


Abstract
Clerocidin (CL) is an effective topoisomerase II-poison, which has been shown to produce DNA depurination and strand breaks per se at the guanine (G) level. To elucidate the roles played by the different functional groups of CL in the reactivity towards nucleic acids, we investigated CL derivatives with key structural modifications. The derivatives were reacted with plasmid, single-/double-stranded DNA and isolated 2′-deoxy-guanosines (dG). We show here that an intact oxirane ring is essential to achieve DNA modification and depurination. Through HPLC/MS and MS/MS techniques we were able to unambiguously characterize adducts obtained by reacting isolated dG and single-/double-stranded DNA with the drugs, indicating beyond reasonable doubt that the structure of a typical adduct is formed by epoxide alkylation at N7 of G with subsequent loss of the pentose unit. Further, we showed that reduction of vicinal carbonyl functions affect drug activity to a large extent. Our findings demonstrate that the characteristic DNA-alkylating properties of CL arise from mutual action of the functional groups present in this molecule. Its oxidation state seems crucial to modulate the rates of reactivity by finely tuning the strain applied on the oxirane ring.
INTRODUCTION
Clerocidin (CL) is a topoisomerase II poison with the characteristic ability to irreversibly trap the DNA–enzyme complex when a guanine is found at the –1 position from the cleavage site (1). In the absence of enzyme, the drug presents intrinsic nucleic acids-alkylating activity, which targets the N7 of guanines in single-stranded DNA, induces depurination and may lead to strand scission at the apurinic site (2).
CL (Fig. 1A) displays several electrophilic groups, which include an α-ketoaldehyde function in equilibrium with the hemi-acetalic form, a strained epoxy ring (region C12-C15), and a second aldehyde at position 4 of the diterpenoid structure. The latter is not believed to contribute directly to the poisoning activity of CL, since congeners devoid of this group, such as terpentecin and UCT4B, are still able to inhibit the eukaryotic enzyme (3–5). On the other hand, structural modifications at the C12-C15 ring system, which were observed upon storage in ethanol, induce the loss of the ability to poison topoisomerase II (1) and nick DNA per se (2), thus implicating the functional groups present in this region in the mechanism of action, both in the presence and in the absence of enzyme.
Figure 1.

Structures of CL and NA derivatives. (A) The dicarbonylic form of the drug (DC) is shown in equilibrium with the closed hemi-acetal species (HA). The numbered atoms outline electrophilic carbons possibly involved in reactions with nucleophilic moieties in the DNA. (B) Intact epoxide naphthalene derivatives. (C) Open epoxide naphthalene derivatives. The carbonyl functions in NA1 and NA4 are unmodified, in NA2 and NA5 they are partially reduced, and in NA3 and NA3open they are fully reduced.
Epoxide alkylation at N7 of guanines is responsible for the activity of compounds, such as the pluramicin family of antibiotics (6,7) and the active metabolite of Aflatoxin B1 (8–10). This observation leads to advance the epoxide at C13 of clerocidin as the most likely candidate to induce DNA damage at guanines, but its direct involvement has not been demonstrated. Similarly, the function of the other parts of the molecule remains to be elucidated and could lead to the rational design of synthetic analogues with improved activity profiles.
This work presents a systematic study of the role of each chemical group in the reactivity of clerocidin towards DNA. Synthetic CL derivatives were designed to include substitution of the diterpenoid moiety and several different modifications at the C12-C15 ring system. First, an aromatic naphthalene was introduced in place of the diterpenoid portion to avoid synthesis of chiral products and to enable the detection by fluorescence spectroscopy. Second, this new class of naphthalene-derivatives was divided into a series of products with an intact (NA1, NA2 and NA3, Fig. 1B) or an open epoxide ring (NA4, NA5 and NA3open, Fig. 1C). Third, to evaluate the contribution of vicinal carbonyls to the alkylation process, the oxidation state of the carbonyls was varied in both series from fully oxidized (CL-like, such as NA1 and NA4), to partially (NA2 and NA5) and fully reduced (NA3 and NA3open). In particular, it should be noted that the oxidation states of C14 in NA2 and NA5 and of both C14 and C15 in NA3 and NA3open are similar to those found in spirocardin A and B, respectively, two closely related CL derivatives with antibacterial activity (11).
The synthetic derivatives were tested for their ability to react with plasmid DNA and single/double-stranded oligonucleotides. Adducts formed with isolated G nucleosides and structured DNA were characterized to obtain unequivocal proof of the involvement of the epoxide ring in DNA alkylation, depurination and strand break. A careful comparison of their reactivity was performed under similar experimental conditions to investigate the possible modulating effects exerted by the carbonyl functions of the C12-C15 ring on the reactivity of the epoxide ring.
MATERIALS AND METHODS
Enzymes, oligonucleotides and chemicals
CL was a kind gift of Leo Pharmaceutical Products (Ballerup, Denmark), while NA compounds were synthesized in our laboratories according to the procedure developed for the synthesis of the highly oxygenated moiety of terpentecin (12,13), and fully characterized by NMR and ESI-MS analy sis (NA1 m/z 270.1, NA2 m/z 272.1, NA3 m/z 274.1, NA4 m/z 288.1, NA5 m/z 290.1, NA3open m/z 292.2). The synthetic procedures will be reported elsewhere. CL and NA analogues were dissolved in absolute ethanol and the concentration determined by measuring absorbance in ethanol at 230 and 282 nm respectively, on a UV/VIS Spectrometer Lambda 12 (Perkin Elmer, MA, USA), using the experimentally determined molar extinction coefficient of 11818 M–1cm–1 for CL and 9493 M–1cm–1 for NA derivatives. Working drug solutions were obtained by diluting fresh stocks in the appropriate buffer. Buffer components, nucleosides and chemicals for MS calibration were purchased from Sigma-Aldrich (MI, USA), while electrophoretic reagents were from Amersham Biosciences Europe (Freiburg, Germany). SV40 plasmid, restriction enzymes and T4 polynucleotide kinase were purchased from Invitrogen (Paisley, UK). [γ-32P]ATP was from Perkin Elmer (MA, USA), while all oligonucleotides were from Eurogentec (Liege, Belgium). HPLC purity solvents were from LabScan (Dublin, Ireland).
Clerocidin nicking of supercoiled SV40
The supercoiled form of SV40 (40 ng/sample) was reacted at 37°C with 100 and 200 µM of CL and NA derivatives in 50 mM phosphate buffer, pH 7.4, in a total volume of 20 µl. The final ethanol concentration was 1.25% (v/v) in all sample and control reactions. After 24 h each reaction was stopped on ice and gel loading buffer [0.025% blue bromophenol, 0.025% xylene cyanol, 2.5% Ficoll (type 400) in water] added. The treated samples were then loaded on a 1% agarose gel in 44.5 mM Tris–borate, 1 mM EDTA, pH 8.3, run and stained with ethidium bromide. The nicked standard was obtained as reported (14), while the linearized standard was obtained by BamHI digestion. Both the nicked and the linearized SV40 were purified by phenol/chloroform extraction followed by ethanol precipitation.
Reaction with oligonucleotides
The ability of CL and NA derivatives to damage single-stranded DNA was tested at the sequencing level using a short oligonucleotide whose sequence spans a strong irreversible site for CL cleavage in the presence of murine topo II, namely position 2640 in the SV40 genome (1). The sequence is CTA TTG CTT TAT TTG TAA CCA TTA TAA GCT (CL2640-up). The oligonucleotide was obtained in desalted and lyophilized form and was gel purified before use. It was then 5′-end-labeled with [γ-32P]ATP by T4 polynucleotide kinase and ethanol precipitated.
Drug reactions with the labeled single-stranded oligonucleotide (4 pmol/sample) were performed at 37°C in 50 mM phosphate buffer, pH 7.4, containing 80 mM KCl and 10 mM MgCl2. After 24 h, samples were ethanol precipitated to eliminate non-reacted drug, then resuspended and split into two aliquots, one of which was kept on ice, while the other was treated at 90°C for 30 min with 1 M piperidine to complete strand scission according to the Maxam and Gilbert protocol (15). Samples were finally lyophilized twice, resuspended in formamide gel loading buffer, heated at 90°C for 5 min and cleavage products resolved in 20% denaturing polyacrylamide gels. For reactions with double-stranded DNA, the labeled oligonucleotide CL2640-up was annealed to equimolar amounts of its complementary cold oligonucleotide (CL2640-down), whose sequence is: AGC TTA TAA TGG TTA CAA ATA AAG CAA TAG. The annealed species was further purified in native polyacrylamide gels and electroeluted using the Biotrap method (Schleicher & Schuell, NH). Reactions with drugs were performed as described above.
HPLC-MS and MS/MS
The following reaction mixtures were prepared to measure reactivity of CL and NA derivatives with dG: (i) 150 nmol of drugs in 25 µl ethanol and 150 nmol of nucleosides in 100 µl HEPES 50 mM, pH 7.4; (ii) 150 nmol of drugs in 25 µl ethanol and 100 µl HEPES 50 mM, pH 7.4; (iii) 150 nmol of nucleosides in 100 µl HEPES 50 mM, pH 7.4 and 25 µl ethanol. Each mixture was incubated at 37°C. At 0, 6, 18, 24 and 48 h of incubation, reaction mixtures were filtrated with 0.45 µm filters (Alltech, Dunboyne, Ireland) and loaded on a HPLC C18 reverse phase column (Extent-C18, 4.6 × 250 mm, Agilent Technologies, CA, USA). The HPLC system consisted of: Series 200 Pumps (Perkin Elmer, CA); UV-1806, UV-Vis Detector (Bio-Rad, Milan, Italy); NCI 900 Network Chromatography Interface (Perkin Elmer, CA, USA). To separate the adducts solvents A [CH3CN:H2O (9:1)/0.05% TFA] and B (H2O/0.05% TFA) were used, delivered at a flow rate of 1 ml/min. For separation of CL and NA1 solutions, the following gradients were used: A 20% for 8 min (equilibration time); A from 20 to 40% in 10 min; A from 40 to 100% in 10 min; A 100% for 5 min, A from 100 to 20% in 5 min. For NA3, NA4 and NA5: A 20% for 8 min (equilibration time); A from 20 to 40% in 25 min; A from 40 to 20% in 5 min. Peaks were detected at 230 nm, collected in Eppendorf tubes, dried at room temperature in Speed Vac UniVapo 100 H (UniEquip, Martinsried, Germany) and stored at 4°C if necessary.
Liquid chromatography mass spectrometry (LC-MS) and direct infusion electrospray ionization (ESI) mass spectrometry were performed, respectively, on a Time-of-Flight (TOF) mass analyser (Mariner ESI-TOF, Applied Biosystems, CA) and on a Fourier-transform mass spectrometer (ESI-FTMS) equipped with a 7 tesla actively shielded superconducting magnet and thermally assisted ESI source (Apex III, Bruker Daltonics, MA). Positive ion mass spectra were acquired on the ESI-TOF instrument by directly injecting 20 µl of analyte solutions in CH3CN:H2O:HCOOH (50:49.5:0.5) at room temperature and with a 0.16 µl/min flow rate. The nozzle temperature was 140°C, while a constant flow of N2 gas was kept at 0.35 l min–1 to facilitate the spray. A three-points external calibration provided typical 100 p.p.m. accuracy. Alternatively, lyophilized samples were dissolved in CH3OH:H2O:CH3COOH (49:49:2) for ESI-FTMS analysis in positive mode. A loop injector connected to a syringe pump was employed to deliver 10 µl samples at a flow rate of 2 µl/min. To facilitate ion desolvation, a heated-metal tubing interface built in house was kept at 120°C throughout the analysis. All data were acquired and processed using the software package XMASS 6.2 (Bruker Daltonics, MA). A resolving power of 150 000 was typically achieved at m/z 500 in broadband mode. A three-points external calibration provided a typical 4 p.p.m. accuracy, or better. Multiple-stage tandem experiments for structural characterization were performed by SORI/CAD with Ar as collision gas.
For reactions with single-stranded and plasmid DNA, CL (0.2 mg) was dissolved in 25 µl ethanol and reacted with 100 µl of ssDNA (3.8 mg) or CL (0.2 mg) was dissolved in 10 µl ethanol and reacted with 40 µl of SV40 supercoiled or linear plasmid DNA (1 µg) in HEPES 50 mM, pH 7.4, at 37°C for 48 h. The crude incubation mixtures were directly injected onto the HPLC and adducts were separated as described above; the fractions whose retention time corresponded to the adduct obtained in reaction of CL with dG, were collected and analyzed by MS as described above.
RESULTS
An intact epoxide is necessary to nick DNA at susceptible Gs
The naphthalene-analogues were initially reacted with a negatively supercoiled plasmid to assess whether the new derivatives retained DNA-cleaving activity typical of the parent drug CL. DNA strand scission can be readily determined from the different electrophoretic migration of supercoiled, nicked and linear forms of the plasmid on agarose gels. Initially four different concentrations were used (50, 100, 200 and 400 µM); at 50 µM CL showed little effect on the supercoiled plasmid, while at 400 µM CL displayed total conversion of the supercoiled plasmid into the nicked and linear forms, as already described (2). Hence, comparison of CL and NA analogues activity was performed at the intermediate concentrations of 100 and 200 µM. As observed in Figure 2, CL induced efficient nicking and linearization of supercoiled DNA at the lowest concentration applied after incubation at 37°C in the absence of topoisomerase II (Fig. 2A, C, E, lanes 4 and 5). Similarly, NA1 converted the supercoiled form of the plasmid (CCC) into the nicked (OC), and linear form (L) in a concentration dependent manner (Fig. 2A, lanes 6 and 7). The effect was comparable to the action of CL on the same substrate (Fig. 2A, lanes 4 and 5; Fig. 2B). Under like conditions, NA2 could nick, but not linearize supercoiled DNA (Fig. 2C, lanes 6 and 7; Fig. 2D), while NA3 was able to nick supercoiled DNA to some extent only at the highest concentration (Fig. 2E, lanes 6 and 7; Fig. 2F). No DNA cleavage could be detected when congeners of the open epoxide series were tested (Fig. 2A, C, E; compare lanes 8 and 9 with control lanes 3; Fig. 2B, D, F). It is clear from these data that an intact oxirane ring must be present for retention of cleavage activity. While carbonyls do not exhibit any strand scission activity per se, their oxidation state appears to have a great influence on the reactivity of the epoxide group.
Figure 2.

Reactivity of CL and NA derivatives on plasmid DNA. The ability to induce cleavage in plasmid DNA of the closed epoxide derivatives was compared to that of their open epoxide analogues. The supercoiled form of SV40 was reacted at 37°C with CL and NA derivatives. After 24 h each reaction was stopped and loaded on 1% agarose gel. (A) NA1 (lanes 6 and 7) and NA4 (lanes 8 and 9); (C) NA2 (lanes 6 and 7) and NA5 (lanes 8 and 9); (E) NA3 (lanes 6 and 7) and NA3open (lanes 8 and 9). In each case CL was used as positive control for DNA cleavage (lanes 4 and 5). (A), (C) and (E) Lanes 4, 6 and 8 correspond to 100 µM of drugs, lanes 5, 7 and 9 correspond to 200 µM of drugs. Lanes 1 and 2 represent controls for nicked (N) and linear (L) DNA respectively; lanes 3 are controls for unreacted supercoiled plasmid (S). CCC refers to the negatively supercoiled form of the plasmid SV40, OC refers to the open circular form and L to the linear structure. (B), (D) and (F) Relative amount (%) of the nicked and linearized forms of SV40 on the total plasmid present in each lane of (A), (C) and (E). CL effect at both drug concentrations is set as 100%, and values for the NA analogues were calculated accordingly.
A previous investigation has shown that CL induces cleavage in plasmid DNA through reactions towards Gs present in locally distorted regions within the supercoiled form of the nucleic acid. In fact, experiments performed on plain single-stranded oligonucleotides and double-stranded oligonucleotides exhibiting bulged segments, revealed an elevated activity exclusively towards unpaired Gs (2). For this reason, a single-stranded oligonucleotide whose sequence corresponds to one of the SV40 hot spots was employed to check whether the naphtalene-derivatives shared the expected alkylating specificity toward accessible G. This 30-nt substrate is characterized by a G in position 2639 of the SV40 sequence, which can be readily alkylated by CL both in the presence and in the absence of enzyme (1,2). Drug analogues were reacted at two different concentrations with the oligonucleotide, under conditions described earlier. The covalent adducts were treated with hot piperidine to complete the strand scission according to the Maxam and Gilbert protocol (15) and to provide a convenient way to assess the extent of G modification (2).
In most cases, a certain degree of substrate alkylation and scission at positions 2639 and 2648 (Fig. 3, G1 and G2, respectively) was detected for the analogues, with somewhat lower efficiency than provided by the CL control (Fig. 3). Two distinct concentrations, 100 and 200 µM, were employed for each compound to highlight possible concentration effects on reactivity. In this regard, NA1 proved to be slightly less active than CL at 100 µM (compare lane 5 with 3, G1 and G2), but exhibited similar activity at 200 µM (compare lane 6 with 4, G1 and G2). Similarly, NA2 proved to be less potent than both CL and NA1 at 100 µM (compare lane 7 with lanes 3 and 5, G1 and G2), whereas it almost matched its fully oxidized derivatives at 200 µM (compare lane 8 with lanes 4 and 6, G1 and G2). NA3 presented almost no cleavage activity at the lower drug concentration, since the yield of modification was comparable to that of the control lane (compare lanes 9 and 2, G1 and G2), whereas extensive cleavage was detected at 200 µM (lane 10, G1 and G2). Reactivity at C appears only after hot piperidine treatment, as already described for CL (2); NA analogues retained essentially the same behavior, with decreasing effects from NA1 to NA3. No modification was detected for NA4, NA5 and NA3open (data not shown), confirming the same inability to react with DNA observed earlier with plasmid SV40. Finally, the NA derivatives were shown to have no effect on double-stranded DNA, as observed for CL (2).
Figure 3.

Reactivity of CL and NA derivatives toward single-stranded DNA. The oligonucleotide CTA TTG CTT TAT TTG TAA CCA TTA TAA GCT was 5′-end-labeled and reacted with CL, NA1, NA2 and NA3 at 100 (odd lanes) or 200 µM (even lanes) in 50 mM phosphate buffer, pH 7.4, 80 mM KCl and 10 mM MgCl2 and ethanol precipitated after 24 h. Each sample was further treated with piperidine 1 M at 90°C for 30 min, vacuum dried, and then loaded on 20% denaturing acrylamide gels. Gels were run at 90 W for 2.5 h and then autoradiographed. Control lanes are shown as C: lane 1 is the non-treated oligo, lane 2 indicates the non-reacted oligo treated in the same conditions as samples reacted with the drugs. Lane 11 is a marker for purines (G+A) obtained by the Maxam and Gilbert reaction (15). Positions of the drug-attacked Gs are indicated on the left side (G1 and G2 refer respectively to positions 2639 and 2648 of the SV40 genome), while the oligonucleotide resolved sequence is reported on the right.
The epoxide moiety reacts with N7 of isolated dGs
The proof in favor of a mechanism involving alkylation of N7 by the epoxide moiety is strong, though indirect. In fact, the lack of alkylating activity shown by the open-oxirane series offers a clear indication that this functional group must be involved in nucleic acids attack. A substrate oligonucleotide including 7-deaza-2′-deoxyguanosine was found to be completely refractory to cleavage by clerocidin, suggesting that N7 must be the target of CL activity (2). However, it could be argued that alternative alkylation at the six-membered ring of guanosine is also possible, but may remain undetected for its inability to induce depurination and strand cleavage. For these reasons, only a direct characterization of the adducts produced by drug reaction can provide an unambiguous and conclusive confirmation of the proposed mechanism.
Samples necessary for adduct characterization were prepared by utilizing 2′-deoxy-guanosine as a substrate in a series of alkylation reactions with the different drug analogues. Adduct formation was monitored by reverse-phase HPLC and HPLC-MS after intervals of 0, 6, 18, 24 and 48 h. Control reactions consisted of drugs and nucleoside treated separately in the same conditions. Complex peak profiles, which are due to multiple solution equilibria involving the C12-C15 ring (16–18), required a differential analysis of chromatograms to correctly identify the fractions containing drug-adducts (Fig. 4A). Essentially one major new peak was detected in each reaction.
Figure 4.
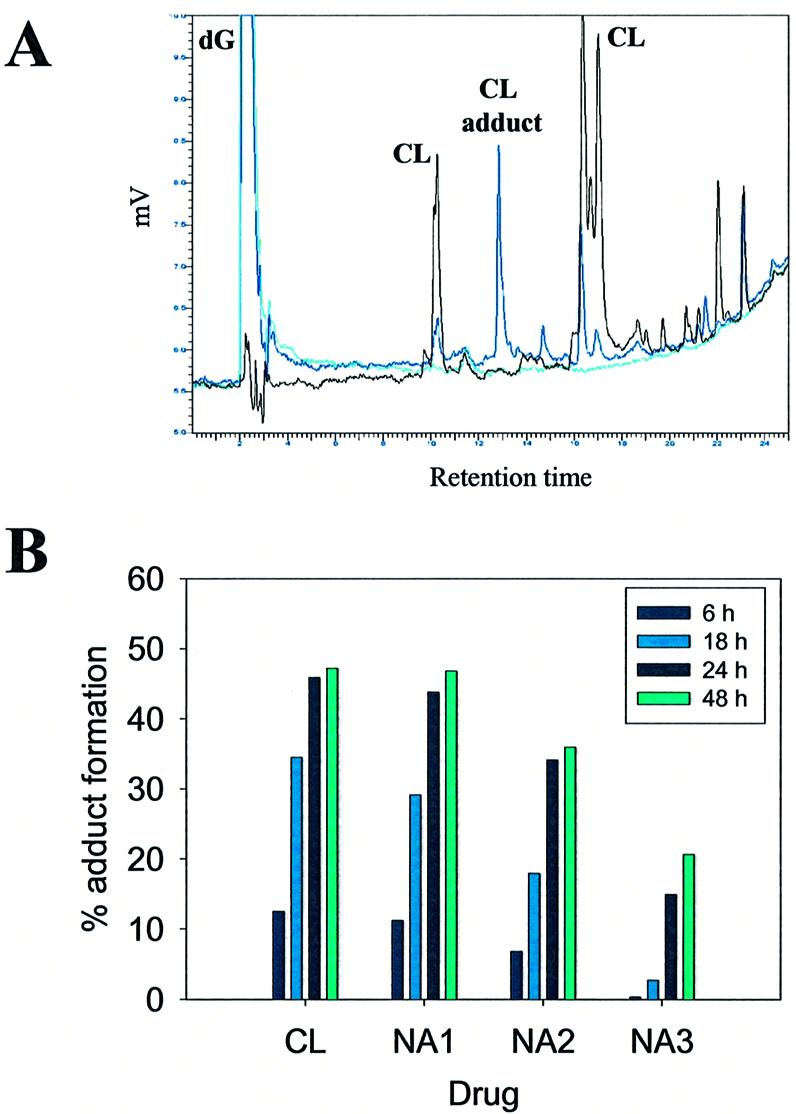
Adduct formation in reactions of CL and derivatives with dG. (A) Chromatogaphic profile for CL-dG reaction. Chromatographic profiles for CL-dG (black line), CL (dark blue line) and dG (light blue line) treated at 37°C for 24 h are shown overlapped. Peak identities are shown above each peak. (B) Relative yields of adduct formation. CL and its NA analogues were reacted with dG for 6, 18, 24 and 48 h. The incubation temperature was set at 37°C for reaction with CL, NA1 and NA2, at 60°C for reaction with NA3. Adduct peaks were identified through comparison of chromatograms obtained from reaction mixtures and free drugs, both treated in the same conditions. For each drug, the percentage of adduct formation was calculated from the ratio of the area of adduct peaks over total peak area.
As expected from the results obtained for the single-stranded oligonucleotide, CL and analogues with intact epoxide (NA1, NA2 and NA3) were able to form stable covalent adducts with guanosine, albeit with different rates. The ratio between the area of the adduct peak and the total area of peaks detected in each chromatogram provides an accurate determination of the yield of reaction (Fig. 4B). In particular, CL and NA1 gave very similar yields upon 24 h reaction at 37°C (45.9 and 43.8% respectively), which were slightly higher than those observed for NA2 (34.1%). To the contrary, no adducts could be detected for NA3 even after 4 days incubation, while increasing the temperature to 60°C for 24 h induced the formation of 14.9% adduct. The same expedient failed to succeed for NA4 and NA5, which could not form G-adducts at either temperature even after extended reaction times. This pattern of modification is in excellent agreement with those observed earlier for the single-stranded substrate under similar experimental conditions.
The fractions corresponding to new peaks formed during the alkylation reactions were collected and analyzed by mass spectrometry based on electrospray ionization coupled with a time-of-flight analyzer (ESI-TOF MS). Molecular masses obtained for these species match 1:1 covalent adducts of 2′-deoxy-guanosine with the different drugs including the loss of the deoxy-ribose moiety (Table 1). The depurination process is a direct and unique consequence of alkylation at N7 of guanines, which is routinely used in biochemical sequencing of DNA based on the Maxam and Gilbert protocol (15) and has been reported for carcinogenic alkylators, such as the diol epoxide of benzo(a)pyrene (19), or epoxide-containing drugs (6,20). A plausible mechanism would involve the initial attack of the epoxide to the nucleophilic N7 of the guanine base. Cleavage of the N-glycosidic bond at N9 is promoted by rearrangement of unshared electrons on the oxygen of the deoxy-ribose ring, or by base-extraction of a proton on C2 and β-elimination across C1-C2 of the pentose. The ensuing electronic rearrangement neutralizes the positive charge on the imidazolinic ring, which was induced by the initial N7 alkylation (Fig. 5A) (21).
Table 1. Identity and m/z values of the adducts obtained reacting CL and the NA derivatives with dG.
| Reactivity with dG | ||
|---|---|---|
| Compound | adduct | m/z |
| CL | CL + Gbase | 500.2 |
| NA1 | NA1 + Gbase | 422.2 |
| NA2 | NA2 + Gbase | 424.2 |
| NA3 | NA3 + Gbase | 426.2 |
| NA4 | no reaction | |
| NA5 | no reaction | |
Figure 5.

(A) Scheme of reaction of CL with dG. Reactions involved in the formation of CL-adduct with G-base are shown. The nucleophilic atom N7 of the purine ring attacks the electron acceptor C13 atom of the epoxide ring, which is activated by the vicinal carbonyl moieties and by the spiro ring system. A positively charged molecule forms, which easily depurinates leading to a stable neutral molecule. (B) Scheme of fragmentation analysis of CL-adduct with G-base molecular ion. Structures corresponding to fragments generated by gas-phase activation of the molecular ion (m/z 500.2) are shown.
The high yields of depurination are generally explained by the elimination of very favorable leaving groups, such as the adducts between drugs and guanine-base detected by mass spectrometry. It should also be noted that adducts retaining the 2′-deoxy-ribose unit were basically not observed, thus effectively ruling out the possibility of alkylation at the six-membered ring, which is not conducive to depurination.
Similar HPLC-MS analysis was performed on the sample mixtures obtained by CL-treatment of plasmid and single-stranded DNA (see above). In each case, we found a peak whose retention time and m/z value corresponded to those observed for the adduct of CL and G-base obtained from the reaction of isolated nucleoside (Fig. 6). These data suggest that application of CL to different nucleic acid substrates (e.g., plasmid, single-stranded DNA, and isolated dG) induces the formation of the same alkylation product. Furthermore, an excellent correlation was found between the rates of CL-reactivity with the different substrates, indicating that the mechanism of base modification and depurination is very likely to remain unchanged whether G is isolated or included in a nucleic acids strand.
Figure 6.

ESI-MS spectrum of ‘CL-Gbase’ adduct. The adduct peak obtained in the reaction of CL with ssDNA showed a retention time corresponding to the CL-adduct with G-base obtained in the reaction of CL with dG. The peak was collected and analyzed by ESI-MS in positive ion mode to determine the molecular masses of the reaction products.
Finally, the analysis of adducts provided by the naphthalene-analogues with dG indicates that reduction of carbonyl functions in NA2 and NA3 does not appear to modify the type of adducts produced, but rather the rates of adduct formation. This pattern of reactivity is consistent with those offered by the same drugs with the other nucleic acid substrates and further supports the hypothesis that modification of the carbonyls on the C12-C15 ring may contribute to modulate the reactivity of the oxirane function.
Characterization of CL-guanine adduct by MS/MS analysis
The fragmentation pattern obtained by gas-phase activation of a selected precursor ion in MS/MS techniques is diagnostic of the precursor structure and can be effectively employed to characterize nucleic acid adducts (22,23). Dissociation experiments were performed off-line on isolated HPLC fractions on a Fourier-transform mass spectrometer (FTMS) (24,25). Product ions were detected in positive ion mode upon activation of CL-guanine adduct (Table 2). Putative ion structures are provided in Figure 5B.
Table 2. Summary of m/z values and relative intensities of ions provided by MS/MS of m/z 500.2 precursor ion, CL-Gbase adduct.
| m/z | Relative intensity (%) |
|---|---|
| 500.2 | 53.6 |
| 482.2 | 10.0 |
| 278.1 | 3.34 |
| 187.1 | 2.5 |
| 152.0 | 100 |
| 135.0 | 8.1 |
The observed fragmentation pattern is in excellent agreement with the proposed structure produced by opening of the epoxide and formation of a covalent bond with N7 of guanine. Gas-phase elimination of water (m/z 482.2) is very facile and characteristic of tertiary alcoholic functions. This process was also noted in the fragmentation of similar CL-adducts with different nucleophiles (not shown) and indicates that opening of the oxirane ring occurs with N7 attacking the secondary carbon C16, rather than the tertiary C13. Due to the relatively higher basicity of the guanine moiety, the diterpenoid portion is lost as neutral and is not detected, while the positive charge is retained by the remaining part, which includes the C12-C15 ring with possible rearrangement (m/z 278.1). Further fragmentation involves the loss of the C12-C15 ring and leaves the protonated form of guanine base (m/z 152.1). Alternative ion structures with the same mass (and thus same elemental composition) are provided as a testament to the chemical complexity of this molecule, but they are nevertheless consistent with the structure proposed for the adduct.
DISCUSSION
CL is an effective topoisomerase II-poison, which has been shown to produce DNA depurination and strand breaks per se. We have investigated CL derivatives with key modifications at the C12-C15 ring system and substitution of the diterpenoid moiety with naphthalene to elucidate the roles of the different functional groups in the reactivity of CL towards nucleic acids. In particular, we discovered that the aromatic ring does not appreciably affect drug activity, sequence specificity and topological requirements, since the naphthalene-derivative with intact C12-C15 portion (NA1) has shown a preference for G and a cleavage activity on plasmid, single- and double-stranded DNA, which are comparable to those of the prototype drug clerocidin. These findings excluded any possible intervention of the aldehyde at position 4 of the diterpenoid ring and focused our attention on the C12-C15 portion of CL.
Next, the examination of carbonyl-reduced (NA2 an NA3) and open epoxide derivatives (NA4, NA5 and NA3open) offered convincing proof that an intact oxirane ring is essential to achieve DNA modification and depurination. In fact, compounds with intact epoxide, but devoid of carbonyl functions, were still able to induce strand breaks in the nucleic acid substrates.
Finally, ESI-TOF MS and MS/MS techniques enabled the unambiguous characterization of adducts obtained by reacting isolated dG nucleosides with the different drugs, indicating beyond reasonable doubt that the structure of a typical adduct is formed by epoxide alkylation of guanine N7 with subsequent loss of the pentose unit. A similar mechanism, which induces depurination and may eventually lead to strand scission, has been demonstrated for other carcinogens and antibiotics bearing epoxide groups (6,7,19,20,26–29).
The same product formed by covalent attachment of CL and guanine base was also detected in reaction mixtures including single- and double-stranded nucleic acids, in the linear and supercoiled form respectively, thus ruling out possible effects due to the different accessibility and chemical environment of target G as free nucleoside in solution, or as part of a DNA strand.
It is very interesting to note that reduction of the carbonyl groups affected drug activity to a large extent. When the carbonyl at C14 was reduced (NA2), the ability of the drug to induce strand breaks into plasmid DNA was moderately lower than the prototype CL; when both functions at C14 and C15 were reduced (NA3), the activity dropped dramatically and an increased reaction temperature was needed to notice any effect at the nucleic acids level. The importance of the presence of the C15 carbonyl is very likely due to the formation of a stable intra-molecular bond with the hydroxyl function at C12 (16). The cyclic hemi-acetal engages the epoxide in a spiro system, which enhances its reactivity towards nucleophilic reagents. Reduction of C14 moderately affects epoxide reactivity, either through destabilization of the hemi-acetalic equilibrium at C15, or through loss of electron withdrawing effects that leaves the epoxide residue less electrophilic.
In conclusion, our findings demonstrate that the characteristic DNA-alkylating properties of CL arise from mutual action of the functional groups present in this molecule. Its oxidation state seems crucial to modulate the rates of reactivity by fine tuning the strain applied and the electron density on the oxirane ring. The diterpenoid ring system does not appear to be essential to the alkylation and depurination activity in vitro, but may cover a fundamental role in positioning the drug in susceptible sites within complex structural contexts in vivo. For this reason, future investigations in the presence of DNA-topoisomerase enzymes will be performed to ascertain whether the DNA-binding mechanism accounts for the prominent activity of clerocidin at the cleavable complex level.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by Associazione Italiana Ricerche sul Cancro (AIRC) and Ministero Istruzione, Università e Ricerca (MIUR).
REFERENCES
- 1.Binaschi M., Zagotto,G., Palumbo,M., Zunino,F., Farinosi,R. and Capranico,G. (1997) Irreversible and reversible topoisomerase II DNA cleavage stimulated by clerocidin: sequence specificity and structural drug determinants. Cancer Res., 57, 1710–1716. [PubMed] [Google Scholar]
- 2.Gatto B., Richter,S., Moro,S., Capranico,G. and Palumbo,M. (2001) The topoisomerase II poison clerocidin alkylates non-paired guanines of DNA: implications for irreversible stimulation of DNA cleavage. Nucleic Acids Res., 29, 4224–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawada S., Yamashita,Y., Fujii,N. and Nakano,H. (1991) Induction of a heat-stable topoisomerase II-DNA cleavable complex by nonintercalative terpenoides, terpentecin and clerocidin. Cancer Res., 51, 2922–2925. [PubMed] [Google Scholar]
- 4.Kawada S.Z., Yamashita,Y., Uosaki,Y., Gomi,K., Iwasaki,T., Takiguchi,T. and Nakano,H. (1992) UCT4B, a new antitumor antibiotic with topoisomerase II mediated DNA cleavage activity, from Streptomyces sp. J. Antibiot. Tokyo, 45, 1182–1184. [DOI] [PubMed] [Google Scholar]
- 5.Kawada S., Yamashita,Y., Ochiai,K., Ando,K., Iwasaki,T., Takiguchi,T. and Nakano,H. (1995) Terpentecin and UCT4B, new family of topoisomerase II targeting antitumor antibiotics produced by Streptomyces: producing organism, fermentation and large scale purification. J. Antibiot. Tokyo, 48, 211–216. [DOI] [PubMed] [Google Scholar]
- 6.Hansen M., Lee,S.-J., Cassady,J. and Hurley,L. (1996) Molecular details of the structure of a psorospermin-DNA covalent/intercalation complex and associated DNA sequence selectivity. J. Am. Chem. Soc., 118, 5553–5561. [Google Scholar]
- 7.Hansen M. and Hurley,L. (1995) Altromycin B threads the DNA helix interacting with both the major and the minor grooves to position itself for site-directed alkylation of guanine N7. J. Am. Chem. Soc., 117, 2421–2429. [Google Scholar]
- 8.Gopalakrishnan S., Harris,T.M. and Stone,M.P. (1990) Intercalation of aflatoxin B1 in two oligodeoxynucleotide adducts: comparative 1H NMR analysis of d(ATCAFBGAT)d(ATCGAT) and d(ATAFBGCAT)2. Biochemistry, 29, 10438–10448. [DOI] [PubMed] [Google Scholar]
- 9.Giri I., Jenkins,M.D., Schnetz-Boutaud,N.C. and Stone,M.P. (2002) Structural refinement of the 8,9-dihydro-8-(N7-guanyl)-9-hydroxy-aflatoxin B(1) adduct in a 5′-Cp(AFB)G-3′ sequence. Chem. Res. Toxicol., 15, 638–647. [DOI] [PubMed] [Google Scholar]
- 10.Jones W.R., Johnston,D.S. and Stone,M.P. (1999) Site-specific synthesis of aflatoxin B(1) adducts within an oligodeoxyribonucleotide containing the human p53 codon 249 sequence. Chem. Res. Toxicol., 12, 707–714. [DOI] [PubMed] [Google Scholar]
- 11.Nakajima N., Okazaki,T., Iwado,S., Kinoshita,T. and Haneishi,T. (1989) New diterpenoid antibiotics, Spirocardins A and B. J. Antibiot. Tokyo, 42, 1741–1748. [DOI] [PubMed] [Google Scholar]
- 12.Takao K., Aiba,Y., Ito,H. and Kobayashi,S. (1996) Synthetic study on antitumor antibiotic terpentecin: construction of the carbobicyclic decalin moiety. Chem. Lett., 11, 931–932. [Google Scholar]
- 13.Takao K. and Kobayashi,S. (1997) Synthetic study on antitumor antibiotic terpentecin: construction of the carbobicyclic decalin moiety. Tetrahedron Lett., 38, 6685–6688. [Google Scholar]
- 14.Wang J.C. (1974) Interactions between twisted DNAs and enzymes. The effects of superhelical turns. J. Mol. Biol., 87, 797–816. [DOI] [PubMed] [Google Scholar]
- 15.Maxam A.M. and Gilbert,W. (1980) Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol., 65, 499–560. [DOI] [PubMed] [Google Scholar]
- 16.Andersen N.R., Lorck,H.O. and Rasmussen,P.R. (1983) Fermentation, isolation and characterization of antibiotic PR-1350. J. Antibiot. Tokyo, 36, 753–760. [DOI] [PubMed] [Google Scholar]
- 17.Andersen N. and Rasmussen,P. (1984) The costitution of Clerocidin. A new antibiotic isolated from Oidiodendron truncatum. Tetrahedron Lett., 25, 465–468. [Google Scholar]
- 18.Andersen N., Lork,H. and Rassmussen,P. (1984) The relative and absolute configuration of clerocidin and its cometabolites. Tetrahedron Lett., 25, 469–472. [Google Scholar]
- 19.Prakash A., Tran,H.-P., Peng,C., Koyalamudi,S. and Dameron,C. (2000) Kinetics of DNA alkylation, depurination and hydrolysis of anti diol epoxide of benzo(a)pyrene and the effect of cadmium on DNA alkylation. Chem. Biol. Interact., 125, 133–150. [DOI] [PubMed] [Google Scholar]
- 20.Neagu I., Koivisto,P., Neagu,C., Kostiainen,R., Stenby,K. and Peltonen,K. (1995) Butadiene monoxide and deoxyguanosine alkylation products at the N7-position. Carcinogenesis, 16, 1809–1813. [DOI] [PubMed] [Google Scholar]
- 21.Kochetkov N.K. and Budowskii,E.I. (1972) Organic Chemistry of Nucleic Acids. Plenum, New York, NY. [Google Scholar]
- 22.Beck J.L., Colgrave,M.L., Ralph,S.F. and Sheil,M.M. (2001) Electrospray ionization mass spectrometry of oligonucleotide complexes with drugs, metals and proteins. Mass Spectrom. Rev., 20, 61–87. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Q., Ding,Z., Creighton,D.J., Ganem,B. and Fabris,D. (2002) Alkylation of nucleic acids by the antitumor agent COMC. Org. Lett., 4, 1459–1462. [DOI] [PubMed] [Google Scholar]
- 24.Comisarow M.B. and Marshall,A.G. (1974) Fourier transform ion cyclotron resonance. Chem. Phys. Lett., 25, 282–283. [Google Scholar]
- 25.Marshall A.G., Hendrickson,C.L. and Jackson,G.S. (1998) Fourier transform ion cyclotron resonance mass spectrometry: a primer. Mass Spectrom. Rev., 17, 1–35. [DOI] [PubMed] [Google Scholar]
- 26.Boogaard P.J., van Sittert,N.J., Watson,W.P. and de Kloe,K.P. (2001) A novel DNA adduct, originating from 1,2-epoxy-3,4-butanediol, is the major DNA adduct after exposure to [2,3-(14)C]-1,3-butadiene,[4-(14)C]-1,2-epoxy-3-butane. Chem. Biol. Interact., 135–136, 687–693. [DOI] [PubMed] [Google Scholar]
- 27.Koivisto P. and Peltonen,K. (2001) N7-guanine adducts of the epoxy metabolites of 1,3-butadiene in mice lung. Chem. Biol. Interact., 135–136, 363–372. [DOI] [PubMed] [Google Scholar]
- 28.Koivisto P., Kilpelainen,I., Rasanen,I., Adler,I.D., Pacchierotti,F. and Peltonen,K. (1999) Butadiene diolepoxide- and diepoxybutane-derived DNA adducts at N7-guanine: a high occurrence of diolepoxide-derived adducts in mouse lung after 1,3-butadiene exposure. Carcinogenesis, 20, 1253–1259. [DOI] [PubMed] [Google Scholar]
- 29.Hara M., Yoshida,M. and Nakamo,H. (1990) Covalent modification and single-strand scission of DNA by a new antitumor antibiotic Kapurimycin A3. Biochemistry, 29, 10449–10455. [DOI] [PubMed] [Google Scholar]