethanol instead of glucose as carbon source (Martínez-Pastor et al., 1996). In this context, we found that expression from both the heterologous STRE-LEU2-lacZ and PDS-LEU2-lacZ genes, while being very low in wild-type cells growing exponentially on glucose (0.5 ± 0.1 and 0.6 ± 0.1 Miller units for STRE-LEU2-lacZ and PDS-LEU2-lacZ, respectively), was significantly enhanced in cells growing exponentially on ethanol (30.0 ± 5.1 and 15.3 ± 3.8 Miller units for STRE-LEU2-lacZ and PDS-LEU2-lacZ, respectively) or glycerol (46.8 ± 15.3 and 25.0 ± 7.8 Miller units for STRE-LEU2-lacZ and PDS-LEU2-lacZ, respectively). Thus, both STRE- and PDS element-driven expression are repressed by the presence of glucose.
Discussion
We have identified the zinc finger protein Gis1 in a screen for dosage-dependent suppressors of the rim15Δ defect in nutrient limitation-induced transcriptional derepression of SSA3. Our results are interpreted most simply in a model in which Gis1 functions in the Ras/cAMP pathway downstream of Rim15 to control transcription of a set of genes whose products are essential for long-term survival following nutrient limitation. Several observations support such a model. First, loss of Gis1 causes a defect in transcriptional derepression following nutrient limitation of various genes, including SSA3, whose expression is known to be negatively regulated by the Ras/cAMP pathway through Rim15. As a result, gis1Δ cells are seriously defective in induction of starvation resistance upon entry into stationary phase. Secondly, deletion of GIS1 partially suppresses the growth defect at elevated temperatures of mutant cells that are compromised for cAPK activity (e.g. cdc25ts, cdc35ts and tpk2ts). Thirdly, overexpression of GIS1 suppresses the defect in transcriptional derepression of SSA3 in nutrient-limited rim15Δ cells, induces transcription of cAPK-repressible genes (e.g. SSA3, HSP12 and HSP26) in exponentially growing wild-type cells and exacerbates the growth defect of strains compromised for cAPK activity. Fourthly, expression driven by the PDS element, which has previously been shown to be negatively regulated by the Ras/cAMP pathway and which confers transcriptional activation following nutrient limitation at the diauxic transition and in stationary phase, is almost entirely dependent on the presence of Gis1.
It was reported recently that most of the growth-related effects of cAPK might be accounted for by Msn2/Msn4-controlled transcription (Smith et al., 1998). In this context, our result showing that loss of Gis1 only partially suppresses the growth defect of cAPK-compromised mutant cells suggests that Gis1 may antagonize cAPK-dependent growth rather weakly. Nevertheless, overexpression of GIS1, in particular under conditions of presumably reduced activity of the Ras/cAMP pathway (e.g. on galactose-containing medium), was found to inhibit growth strongly. This indicates that one role for Gis1 is indeed to stimulate the expression of growth-inhibitory genes and that cAPK-dependent control of growth is the result of a coordinated regulation of various, different transcription factors. Accordingly, cAPK might regulate growth through its direct and/or indirect modulation of transcriptional activators including Msn2, Msn4 and Gis1, as well as by its action on transcriptional repressors such as, for instance, Sok2 (Ward et al., 1995; Smith et al., 1998). Interestingly, although there is as yet no evidence that cAPK phosphorylates Msn2 directly, recent results suggest that cAPK exerts its effect on Msn2-dependent gene expression by directly inhibiting Msn2 function (Görner et al., 1998). In this context, our model posits that Gis1 acts downstream of a protein kinase, Rim15, which is negatively controlled by cAPK-dependent phosphorylation (Reinders et al., 1998). One might predict, therefore, that Gis1 activity is modulated directly by Rim15-dependent phosphorylation (Figure 7). In accordance with such a model, we found that transcriptional activity of a LexA–Gis1 hybrid protein is partially dependent on the presence of Rim15. Nevertheless, both the one-hybrid assay and the rim15Δ suppression experiments indicate that Gis1 may become largely independent of Rim15 when overproduced from the strong constitutive ADH1 or inducible GAL1 promoters. This effect may be due to the fact that GIS1 overexpression yields protein levels that exceed a certain threshold value beyond which Gis1 becomes independent of upstream regulatory proteins. Alternatively, Gis1 may be controlled via additional Ras/cAMP/Rim15-independent mechanisms (Figure 7). Further studies with particular focus on the biochemical characterization of Gis1, which thus far has been hampered by excessive proteolysis of Gis1 (data not shown; see also Jang et al., 1999), should allow a more detailed assessment of this issue.

Fig. 7. Model for cAPK regulation of nutrient limitation-induced gene expression. The relative importance of Gis1 and Msn2/Msn4 for transcriptional activation of HSP12, HSP26 and SSA3 upon nutrient limitation is illustrated by the different shading (i.e. darker shading representing stronger dependence on the corresponding zinc finger protein; for studies of Msn2/Msn4-dependent transcription, see Martínez-Pastor et al., 1996). Notably, HSP12 and HSP26 have several STREs in their promoter regions, while the SSA3 promoter contains no conserved STRE, but several PDS elements (indicated in brackets). Arrows and bars denote positive and negative interactions, respectively. Dashed arrows and bars refer to potential interactions. For further details, see the text.
Loss of Gis1 causes a defect in nutrient limitation-induced derepression/activation of SSA3, HSP12 and HSP26. Thus, while Gis1 has recently been found to function as a damage-responsive repressor of PHR1 (Jang et al., 1999), we show here that it formally functions as an activator of SSA3, HSP12 and HSP26. Such a dual role in transcriptional activation/repression is not uncommon amongst transcriptional regulators and may be based on specific protein–protein interactions, promoter sequence contexts or physiological conditions (e.g. the repressor–activator Ume6; Krishnamurthy et al., 1997; Sweet et al., 1997). Notably, in this context, both our one-hybrid experiments and our overexpression studies suggest that Rph1, unlike its homolog Gis1, may function as a repressor of STRE- and PDS element-controlled genes. Together, these findings support the previous notion that Rph1 and Gis1, in addition to their common role in regulation of PHR1 expression, may also exert distinct roles in transcriptional control (Balciunas and Ronne, 1999).
Interestingly, in vitro footprinting and binding competition studies indicated that Rph1 and Gis1 bind, like Msn2 and Msn4, to STRE-like sequences, suggesting that Gis1 and Msn2/Msn4 may overlap functionally in vivo (Treger et al., 1998; Jang et al., 1999). Surprisingly, however, we found that both msn2 msn4 and gis1Δ mutants were defective specifically for STRE-driven and PDS element-driven expression, respectively. Thus, Msn2/Msn4 and Gis1 are not functionally equivalent in vivo and are likely to discriminate between the two very similar STRE and PDS promoter elements. Such a conclusion is supported further by our findings that gis1Δ mutants are seriously defective for nutrient limitation-induced transcriptional activation of the PDS element-controlled SSA3 gene, while exhibiting only a minor defect in transcriptional activation of the STRE-controlled HSP12 and HSP26 genes under the same conditions. Nevertheless, the observed minor effect of loss of Gis1 on HSP12 and HSP26 expression suggests that Gis1 can, possibly in a proper promoter context, also mediate partial STRE-dependent activation. Such context specificity may be masked in heterologous promoter studies because of the absence of potential ancillary sequences that modulate STRE-driven transcription. Therefore, detailed reinvestigation of the HSP12 and HSP26 promoters in the context of the present results should allow us to understand the relationship between STREs and their dependence upon the Msn2, Msn4 and Gis1 transcriptional regulators following nutrient limitation.
A further interesting point with respect to the heterologous promoter studies is the finding that rim15Δ mutants were partially reduced for nutrient limitation-induced activation of STRE-lacZ. One explanation for this result may be that Rim15 is involved in Gis1-independent, possibly Msn2/Msn4-dependent, control of STRE. Alternatively, Rim15 could, as suggested previously (Reinders et al., 1998), be involved in mechanisms of feedback inhibition of the Ras/cAMP pathway. In this scenario, loss of Rim15 could cause overactivation of the Ras/cAMP pathway and, consequently, inhibition of Msn2/4 function. Notably, both explanations are consistent with our model that Rim15 acts upstream of Gis1 and may overall, therefore, cause a more serious physiological defect than loss of Gis1 (Figure 7).
A final issue that remains to be addressed is that Gis1 has previously been implicated in a mechanism of positive regulation of the Ras/cAMP pathway (Balciunas and Ronne, 1999). Accordingly, and at variance with the results presented here, the authors found that overexpression of GIS1 suppressed a cdc25-5ts mutation. It must be noted, however, that the experimental set-up in that report differed in at least two important aspects from our studies. First, the cdc25-5ts mutant under study, in contrast to the one used here, harbored an additional mutation in the PDE2 gene whose product, phosphodiesterase, negatively regulates Ras/cAMP signaling (Sass et al., 1986). Secondly, overexpression of GIS1 was achieved by expression of GIS1 under its own promoter from a 2µ plasmid. Presumably, this may yield much lower Gis1 protein levels than expression of GIS1 from the strong ADH1 and GAL1 promoters, as done in the present study. One might speculate, therefore, that moderate overproduction of Gis1 in a cdc25-5ts pde2 mutant, while not reaching the threshold level for growth inhibition, causes transcriptional activation of one or more genes that act as specific suppressors of the cdc25-5ts mutation. One such candidate gene encodes Tfs1, which, when overproduced, shows allele-specific suppression of cdc25-1ts and cdc25-5ts (Robinson and Tatchell, 1991). Intriguingly, we found the TFS1 gene to contain a PDS element consensus site at position –136 in its promoter region, which may confer the reported induction upon nutrient limitation via the transcriptional activator Gis1 (DeRisi et al., 1997). Despite the admittedly speculative nature of this model, it may reconcile the obviously contradictory observations illustrated above, and leads to several predictions that can be tested in future studies.
Materials and methods
Strains, media, and microbiological and recombinant DNA methods
The S.cerevisiae strains used in this study are listed in Table III. To construct gis1Δ, rph1Δ and rim15Δ mutants, the complete GIS1, RPH1 and RIM15 coding regions were deleted by the PCR method (Baudin et al., 1993) using the Expand™ High Fidelity PCR System (Roche Diagnostics) and either plasmid pRS303 (HIS3; Sikorski and Hieter, 1989), pRS304 (TRP1; Sikorski and Hieter, 1989) or pFA6a (kanMX2; Wach et al., 1994) as template. The PCR products that contained flanking sequences of GIS1, RPH1 and RIM15 separated by the HIS3, TRP1 or kanMX2 modules were extracted with phenol/chloroform, precipitated and used for transformations. The gis1Δ::HIS3 deletion cassette was transformed into strain AR2 to construct IP6; the gis1Δ::kanMX2 deletion cassette was transformed into strains OL86, PD6517 and SGY446 to construct NB19, NB21 and NB23, respectively; and the rph1Δ::TRP1 deletion cassette was transformed into strain IP6 to construct CDV100. Strains CDV101 and CDV104 were constructed by crossing haploid segregants from CDV100 as follows: CDV100-10D × CDV100-4D (CDV101) and CDV100-6C × CDV100-10A (CDV104).
Table III. List of yeast strains.
| Strain | Genotype | Source | |
|---|---|---|---|
| YEF473 | MATa/α | his3/his3 leu2/leu2 lys2/lys2 trp1/trp1 ura3/ura3 | Bi and Pringle (1996) |
| AR1 | MATa/α | his3/his3 leu2/leu2 lys2/lys2 trp1/trp1 ura3/ura3 | Reinders et al. (1998) |
| rim15Δ::kanMX2/RIM15 | |||
| AR1-1A | MATa | his3 leu2 lys2 trp1 ura3 | segregant from AR1 |
| AR1-1C | MATa | his3 leu2 lys2 trp1 ura3 rim15Δ::kanMX2 | segregant from AR1 |
| AR2 | MATa/α | his3/his3 leu2/leu2 lys2/lys2 trp1/trp1 ura3/ura3 | Reinders et al. (1998) |
| rim15Δ::kanMX2/rim15Δ::kanMX2 | |||
| OL86 | MATα | leu2 trp1 ade2 cdc25-5 | Camonis et al. (1986) |
| PD6517 | MATα | ade8 leu2 trp1 cdc35-10 | Becher dos Passos et al. (1992) |
| SGY446 | MATα | tpk1Δ::ADE8 tpk2-63(Ts) tpk3::TRP1 ura3-52 | Smith et al. (1998) |
| his3 leu2-3,112 trp1 ade8 | |||
| ASY18 | MATα | tpk1Δ::ADE8 TPK2 tpk3::TRP1 ura3-52 his3 | Smith et al. (1998) |
| leu2-3,112 trp1 ade8 | |||
| NB19 | MATα | leu2 trp1 ade2 cdc25-5 gis1Δ::kanMX2 | this study |
| NB21 | MATα | ade8 leu2 trp1 cdc35-10 gis1Δ::kanMX2 | this study |
| NB23 | MATα | tpk1Δ::ADE8 tpk2-63(Ts) tpk3::TRP1 ura3-52 | this study |
| his3 leu2-3,112 trp1 ade8 gis1Δ::kanMX2 | |||
| IP6 | MATa/α | his3/his3 leu2/leu2 lys2/lys2 trp1/trp1 ura3/ura3 | this study |
| gis1Δ::HIS3/GIS1 rim15Δ::kanMX2/RIM15 | |||
| CDV100 | MATa/α | his3/his3 leu2/leu2 lys2/lys2 trp1/trp1 ura3/ura3 | this study |
| gis1Δ::HIS3/GIS1 rim15Δ::kanMX2/RIM15 | |||
| rph1Δ::TRP1/RPH1 | |||
| CDV100-10D | MATa | his3 leu2 lys2 trp1 ura3 gis1Δ::HIS3 | segregant from CDV100 |
| CDV100-4D | MATα | his3 leu2 lys2 trp1 ura3 gis1Δ::HIS3 | segregant from CDV100 |
| CDV100-6C | MATa | his3 leu2 lys2 trp1 ura3 gis1Δ::HIS3 | segregant from CDV100 |
| rim15Δ::kanMX2 | |||
| CDV100-10A | MATα | his3 leu2 lys2 trp1 ura3 gis1Δ::HIS3 | segregant from CDV100 |
| rim15Δ::kanMX2 | |||
| CDV101 | MATa/α | his3/his3 leu2/leu2 lys2/lys2 trp1/trp1 ura3/ura3 | CDV100-10D × |
| gis1Δ::HIS3/gis1Δ::HIS3 | CDV100-4D | ||
| CDV104 | MATa/α | his3/his3 leu2/leu2 lys2/lys2 trp1/trp1 ura3/ura3 | CDV100-6C × |
| gis1Δ::HIS3/gis1Δ::HIS3 rim15Δ::kanMX2/ | CDV100-10A | ||
| rim15Δ::kanMX2 | |||
| NB14 | MATa | his3 leu2 lys2 trp1 ura3 GIS1-Myc13:TRP1 | this study |
| NB15 | MATa | his3 leu2 lys2 trp1 ura3 rim15Δ::kanMX2 | this study |
| GIS1-Myc13:TRP1 | |||
| W303-1A | MATa | ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 | Thomas and Rothstein (1989) |
| Wmsn2msn4 | MATa | ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 | Martínez-Pastor et al. (1996) |
| ura3-1 msn2-Δ3::HIS3 msn4-1::TRP1 | |||
| CDV120 | MATa | ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 | this study |
| ura3-1 URA3::STRE-lacZ | |||
| CDV121 | MATa | ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 | this study |
| ura3-1 URA3::STRE-lacZ rim15Δ::kanMX2 | |||
| CDV122 | MATa | ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 | this study |
| ura3-1 URA3::STRE-lacZ gis1Δ::kanMX2 | |||
| CDV123 | MATa | ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 | this study |
| URA3::STRE-lacZ msn2-Δ3::HIS3 msn4-1::TRP1 | |||
| CDV124 | MATa | ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 1 | this study |
| ura3-URA3::STRE-lacZ msn2-Δ3::HIS3 | |||
| msn4-1::TRP1 gis1Δ::kanMX2 | |||
| IP17 | MATa | ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 | this study |
| ura3-1 URA3::STRE-lacZ rph1Δ::TRP1 | |||
| CDV125 | MATa | ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 | this study |
| ura3-1 URA3::PDS-lacZ | |||
| CDV126 | MATa | ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 | this study |
| ura3-1 URA3::PDS-lacZ rim15Δ::kanMX2 | |||
| CDV127 | MATa | ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 | this study |
| ura3-1 URA3::PDS-lacZ gis1Δ::kanMX2 | |||
| CDV128 | MATa | ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 | this study |
| URA3::PDS-lacZ msn2-Δ3::HIS3 msn4-1::TRP1 | |||
| CDV129 | MATa | ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 | this study |
| ura3-1 URA3::PDS-lacZ msn2-Δ3::HIS3 | |||
| msn4-1::TRP1 gis1Δ::kanMX2 | |||
| IP13 | MATa | ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 | this study |
| ura3-1 URA3::PDS-lacZ rph1Δ::TRP1 |
Linearized, NcoI-cut integrative vectors pCTT1-18/7x and pLS9-PDS were transformed into strain W303-1A to construct CDV120 and CDV125, respectively, co-transformed with the rim15Δ::kanMX2 deletion cassette into strain W303-1A to construct CDV121 and CDV126, respectively, co-transformed with the gis1Δ::kanMX2 deletion cassette into strain W303-1A to construct CDV122 and CDV127, respectively, transformed into strain Wmsn2msn4 to construct CDV123 and CDV128, respectively, co-transformed with the gis1Δ::kanMX2 deletion cassette into strain Wmsn2msn4 to construct CDV124 and CDV129, and co-transformed with the rph1Δ::TRP1 deletion cassette into strain W303-1A to construct IP17 and IP13, respectively.
Transformants that had GIS1 replaced by gis1Δ::HIS3 or gis1Δ::kanMX2, RPH1 replaced by rph1Δ::TRP1, or RIM15 replaced by rim15Δ::kanMX2 were confirmed by PCR and/or Southern blot analysis (data not shown). NB14 and NB15 were generated by PCR-based, chromosomal myc13 tagging of GIS1 in AR1-1A and AR1-1C, respectively, using pFA6a-13Myc-TRP1 as template (Longtine et al., 1998).
Plasmid manipulations were performed in Escherichia coli strain DH5α (Gibco-BRL) using standard procedures (Sambrook et al., 1989). Yeast and E.coli media, including the rich, glucose-containing medium (YPD), the defined media (SD with appropriate supplements) and sporulation medium, were prepared by standard recipes (Sambrook et al., 1989; Rose et al., 1990). Standard procedures of yeast genetics and molecular biology were used (Sambrook et al., 1989; Guthrie and Fink, 1991). Yeast transformations were performed using a modification of the Li+ ion method (Gietz et al., 1992). Sporulation experiments were performed essentially as described (Reinders et al., 1998).
Plasmid constructions
For construction of the galactose-inducible GAL1-GIS1 and GAL1-RPH1 alleles, the full-length GIS1 and RPH1 coding sequences were amplified using the Expand Long Template PCR System (Boehringer Mannheim) and genomic DNA of strain AR2 (Table III) as template. SalI restriction sites were introduced immediately upstream of both ATG start codons, and NotI restriction sites were introduced 227 and 220 bp downstream of the GIS1 and RPH1 stop codons, respectively. The PCR products were cloned at the SalI–NotI sites of YCpIF2 (Foreman and Davis, 1994) to yield YCpIF2-GIS1 and YCpIF2-RPH1. Plasmids YCpADH1-GIS1 and YCpADH1-RPH1 were constructed by replacing the GAL1 promoter-containing ApaI–SalI fragments of YCpIF2-GIS1 and YCpIF2-RPH1, respectively, with a PCR-generated ApaI–SalI fragment containing the 854 nucleotides upstream of and including the ADH1 start codon. Plasmids YCpIF2-RIM15, YCpADH1 and YCpADH1-RIM15 were described earlier (Reinders et al., 1998). To fuse Gis1 and Rph1 to the LexA DBD coding sequence in plasmid pEG202 (Zervos et al., 1993), GIS1 and RPH1 full-length coding sequences were amplified as described above and cloned at the SalI–NotI sites of a modified version of plasmid pEG202 that contains a unique SalI site in its polylinker. The constructs contain four (EFVD) additional amino acids between the LexA DBD and the first amino acid (M) of the fused protein. Plasmid pLS9-PDS was constructed by cloning a synthetic double-stranded oligonuceotide consisting of 37 bp identical to the SSA3-PDS region (–206 to –170), plus AATT overhangs and an additional HindIII restriction site upstream of the SSA3-PDS region, into the EcoRI site of pLS9 (Sarokin and Carlson, 1986). Plasmid pCTT1-18/7x has been described in Marchler et al. (1993).
Preparation and northern analysis of RNA
Extraction of total cellular RNA was performed as described previously (Piper, 1994). For northern analysis, 10 µg of total RNA were separated on 1.1% agarose gels containing 0.65 M formaldehyde, transferred to nitrocellulose membranes (Protran BA 83, 0.2 µm; Schleicher and Schuell, Germany) in 20× SSC and hybridized with [32P]dATP-labeled DNA fragments that were amplified by PCR from genomic DNA (of strain AR2; Table III) and labeled by using the Prime-It Random Primer Labeling Kit (Stratagene). The primers used for PCR to generate SSA3, HSP12, HSP12 and SSB1 DNA fragments have been described in Reinders et al. (1998); those used to generate the 2.78 kb TPS2 DNA fragment were as follows: 5′-ACCACTGCCCCGGGCAATTCTCCAAAGAAGAGACAG-3′ (forward) and 5′-ACTATTATATATATGCCCGGGTCGCAACACCACATA-3′ (reverse).
Protein analyses
Protein concentrations were measured either by the Bio-Rad protein assay according to the manufacturer’s instructions or by a modified Lowry assay (Peterson, 1977) using bovine serum albumin (BSA) as standard. For immunodetection of Gis1-myc13, total cellular proteins were extracted by vortexing cells in lysis buffer [0.2 M Tricine (Na+) pH 7.0, and one tablet of complete protease inhibitor cocktail (Boehringer Mannheim) per 50 ml] in the presence of acid-washed glass beads (0.4 mm diameter; Merck), run on SDS–polyacrylamide gels (10% w/v acrylamide) and transferred electrophoretically to nitrocellulose membranes as described previously (Reinders et al., 1999). Blots were stained with Ponceau S to ensure equal protein loading. The membranes were incubated with 1:5000 diluted monoclonal mouse anti-myc (MIgG1) antibodies (Invitrogen), followed by alkaline phosphatase-conjugated rabbit anti-mouse IgG secondary antibodies (Biosource, USA). Reactive bands were visualized by an enzyme-catalyzed color reaction with 5-bromo-4-chloro-3-indolyl phosphate and nitroblue tetrazolium salt (Fluka, Switzerland).
Acknowledgments
Acknowledgements
We thank P.Estruch, H.Ronne and S.Garrett for strains, and E.A.Craig, M.Carlson and H.Ruis for plasmids. We are grateful to D.Balciunas, T.Boller and A.Wiemken for critically reading the manuscript. This work was supported by the Swiss National Science Foundation, grant 3100-052245.97/1 to C.D.V.
References
- Balciunas D. and Ronne,H. (1999) Yeast genes GIS1–4: multicopy suppressors of the Gal– phenotype of snf1 mig1 srb8/10/11 cells. Mol. Gen. Genet., 262, 589–599. [DOI] [PubMed] [Google Scholar]
- Baudin A., Ozier-Kalogeropoulos,O., Denouel,A., Lacroute,F. and Cullin,C. (1993) A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res., 21, 3329–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher dos Passos J., Vanhalewyn,M., Brandao,R.L., Castro,I.M., Nicoli,J.R. and Thevelein,J.M. (1992) Glucose-induced activation of plasma membrane H+-ATPase in mutants of the yeast Saccharomyces cerevisiae affected in cAMP metabolism, cAMP-dependent protein phosphorylation and the initiation of glycolysis. Biochim. Biophys. Acta, 1136, 57–67. [DOI] [PubMed] [Google Scholar]
- Bi E. and Pringle,J.R. (1996) ZDS1 and ZDS2, genes whose products may regulate Cdc42p in Saccharomyces cerevisiae. Mol. Cell. Biol., 16, 5264–5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhm S., Frishman,D. and Mewes,H.W. (1997) Variations of the C2H2 zinc finger motif in the yeast genome and classification of yeast zinc finger proteins. Nucleic Acids Res., 25, 2464–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boorstein W.R. and Craig,E.A. (1990) Regulation of a yeast HSP70 gene by a cAMP responsive transcriptional control element. EMBO J., 9, 2543–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boy-Marcotte E., Perrot,M., Bussereau,F., Boucherie,H. and Jacquet,M. (1998) Msn2p and Msn4p control a large number of genes induced at the diauxic transition which are repressed by cyclic AMP in Saccharomyces cerevisiae. J. Bacteriol., 180, 1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broach J.R. and Deschenes,R.J. (1990) The function of RAS genes in Saccharomyces cerevisiae.Adv. Cancer Res., 54, 79–139. [DOI] [PubMed] [Google Scholar]
- Camonis J.H., Kalékine,M., Gondré,B., Garreau,H., Boy-Marcotte,E. and Jacquet,M. (1986) Characterization, cloning and sequence analysis of the CDC25 gene which controls the cyclic AMP level of Saccharomyces cerevisiae.EMBO J., 5, 375–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry J.R., Johnson,T.R., Dollard,C., Shuster,J.R. and Denis,C.L. (1989) Cyclic AMP-dependent protein kinase phosphorylates and inactivates the yeast transcriptional activator ADR1. Cell, 56, 409–419. [DOI] [PubMed] [Google Scholar]
- DeRisi J.L., Iyer,V.R. and Brown,P.O. (1997) Exploring the metabolic and genetic control of gene expression on a genomic scale. Science, 278, 680–686. [DOI] [PubMed] [Google Scholar]
- Engelberg D., Zandi,E., Parker,C.S. and Karin,M. (1994) The yeast and mammalian Ras pathways control transcription of heat shock genes independently of heat shock transcription factor. Mol. Cell. Biol., 14, 4929–4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estruch F. and Carlson,M. (1993) Two homologous zinc finger genes identified by multicopy suppression in a SNF1 protein kinase mutant of Saccharomyces cerevisiae.Mol. Cell. Biol., 13, 3872–3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foreman P.K. and Davis,R.W. (1994) Cloning vectors for the synthesis of epitope-tagged, truncated and chimeric proteins in Saccharomyces cerevisiae. Gene, 144, 63–68. [DOI] [PubMed] [Google Scholar]
- Garrett S. and Broach,J. (1989) Loss of Ras activity in Saccharomyces cerevisiae is suppressed by disruptions of a new kinase gene, YAK1, whose product may act downstream of the cAMP-dependent protein kinase. Genes Dev., 3, 1336–1348. [DOI] [PubMed] [Google Scholar]
- Garrett S., Menold,M.M. and Broach,J.R. (1991) The Saccharomyces cerevisiae YAK1 gene encodes a protein kinase that is induced by arrest early in the cell cycle. Mol. Cell. Biol., 11, 4045–4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz D., St. Jean,A., Woods,R.A. and Schiestl,R.H. (1992) Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res., 20, 1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görner W., Durchschlag,E., Martínez-Pastor,M.T., Estruch,F., Ammerer,G., Hamilton,B., Ruis,H. and Schüller,C. (1998) Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev., 12, 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C. and Fink,G.R. (1991) Guide to yeast genetics and molecular biology. Methods Enzymol., 194. [PubMed] [Google Scholar]
- Gyuris J., Golemis,E., Chertkov,H. and Brent,R. (1993) Cdi1, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell, 75, 791–803. [DOI] [PubMed] [Google Scholar]
- Hall D.D., Markwardt,D.D., Parviz,F. and Heideman,W. (1998) Regulation of the Cln3-Cdc28 kinase by cAMP in Saccharomyces cerevisiae.EMBO J., 17, 4370–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang Y.K., Wang,L. and Sancar,G.B. (1999) RPH1 and GIS1 are damage-responsive repressors of PHR1.Mol. Cell. Biol., 19, 7630–7638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C. and Struhl,K. (1994) Protein kinase A mediates growth-regulated expression of yeast ribosomal protein genes by modulating RAP1 transcriptional activity. Mol. Cell. Biol., 14, 1920–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy M., Xiao,Y. and Mitchell,A.P. (1997) Interaction of yeast repressor–activator protein Ume6p with glycogen synthase kinase 3 homolog Rim11p. Mol. Cell. Biol., 17, 7230–7236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine M.S., McKenzie,A.,III, Demarini,D.J., Shah,N.G., Wach,A., Brachat,A., Philippsen,P. and Pringle,J.R. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae.Yeast, 14, 953–961. [DOI] [PubMed] [Google Scholar]
- Mager W.H. and De Kruijff,A.J. (1995) Stress-induced transcriptional activation. Microbiol. Rev., 59, 506–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler G., Schüller,C., Adam,G. and Ruis,H. (1993) A Saccharomyces cerevisiae UAS element controlled by protein kinase A activates transcription in response to a variety of stress conditions. EMBO J., 12, 1997–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Pastor M.T., Marchler,G., Schüller,C., Marchler-Bauer,A., Ruis,H. and Estruch,F. (1996) The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress-response element (STRE). EMBO J., 15, 2227–2235. [PMC free article] [PubMed] [Google Scholar]
- Miller J.H. (1972) Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Neumann-Silberberg F.S., Bhattacharya,S. and Broach,J.R. (1995) Nutrient availability and the Ras/cyclic AMP pathway both induce expression of ribosomal protein genes in Saccharomyces cerevisiae but by different mechanisms. Mol. Cell. Biol., 15, 3187–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrou J.L., Enjalbert,B. and François,J. (1999) STRE- and cAMP-independent transcriptional induction of Saccharomyces cerevisiae GSY2 encoding glycogen synthase during diauxic growth on glucose. Yeast, 15, 1471–1484. [DOI] [PubMed] [Google Scholar]
- Peterson G.L. (1977) A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal. Biochem., 83, 346–356. [DOI] [PubMed] [Google Scholar]
- Piper P.W. (1994) Measurement of transcription. In Johnston,J.R. (ed.), Molecular Genetics of Yeast. A Practical Approach. IRL Press at Oxford University Press, Oxford, UK, pp. 135–146. [Google Scholar]
- Ramer S.W., Elledge,S.J. and Davis,R.W. (1992) Dominant genetics using a yeast genomic library under the control of a strong inducible promoter. Proc. Natl Acad. Sci. USA, 89, 11589–11593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinders A., Bürckert,N., Boller,T., Wiemken,A. and De Virgilio,C. (1998) Saccharomyces cerevisiae cAMP-dependent protein kinase controls entry into stationary phase through the Rim15p protein kinase. Genes Dev., 12, 2943–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinders A., Romano,I., Wiemken,A. and De Virgilio,C. (1999) The thermophilic yeast Hansenula polymorpha does not require trehalose synthesis for growth at high temperatures but does for normal acquisition of thermotolerance. J. Bacteriol., 181, 4665–4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson L.C. and Tatchell,K. (1991) TFS1: a suppressor of cdc25 mutations in Saccharomyces cerevisiae.Mol. Gen. Genet., 230, 241–250. [DOI] [PubMed] [Google Scholar]
- Rose M.D., Winston,F. and Hieter,P. (1990) Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Ruis H. and Schüller,C. (1995) Stress signaling in yeast. BioEssays, 17, 959–965. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Sarokin L. and Carlson,M. (1986) Short repeated elements in the upstream regulatory region of the SUC2 gene of Saccharomyces cerevisiae. Mol. Cell. Biol., 6, 2324–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sass P., Field,J., Nikawa,J., Toda,T. and Wigler,M. (1986) Cloning and characterization of the high-affinity cAMP phosphodiesterase of Saccharomyces cerevisiae.Proc. Natl Acad. Sci. USA, 83, 9303–9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt A.P. and McEntee,K. (1996) Msn2p, a zinc finger DNA-binding protein, is the transcriptional activator of the multistress response in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 93, 5777–5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski R.S. and Hieter,P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics, 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A., Ward,M.P. and Garrett,S. (1998) Yeast PKA represses Msn2p/Msn4p-dependent gene expression to regulate growth, stress response and glycogen accumulation. EMBO J., 17, 3556–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet D.H., Kang,Y.K. and Sancar,G.B. (1997) Role of UME6 in transcriptional regulation of a DNA repair gene in Saccharomyces cerevisiae.Mol. Cell. Biol., 17, 6223–6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadi D., Hasan,R.N., Bussereau,F., Boy-Marcotte,E. and Jacquet,M. (1999) Selection of genes repressed by cAMP that are induced by nutritional limitations in Saccharomyces cerevisiae.Yeast, 15, 1733–1745. [DOI] [PubMed] [Google Scholar]
- Tatchell K. (1986) RAS genes and growth control in Saccharomyces cerevisiae.J. Bacteriol., 166, 364–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevelein J.M. and de Winde,J.H. (1999) Novel sensing mechanisms and targets for the cAMP-protein kinase A pathway in the yeast Saccharomyces cerevisiae.Mol. Microbiol., 33, 904–918. [DOI] [PubMed] [Google Scholar]
- Thomas B.J. and Rothstein,R. (1989) Elevated recombination rates in transcriptionally active DNA. Cell, 56, 619–630. [DOI] [PubMed] [Google Scholar]
- Treger J.M., Schmitt,A.P., Simon,J.R. and McEntee,K. (1998) Transcriptional factor mutations reveal regulatory complexities of heat shock and newly identified stress genes in Saccharomyces cerevisiae. J. Biol. Chem., 273, 26875–26879. [DOI] [PubMed] [Google Scholar]
- Varela J.C.S., Praekelt,U.M., Meacock,P.A., Planta,R.J. and Mager,W.H. (1995) The Saccharomyces cerevisiae HSP12 gene is activated by the high-osmolarity glycerol pathway and negatively regulated by protein kinase A. Mol. Cell. Biol., 15, 6232–6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidan S. and Mitchell,A.P. (1997) Stimulation of yeast meiotic gene expression by the glucose-repressible protein kinase Rim15p. Mol. Cell. Biol., 17, 2688–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A., Brachat,A., Pöhlmann,R. and Philippsen,P. (1994) New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae.Yeast, 10, 1793–1808. [DOI] [PubMed] [Google Scholar]
- Ward M.P., Gimeno,C.J., Fink,G.R. and Garrett,S. (1995) SOK2 may regulate cyclic AMP-dependent protein kinase-stimulated growth and pseudohyphal development by repressing transcription. Mol. Cell. Biol., 15, 6854–6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner-Washburne M., Becker,J., Kosic-Smithers,J. and Craig,E.A. (1989) Yeast Hsp70 RNA levels vary in response to the physiological status of the cell. J. Bacteriol., 171, 2680–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner-Washburne M., Braun,E., Johnston,G.C. and Singer,R.A. (1993) Stationary phase in the yeast Saccharomyces cerevisiae.Microbiol. Rev., 57, 383–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zervos A.S., Gyuris,J. and Brent,R. (1993) Mxi1, a protein that specifically interacts with Max to bind Myc–Max recognition sites. Cell, 72, 223–232. [DOI] [PubMed] [Google Scholar]