


Abstract
Cells with non-functional poly(ADP-ribose) polymerase (PARP-1) show increased levels of sister chromatid exchange, suggesting a hyper recombination phenotype in these cells. To further investigate the involvement of PARP-1 in homologous recombination (HR) we investigated how PARP-1 affects nuclear HR sites (Rad51 foci) and HR repair of an endonuclease-induced DNA double-strand break (DSB). Several proteins involved in HR localise to Rad51 foci and HR-deficient cells fail to form Rad51 foci in response to DNA damage. Here, we show that PARP-1 mainly does not localise to Rad51 foci and that Rad51 foci form in PARP-1–/– cells, also in response to hydroxyurea. Furthermore, we show that homology directed repair following induction of a site-specific DSB is normal in PARP-1-inhibited cells. In contrast, inhibition or loss of PARP-1 increases spontaneous Rad51 foci formation, confirming a hyper recombination phenotype in these cells. Our data suggest that PARP-1 controls DNA damage recognised by HR and that it is not involved in executing HR as such.
INTRODUCTION
Poly(ADP-ribose) polymerase (PARP-1) is an abundant nuclear protein in mammalian cells that catalyses the formation of poly(ADP-ribose) (PAR) polymers using NAD+ as substrate. Upon DNA damage, PARP-1 binds rapidly to a DNA single-strand break and catalyses the addition of negatively charged PAR chains to itself (automodification) and other proteins [reviewed in (1,2)]. The binding of PARP-1 to DNA single-strand breaks (SSBs) is believed to protect DNA lesions from further processing until PARP-1 is dissociated from the break by the accumulated negative charge resulting from PAR polymers (3,4).
Although PARP-1 has been implicated in several nuclear processes, such as modulation of chromatin structure, DNA replication, DNA repair and transcription, PARP-1 knockout mice develop normally (5). Cells isolated from these mice exhibit a hyper recombination phenotype and genetic instability in the form of increased levels of sister chromatid exchange (SCE), micronuclei and tetraploidy (6–8). Genetic instability may also occur in these PARP-1 knockout mice through telomere shortening, increased frequency of chromosome fusion and aneuploidy (9), although all of these results could not be repeated in another set of PARP-1 knockout mice (10). In the former mice knockout, PARP-1 null mutation also rescued impaired V(D)J recombination in SCID mice (11). Also, overexpression of PARP-1 has been shown to suppress SCE induced by DNA damaging treatment, indicating that PARP-1 activity influences recombination (12).
Overall these results support the view suggested by Lindahl et al. that PARP-1 has a protective role against recombination (4). They proposed that binding of PARP-1 to a DNA break prevents the recombination machinery from recognising and processing DNA lesions or, alternatively, that the negative charges accumulated following poly(ADP)-ribosylation repel adjacent recombinogenic DNA sequences. Only the latter model is consistent with inhibition of PARP-1 itself and expression of a dominant negative mutant PARP-1, inducing SCE, gene amplification and homologous recombination (HR) (13–17). Although several lines of evidence show that loss of PARP-1 activity causes a hyper recombination phenotype, it is not clear whether PARP-1 affects only the number of lesions that trigger HR or if PARP-1 is involved in carrying out HR per se.
Proteins involved in HR are found in nuclear foci in a small portion of normally growing cells in S phase of the cell cycle (18–23). These nuclear foci have been shown to be located in post-replicative chromatin in untreated cells in S-phase of the cell cycle (24), which may reflect the role of HR at replication forks. RAD51 is a stable component of these nuclear foci and is commonly used to visualise them (25). The accumulation of RAD51 foci after DNA damage is generally thought to reflect the assembly of HR repair complexes to repair DNA lesions (26).
The results presented here show that PARP-1 controls the number of nuclear HR sites (Rad51 foci) and that a HR event, using gene conversion, is unaffected by inhibition of PARP-1.
MATERIALS AND METHODS
Cell culture
The SW480, DLD-1 and HCT116 cell lines originated from the American Type Culture Collection (Manassas, VA). The A11 and A19 cell lines were a generous gift from Zhao-Qi Wang (27). All cell lines in this study were grown in Dulbecco’s modified Eagle’s Medium (DMEM) with 10% fetal bovine serum and penicillin (100 U/ml) and streptomycin sulphate (100 µg/ml) at 37°C under an atmosphere containing 5% CO2.
Immunofluorescence
Cells were plated onto coverslips and grown for 4 h before treatments or overnight if treatments were not used. The medium was removed, the coverslips rinsed once in PBS (37°C) and fixed in 3% paraformaldehyde in PBS-T (PBS containing 0.1% Triton X-100) for 20 min. The coverslips were rinsed once in PBS-T prior to incubation with primary antibody for 16 h at 4°C. The primary antibodies used in this study were: mouse monoclonal anti PARP-1 (F-2, Santa Cruz) at a dilution of 1:1000, rabbit polyclonal anti-PARP-1 (H-250, Santa Cruz) at a dilution of 1:1000, rabbit polyclonal anti-Rad51 (28) at a dilution of 1:1000 and rabbit polyclonal anti-Rad51 (H-92, Santa Cruz) at a dilution of 1:1000. The coverslips were rinsed 4 × 15 min in PBS-T followed by a 1-h incubation at room temperature with the appropriate secondary antibody and then rinsed 4 × 15 min in PBS-T. The secondary antibodies used in this study were Cy-3-conjugated goat anti-rabbit IgG antibody (Zymed) at a concentration of 1:500, Alexa 488 goat anti-mouse IgG antibody (Molecular Probes) at a concentration of 1:500. Antibodies were diluted in PBS containing 3% bovine serum albumin. DNA was stained with 1 µg/ml To Pro (Molecular Probes). Coverslips were mounted with SlowFade Antifade Kit (Molecular Probes).
Images were obtained with a Zeiss LSM 510 inverted confocal microscope using planapochromat 63×/NA 1.4 oil immersion objective and excitation wavelengths 546 and 630 nm. Through focus maximum projection images were acquired from optical sections 0.50 µm apart and with a section thickness of 1.0 µm. Images were processed using Adobe PhotoShop (Abacus Inc.).
The frequencies of cells containing Rad51 foci were determined in at least two separate experiments. At least 300 nuclei were counted on each slide. Nuclei containing more than 10 foci were classified as positive.
Recombination assay
The SCneo recombination substrate and the pCMV3nls-I-SceI expression vector were kind gifts from Dr Maria Jasin (29). SPD8 cells (7.5 × 106) were electroporated (voltage: 2.5 kV/cm; capacitance setting: 25 µF) with 15 µg uncut SCneo substrate and plated in a non-selective medium. Hygromycin (final concentration 0.1 mM) was added to the medium 48 h later and hygR colonies were isolated and expanded. Southern blotting was performed on genomic DNA (10 µg) isolated from each clone, the 1.1 kb XhoI–BamHI fragment radiolabelled with [α-32P]dCTP was used as probe and detection was carried out by autoradiography. For the recombination assay 1.5 × 106 S8SN.11 cells were inoculated overnight before being transiently transfected for 5 h with the pCMV3nls-I-SceI expression vector (10 ng) using lipofectamine according to manufacturer’s protocol (final concentration 10 µg/ml; Lipofectamine2000™, Invitrogen). Treatment with 1,5-dihydroxyisoquinoline (ISQ, Sigma) was performed during transfection or 24 h after transfection. The cells were trypsinised and counted 24 h after transfection. The recombination frequency was determined by seeding 1 × 105 cells (after I-SceI induction) or 1 × 106 cells (for control) per dish (Ø 100 mm) in media containing 1 mg/ml G418. The cloning efficiency was determined by plating two dishes with 500 cells each. After 7 days, cells on the cloning plates were fixed with methylene blue [4 g methylene blue (Merck)/l methanol] and the colonies scored. In the case of the selection plates, fixation and scoring were performed 4 days later. Colonies consisting of more than 50 cells were subsequently counted. All experiments were repeated independently three times.
RESULTS
PARP-1 does not principally localise in Rad51 foci
Several proteins with a role in HR have been found to localise in Rad51 foci (18–23,25). Since PARP-1-deficient mice exhibit genetic instability and since PARP-1 cooperates with DNA-PKcs to minimise genomic damage caused by DNA strand breaks (11), PARP-1 might co-operate in executing HR repair and possibly co-localise in Rad51 foci. To test this hypothesis we localised PARP-1 and Rad51 in the three human tumour cell lines DLD-1, SKUT-1 and SW480 with and without treatments with hydroxyurea. PARP-1 localised throughout the nucleus in the SW480 cell line. PARP-1 also stained in the nucleus in the DLD-1 and SKUT-1 cell lines, but concentrated in nucleoli, which also has been reported for the MDBK, HeLa and CHO cell lines (30). In general, PARP-1 did not co-localise in Rad51 foci in any of the cell lines used (Fig. 1). Two different polyclonal antibodies towards PARP-1 failed to co-localise in Rad51 foci (data not shown).
Figure 1.

Localisation of PARP-1 and Rad51 in the human cell lines SKUT-1, DLD-1 and SW480. In merged images, DNA is displayed in blue, anti-Rad51 in red and anti-PARP-1 in green.
PARP-1 is not required for assembly of spontaneous or induced Rad51 foci
HR is deregulated in PARP-1 deficient cells as well as in cells deficient for XRCC2, XRCC3 or BRCA2 (29,31,32). Cells deficient in the latter genes fail to form spontaneous or DNA damage-induced Rad51 foci (32,33). Thus, although PARP-1 mainly failed to localise to Rad51 foci, PARP-1 activity might still be a requirement for Rad51 foci to form. To test this, we used a set of immortalised mouse embryonic fibroblasts isolated from two littermates with either a functional PARP-1 (A19) or with a truncation in exon 2 in both alleles of PARP-1 (A11) (27). Rad51 foci formation was visualised in both PARP-1 proficient and deficient cells (Fig. 2). An increased number of Rad51 foci were also observed after hydroxyurea treatments in both cell lines, in agreement with previous reports that hydroxyurea induces Rad51 foci (34).
Figure 2.
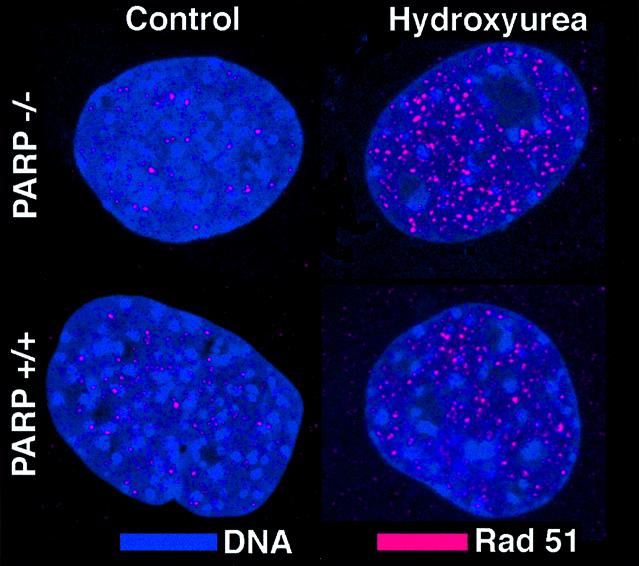
DNA (blue) and Rad51 foci (red) are visualised in A11 (PARP–/–) or A19 (PARP+/+) cells with or without a 24-h hydroxyurea treatment (0.2 mM).
Homology-directed repair of a single DNA double-strand break is normal when inhibiting PARP-1
HR is involved in the repair of double-strand breaks (DSBs) in mammalian cells. To test if inhibition of PARP-1 affects HR repair of a DSB, we used an assay developed by Jasin and coworkers (29) that measures the homology-based recombination between two defective G418 genes following introduction of a DNA DSB by transiently expressing the restriction endonuclease I-SceI. First, we transfected a V79 Chinese hamster cell line (SPD8) with the recombination reporter SCneo (Fig. 3A) and isolated hygromycin-resistant clones. DNA isolated from the hygromycin-resistant S8SN.11 clone was analysed by Southern blotting. Cleavage with HindIII and XhoI produced a 4 kb fragment indicating that the full construct was integrated. Additional cleavage with NcoI shows that the internal structure of the construct was preserved. Cleavage with HindIII or XhoI alone produced a single band, showing that the SCneo reporter was integrated as a single copy in S8SN.11 cells (Fig. 3C).
Figure 3.

Recombination substrate integrated in V79(SPD8) cells. (A) Structure of SCneo (29). (B) Predicted HR products resulting in G418 resistance. (C) Southern blot on DNA isolated from the SCneo transfected cell line S8SN.11 and probed with the S2neo fragment.
Following transfection of the I-SceI expression vector pCMV3xnlsI-SceI, S8SN.11 cells containing an intact single copy of SCneo produced G418R colonies at a frequency of 1.4–4.1 × 10–3. S8SN.11 cells spontaneously produced G418R colonies at a frequency of 0.3–1.7 × 10–5 (Fig. 4), which is similar to a result previously obtained in hamster V79 hamster cells (29).
Figure 4.

Recombination frequency in the S8SN.11 cell line with or without inhibition of PARP-1 using ISQ. A DSB was induced in the SCneo substrate by transient transfection with the pCMV3xnlsI-SceI vector.
To test whether PARP-1 influences the repair of an endonuclease-induced DSB by HR, we co-treated S8SN.11 at the time of transfection with the PARP-1 inhibitor ISQ. Although PARP-1 was inhibited in S8SN.11 cells they were still proficient in forming G418R colonies by HR repair (Fig. 4). Given that HR repair is probably a slow event (34), we also post-treated cells in the presence of ISQ for 24 h. This treatment did not affect the HR repair of a DSB in the S8SN.11 cell line (Fig. 4). Unfortunately, we were unable to confirm these results in PARP-1–/– cells, since they are resistant to G418 as a result of the PARP-1 knockout.
Loss of PARP-1 activity induces Rad51 foci formation
PARP-1 deficiency confers genetic instability (5) and inhibition of the enzymatic activity of the protein has been shown in certain systems to induce HR (14–17). We wanted to investigate whether loss or inhibition of PARP-1 provides a lesion or another substrate that would provoke a repair response by proteins involved in HR. To investigate this, we examined the formation of Rad51 foci in A19 (PARP-1+/+) and A11 (PARP-1–/–) cells treated with 3-AB, ISQ or 8-hydroxy-2-methylquinazolinone (NU1025). In PARP-1-deficient A11 cells, Rad51 foci were 30-fold more abundant than in the corresponding PARP-1 proficient A19 cells. Furthermore, we found an increased number of cells with Rad51 foci following treatments with 3-AB, ISQ and NU1025 only in the A19 cells (Fig. 5).
Figure 5.

Percentage cells containing more than 10 Rad51 foci in A11 (PARP–/–) or A19 (PARP+/+) cells following a 24-h treatment with 3-AB (2 mM), ISQ (0.6 mM) or NU1025 (0.1 mM). At least 300 nuclei were counted for each treatment and experiment. Error bars designate standard error from at least three experiments.
DISCUSSION
Although PARP-1–/– cells show a hyper recombination phenotype (6–8), the underlying mechanisms have not been revealed. In hypothesis, either PARP-1 could control the lesions that are recognised by HR or PARP-1 could participate in carrying out HR. Several proteins with suggested function in executing HR repair (e.g. BLM, BRCA1, BRCA2, RAD52, RAD54, RPA, WRN) co-localise with Rad51 at sites of HR repair (18–23,25). Since we observed that PARP-1 mainly does not localise to Rad51 foci (Fig. 1) our result suggests that PARP-1 does not execute HR as such.
Cells deficient in a protein with a role in carrying out HR often fail to form RAD51 foci, as in the case of the Rad51 paralogs (33). Since we found that PARP-1–/– cells were proficient for both spontaneous and hydroxyurea-induced Rad51 foci formation (Fig. 2) our results show that PARP-1 is not required for either spontaneous or induced HR sites to form.
Cells deficient in HR proteins (e.g. XRCC2 or XRCC3) fail to use homology-directed repair of a DSB (29,31). Using the same system as in these reports, we found that inhibition of PARP-1 had no effect on HR repair of a DSB (Fig. 4). Altogether, these results suggest that PARP-1 does not carry out HR repair of a DSB and favour the hypothesis that PARP-1 has a controlling role of lesions recognised by HR.
In support for this controlling hypothesis, we found that inhibition or loss of PARP-1 induces Rad51 foci formation (Fig. 5). This is in agreement with previous studies that indicate that inhibition of PARP-1 induces HR (14–17) and that loss of PARP-1 increases levels of SCE and micronuclei (6–8). Furthermore, we found that 3-AB, ISQ or NU1025 increased the number of cells with Rad51 foci only in those cells that have a functional PARP-1. These results show that the induction of Rad51 foci following 3-AB, ISQ or NU1025 treatment is a specific consequence of inhibition of PARP-1.
Our results showing that HR repair of a DSB is unaffected by inhibition of PARP-1 may contrast with earlier findings that PARP-1 increases spontaneous SCE events (6–8). Also, these results may diverge from the result that SCE induced by an alkylating agent is repressed by overexpression of PARP-1 (12) and that SCE is more potently induced in PARP-1–/– cells (6) and in cells expressing a DNA binding domain of PARP-1 (16). However, the HR assay used here exclusively involves a gene conversion type of HR repair mechanism and SCE has never been found in connection with this HR repair event (35). In contrast, SCE has been shown to be involved in the HR repair of SSBs converted into DSBs at replication forks (36).
We speculate that the reason why PARP-1 activity affects SCE, but not gene conversion can be explained by elevated levels of SSBs that increase the amount of SCE but not the amount of gene conversion events. PARP-1 has an important role in SSB repair and base excision repair (BER) (37). Loss of PARP-1 activity may increase the amount of endogenous or induced SSBs. These may be converted into DSBs at replication forks, which may trigger SCE (36), but not the gene conversion event investigated here.
This hypothesis is supported by the fact that overexpression of PARP-1 decreases the amount of alkylating-induced SCE (12). In this case overexpression of PARP-1 may increase the repair rate of alkylating damage and, thus, fewer SSBs would be converted into DSBs at replication forks, resulting in less induced SCE. This would also explain why SCE is more potently induced in PARP-1–/– cells (6) and in cells expressing a DNA binding domain of PARP-1 (16), since absence of PARP-1 activity would increase the amount of SSBs that are converted into DSBs and trigger SCE.
The idea that a defect in BER may trigger SCE is not new. Thompson et al. reported in 1982 that the EM9 cell line, with a BER defect, has an increased level of SCE (38), which was later found to be due to a deficient XRCC1 gene (39). These data further support our hypothesis that the increased level of HR and SCE found in PARP-1-deficient cells is related to a general defect in BER, rather than that PARP-1 is involved in catalysing HR per se. Future experiments should be aimed to test this hypothesis directly.
Acknowledgments
ACKNOWLEDGEMENTS
We wish to thank Dr Nicola Curtin for providing NU1025, Dr Maria Jasin for providing the SCneo construct, Drs Zhao-Qi Wang and Deborah Barnes for providing A11 and A19 cell lines, Drs Fiona Benson and Stephen West for anti-Rad51 antibody, and Drs Mark Meuth and Helen Bryant for critical reading of this manuscript. The Swedish Cancer Society, the Swedish National Board for Laboratory Animals and Yorkshire Cancer Research provided financial support for this investigation.
REFERENCES
- 1.d'Amours D., Desnoyers,S., D’Silva,I. and Poirier,G.G. (1999) Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J., 342, 249–268. [PMC free article] [PubMed] [Google Scholar]
- 2.Herceg Z. and Wang,Z.Q. (2001) Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat. Res., 477, 97–110. [DOI] [PubMed] [Google Scholar]
- 3.Satoh M.S. and Lindahl,T. (1992) Role of poly(ADP-ribose) formation in DNA repair. Nature, 356, 356–358. [DOI] [PubMed] [Google Scholar]
- 4.Lindahl T., Satoh,M.S., Poirier,G.G. and Klungland,A. (1995) Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem. Sci., 20, 405–411. [DOI] [PubMed] [Google Scholar]
- 5.Shall S. and de Murcia,G. (2000) Poly(ADP-ribose) polymerase-1: what have we learned from the deficient mouse model? Mutat. Res., 460, 1–15. [DOI] [PubMed] [Google Scholar]
- 6.Ménissier de Murcia J., Niedergang,C., Trucco,C., Ricoul,M., Dutrillaux,B., Mark,M., Oliver,F.J., Masson,M., Dierich,A., LeMeur,M., Walztinger,C., Chambon,P. and de Murcia,G. (1997) Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl Acad. Sci. USA, 94, 7303–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Z.Q., Stingl,L., Morrison,C., Jantsch,M., Los,M., Schulze-Osthoff,K. and Wagner,E.F. (1997) PARP is important for genomic stability but dispensable in apoptosis. Genes Dev., 11, 2347–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simbulan-Rosenthal C.M., Haddad,B.R., Rosenthal,D.S., Weaver,Z., Coleman,A., Luo,R., Young,H.M., Wang,Z.Q., Ried,T. and Smulson,M.E. (1999) Chromosomal aberrations in PARP(-/-) mice: genome stabilization in immortalized cells by reintroduction of poly(ADP-ribose) polymerase cDNA. Proc. Natl Acad. Sci. USA, 96, 13191–13196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.d'Adda di Fagagna F., Hande,M.P., Tong,W.M., Lansdorp,P.M., Wang,Z.Q. and Jackson,S.P. (1999) Functions of poly(ADP-ribose) polymerase in controlling telomere length and chromosomal stability. Nature Genet., 23, 76–80. [DOI] [PubMed] [Google Scholar]
- 10.Samper E., Goytisolo,F.A., Menissier-de-Murcia,J., Gonzalez-Suarez,E., Cigudosa,J.C., de Murcia,G. and Blasco,M.A. (2001) Normal telomere length and chromosomal end capping in poly(ADP-ribose) polymerase-deficient mice and primary cells despite increased chromosomal instability. J. Cell Biol., 154, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morrison C., Smith,G.C., Stingl,L., Jackson,S.P., Wagner,E.F. and Wang,Z.Q. (1997) Genetic interaction between PARP and DNA-PK in V(D)J recombination and tumorigenesis. Nature Genet., 17, 479–482. [DOI] [PubMed] [Google Scholar]
- 12.Meyer R., Muller,M., Beneke,S., Kupper,J.H. and Burkle,A. (2000) Negative regulation of alkylation-induced sister-chromatid exchange by poly(ADP-ribose) polymerase-1 activity. Int. J. Cancer, 88, 351–355. [DOI] [PubMed] [Google Scholar]
- 13.Kupper J.H., Muller,M. and Burkle,A. (1996) Trans-dominant inhibition of poly(ADP-ribosyl)ation potentiates carcinogen induced gene amplification in SV40-transformed Chinese hamster cells. Cancer Res., 56, 2715–2717. [PubMed] [Google Scholar]
- 14.Magnusson J. and Ramel,C. (1990) Inhibitor of poly(ADP-ribose)transferase potentiates the recombinogenic but not the mutagenic action of alkylating agents in somatic cells in vivo in Drosophila melanogaster. Mutagenesis, 5, 511–514. [DOI] [PubMed] [Google Scholar]
- 15.Waldman A.S. and Waldman,B.C. (1991) Stimulation of intrachromosomal homologous recombination in mammalian cells by an inhibitor of poly(ADP-ribosylation). Nucleic Acids Res., 19, 5943–5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schreiber V., Hunting,D., Trucco,C., Gowans,B., Grunwald,D., de Murcia,G. and de Murcia,J.M. (1995) A dominant-negative mutant of human poly(ADP-ribose) polymerase affects cell recovery, apoptosis and sister chromatid exchange following DNA damage. Proc. Natl Acad. Sci. USA, 92, 4753–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Semionov A., Cournoyer,D. and Chow,T.Y. (1999) Inhibition of poly(ADP-ribose)polymerase stimulates extrachromosomal homologous recombination in mouse Ltk-fibroblasts. Nucleic Acids Res., 27, 4526–4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tashiro S., Kotomura,N., Shinohara,A., Tanaka,K., Ueda,K. and Kamada,N. (1996) S phase specific formation of the human Rad51 protein nuclear foci in lymphocytes. Oncogene, 12, 2165–2170. [PubMed] [Google Scholar]
- 19.Scully R., Chen,J., Ochs,R.L., Keegan,K., Hoekstra,M., Feunteun,J. and Livingston,D.M. (1997) Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell, 90, 425–435. [DOI] [PubMed] [Google Scholar]
- 20.Mizuta R., LaSalle,J.M., Cheng,H.L., Shinohara,A., Ogawa,H., Copeland,N., Jenkins,N.A., Lalande,M. and Alt,F.W. (1997) RAB22 and RAB163/mouse BRCA2: proteins that specifically interact with the RAD51 protein. Proc. Natl Acad. Sci. USA, 94, 6927–6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Golub E.I., Gupta,R.C., Haaf,T., Wold,M.S. and Radding,C.M. (1998) Interaction of human Rad51 recombination protein with single-stranded DNA binding protein, RPA. Nucleic Acids Res., 26, 5388–5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu L., Davies,S.L., Levitt,N.C. and Hickson,I.D. (2001) Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J. Biol. Chem., 276, 19375–19381. [DOI] [PubMed] [Google Scholar]
- 23.Sakamoto S., Nishikawa,K., Heo,S.J., Goto,M., Furuichi,Y. and Shimamoto,A. (2001) Werner helicase relocates into nuclear foci in response to DNA damaging agents and co-localizes with RPA and Rad51. Genes Cells, 6, 421–430. [DOI] [PubMed] [Google Scholar]
- 24.Tashiro S., Walter,J., Shinohara,A., Kamada,N. and Cremer,T. (2000) Rad51 accumulation at sites of DNA damage and in postreplicative chromatin. J. Cell Biol., 150, 283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Essers J., Houtsmuller,A.B., van Veelen,L., Paulusma,C., Nigg,A.L., Pastink,A., Vermeulen,W., Hoeijmakers,J.H. and Kanaar,R. (2002) Nuclear dynamics of RAD52 group homologous recombination proteins in response to DNA damage. EMBO J., 21, 2030–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haaf T., Golub,E.I., Reddy,G., Radding,C.M. and Ward,D.C. (1995) Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc. Natl Acad. Sci. USA, 92, 2298–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z.Q., Auer,B., Stingl,L., Berghammer,H., Haidacher,D., Schweiger,M. and Wagner,E.F. (1995) Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev., 9, 509–520. [DOI] [PubMed] [Google Scholar]
- 28.Benson F.E., Stasiak,A. and West,S.C. (1994) Purification and characterisation of the human Rad51 protein, an analogue of E.coli RecA. EMBO J., 13, 5764–5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson R.D., Liu,N. and Jasin,M. (1999) Mammalian XRCC2 promotes the repair of double-strand breaks by homologous recombination. Nature, 401, 397–399. [DOI] [PubMed] [Google Scholar]
- 30.Desnoyers S., Kaufmann,S.H. and Poirier,G.G. (1996) Alteration of the nucleolar localization of poly(ADP-ribose) polymerase upon treatment with transcription inhibitors. Exp. Cell Res., 227, 146–153. [DOI] [PubMed] [Google Scholar]
- 31.Pierce A.J., Johnson,R.D., Thompson,L.H. and Jasin,M. (1999) XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev., 13, 2633–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kraakman van der Zwet M., Overkamp,W.J., van Lange,R.E., Essers,J., van Duijn-Goedhart,A., Wiggers,I., Swaminathan,S., van Buul,P.P., Errami,A., Tan,R.T., Jaspers,N.G., Sharan,S.K., Kanaar,R. and Zdzienicka,M.Z. (2002) Brca2 (XRCC11) deficiency results in radioresistant DNA synthesis and a higher frequency of spontaneous deletions. Mol. Cell. Biol., 22, 669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takata M., Sasaki,M.S., Tachiiri,S., Fukushima,T., Sonoda,E., Schild,D., Thompson,L.H. and Takeda,S. (2001) Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol. Cell. Biol., 21, 2858–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lundin C., Erixon,K., Arnaudeau,C., Schultz,N., Jenssen,D., Meuth,M. and Helleday,T. (2002) Different roles for non-homologous end joining and homologous recombination following replication arrest in mammalian cells. Mol. Cell. Biol., 22, 5869–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson R.D. and Jasin,M. (2000). Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J., 19, 3398–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arnaudeau C., Lundin,C. and Helleday,T. (2001) DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism. J. Mol. Biol., 307, 1235–1245. [DOI] [PubMed] [Google Scholar]
- 37.Dantzer F., Schreiber,V., Niedergang,C., Trucco,C., Flatter,E., De La Rubia,G., Oliver,J., Rolli,V., Menissier-de Murcia,J. and de Murcia,G. (1999) Involvement of poly(ADP-ribose) polymerase in base excision repair. Biochimie, 81, 69–75. [DOI] [PubMed] [Google Scholar]
- 38.Thompson L.H., Brookman,K.W., Dillehay,L.E., Carrano,A.V., Mazrimas,J.A., Mooney,C.L. and Minkler,J.L. (1982) A CHO-cell strain having hypersensitivity to mutagens, a defect in DNA strand-break repair and an extraordinary baseline frequency of sister-chromatid exchange. Mutat. Res., 195, 427–440. [DOI] [PubMed] [Google Scholar]
- 39.Caldecott K.W., Tucker,J.D. and Thompson,L.H. (1992) Construction of human XRCC1 minigenes that fully correct the CHO DNA repair mutant EM9. Nucleic Acids Res., 20, 4575–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]