


Abstract
RNA interference (RNAi) is mediated by small interfering (si) RNAs that target and degrade mRNA in a sequence-specific manner. Cellular expression of siRNA can be achieved by the use of expression cassettes driven by RNA polymerase III (pol III) promoters. Here, we demonstrate that a modified tRNAmet-derived (MTD) promoter effectively drives the cellular expression of HIV-1-specific siRNA. We observed up to 56% greater inhibition of virus production when the MTD promoter was used to drive the expression of short hairpin (sh) RNA targeting the HIV-1 transactivator protein tat compared to cassettes containing other pol III promoters such as H1, U6+1 and U6+27. We conclude that the MTD promoter is ideally suited to drive intracellular expression of HIV-1 specific siRNA and may serve as an important component of future RNAi vector delivery systems.
INTRODUCTION
Recent publications have described vector systems using RNA polymerase III (Pol III) promoters to express functional siRNA for silencing a variety of target genes (1–4). Pol III transcripts are abundant in human cells, thus forming the rationale for using Pol III promoters to express high levels of RNA molecules such as ribozymes, antisense compounds and siRNA (2,5). To date, two different Pol III promoters, the human U6 small nuclear (sn) RNA and the human H1 promoter have been widely used in siRNA expression vectors (1,6). U6 snRNA plays a central role in processing premature RNA species (7). The H1 transcript is a component of human nuclear RNase P, an enzyme that cleaves tRNA precursors to produce mature 5′-termini (8). Pol III promoters are classified into three different categories (Type I, II and III) based upon the composition of the promoter elements and their position relative to the transcriptional start site (9). Type III promoters such as U6 and H1 have a compact and relatively simple organization and are located upstream of the transcribed region. Transcription is terminated within a stretch of four to six uridines yielding small RNA transcripts with a defined 3′ overhang. This structural feature was shown to be critical for the function of siRNA synthesized in vitro (10).
Type II promoters, such as tRNA promoters, have been widely used to express ribozymes and antisense RNA (11–15). Previous studies have shown that the tRNA promoter is more effective in driving the expression of ribozymes than other Pol II and Pol III promoters (16). Optimization of the met tRNA transcription unit increased the expression of transcripts by >100-fold (5,15). In this study, we compared the utility of type II and type III RNA Pol III promoters in mediating HIV-1 specific RNAi. We incorporated a modified human tRNAmet-derived (MTD) promoter into a short hairpin RNA (shRNA) expression cassette. We designed siRNA that targeted exon-1 of the HIV-1 tat gene which encodes a transactivator protein critical for viral replication (17). The expression levels and antiviral efficacy of tat shRNA driven by the MTD promoter were determined and compared to those obtained using other promoters including H1, U6, U6+1 and U6+27. Our experiments demonstrate that the MTD promoter was most effective in mediating HIV-1 specific RNAi.
MATERIALS AND METHODS
siRNA sequences
siRNA with the following sense and antisense sequences were used: tat (sense), 5′-CUG CUU GUA CCA AUU GCU AdTdT-3′; tat (antisense), 5′-UAG CAA UUG GUA CAA GCA GdTdT-3′. Synthetic RNA was purchased from Dharmacon Research (Lafayette, CO, USA) and was deprotected and annealed according to the manufacturer’s instructions.
Construction of shRNA expression cassettes
The human H1 promoter and different versions of the human U6 promoter (U6, U6+1, U6+27) were PCR-amplified from genomic DNA isolated from HeLa cells. Primers used for the generation of the H1 promoter were H1S, 5′-CCA TGG AAT TCG AAC GCT GA-3′, and H1AS, 5′-GGG AAA GAG TGG TCT CAT AC-3′ and those used for the various U6 promoters were: U6S, 5′-AAG GTC GGG CAG GAA GA-3′, U6AS, 5′-GGT GTT TCG TCC TTT CCA C-3′, U6+1AS, 5′-CGG TGT TTC GTC CTT TCC AC-3′ and U6+27AS, 5′-TAG TAT ATG TGC TGC CGA AGC GAG CAC GGT GTT TCG TCC T-3′, respectively. The MTD promoter was generated by overlap extension PCR (18) using the following primers: TS, 5′-TGC TGG GCC CAT AAC CCA GAG GTC GAT GGA TCG AAA CCT GCG CCA CTC CTG ATG AGC CTC TAG ACA CTC TCG AGG GCG AT-3′, TAS, 5′-ATC GCC CTC GAG AGT GTC TAG AGG CTC ATC AGG AGT GGC GCA GGT TTC GAT C CA TCG ACC TCT GGG TTA TGG GCC CAG CA-3′, Not-TS, 5′-GCG GCC GCG GCA GAA CAG TCG AGT GG C GCA GCG GAA GCG TGC TGG GCC CAT AAC-3′, and Not-TAS, 5′-GCG GCC GCA TCG ATG TTA GGA GAT CTA AAC AGA ACA GTA TCG CCC TCG AGA GTG TCT AG-3′.
To generate the H1 and U6 promoter cassettes with appropriate restriction sites, second round PCRs were performed on the respective first round PCR products. For the H1 promoter cassette, the primers were NH1, 5′-GTG ACA GCG GCC GCC CAT GGA ATT CGA ACG CT-3′ and H1MCS, 5′-CAA CAG GCG GCC GCA TCG ATG TTA GGA GAT CTA AAA ACT CGA GAT TTC ATC TAG AGG GAA AGA GTG GTC TCA-3′. For the U6 promoter cassette, PCR was performed with primers NU6, 5′-GTG ACA GCG GCC GCA AGG TCG GGC AGG AAG A-3′ and U6MCS, 5′-CAA CAG GCG GCC GCA TCG ATG TTA GGA GAT CTA AAA ACT CGA GAT TTC ATC TAG AGG TGT TTC GTC CTT TCC-3′.
PCR products containing each promoter were cloned into the NotI site of the shuttle vector pCMV-MCS (Stratagene, Valencia, CA, USA) yielding the constructs pCMV-MTD-MCS, pCMV-H1-MCS, pCMV-U6-MCS, pCMV-U6+1-MCS and pCMV-U6+27-MCS. A neomycin resistance cassette, including the SV40 early promoter and the SV40 polyadenylation signal, was inserted into the BglII and ClaI sites of each of the pCMV-MCS constructs. The resulting siRNA expression cassettes were subcloned into the NotI site of pAAV-MCS (Stratagene).
A CMV promoter-driven shRNA expression cassette was generated by subcloning the CMV promoter, including the multiple cloning site, polyadenylation signal and neomycin selection cassette from pcDNA3.1 (Invitrogen, Carlsbad, CA, USA), into Mlu and Bst 1107 sites previously introduced into pAAV-MCS. Two complementary oligonucleotides containing 21-nt tat sequences 5′-CTA GAA CTG CTT GTA CCA ATT GCT ATT AGG ATC AAT AGC AAT TGG TAC AAG CAG TTTTTC-3′ and 5′-TCG AGA AAA ACT GCT TGT ACC AAT T GC TAT TGA TCC TAA TAG CAA TTG GTA CAA GCA GTT CTA GT-3′ were annealed and cloned into the XbaI and XhoI sites of pAAV-MTD-MCS-Neo, pAAV-H1-MCS-Neo, pAAV-U6-MCS-Neo, pAAV-U6+1-MCS-Neo, pAAV-U6+27-MCS-Neo and pAAV-pCMV-MCS-Neo. In addition, a control vector, pAAV-MTD-luc-Neo, was generated which synthesizes shRNA targeting the luciferase gene. All Pol III shRNA expression cassettes included a hexaloop (AGGATC) separating tat sense and antisense sequences followed by a 5T termination signal (Fig. 1).
Figure 1.

Schematic representation of the various shRNA expression units. Unique restriction sites in the AAV-DNA vector allowed the exchange of different promoters. Tat siRNA is expressed in the form of a hairpin transcript consisting of a 21-nt sense and 21-nt antisense sequence separated by a hexaloop. The five thymidine stretches serve as a RNA pol III termination signal whereas the pABGH polyadenylation sequence terminates RNA pol II transcription. Additional components of the recombinant AAV-DNA vector include inverted terminal repeats (ITRs) and a neomycin selection cassette.
Cell culture and transient transfections
293T cells were trypsinized and plated at 4 × 105 cells per well in six-well plates 24 h prior to transfection in DMEM containing 10% fetal bovine serum (FBS). Either synthetic RNA (200 pmol) or DNA (1 µg) from the various shRNA expression cassettes was co-transfected with 1 µg HIV-1NL4.3 using 6 µl Lipofectamine 2000 (Invitrogen) per reaction. Cell-free supernatant was collected 2 days later and HIV-1 p24 antigen levels were quantified by ELISA (Beckman-Coulter, Fullerton, CA, USA) according to the manufacturer’s instructions.
Real-time RT–PCR
Total cellular RNA was extracted using Trizol reagent (Invitrogen) and treated with RQ1 DNase (Promega, Madison, WI) according to the manufacturer’s protocol. DNase-treated RNA (1 µg) was added to a reverse transcription (RT) reaction containing Powerscript Reverse Transcriptase (Clontech, Palo Alto, CA), 1 mM of each dNTP, 1× First-Strand buffer (Clontech), 200 ng random hexamers (Promega), 10 U RNasin (Promega). Reverse transcription was performed at 42°C for 1 h followed by heat-inactivation of the RT enzyme at 70°C for 15 min. cDNA (2 µl) was added to 48 µl of PCR mix containing 1× Titanium Taq PCR buffer (Clontech), 20 pmol of sense primer TS, 5′-CAT CCA GGA AGT CAG CCT A-3′, and antisense primer TAS, 5′-TTC CTG CCA TAG GAG ATG C-3′, 1 mM dNTPs, SYBR Green I (1:75 000), 10 nM fluorescein, and 1× Titanium Taq polymerase (Clontech). For RNA normalization, a GAPDH-PCR was performed with the same PCR reagents except for the primers which were G1, 5′-GAT TTC TCC CCC TTC TGC TGA TG-3′ and G2, 5′-CCT TGG CTG GGG GTG CTA A-3′. An external standard curve was created using RNA transcripts of tat exon-1 which were generated with the MEGAscript in vitro transcription kit (Ambion, Austin, TX). Real-time PCR was carried out in an iCycler (Bio-Rad, Hercules, CA) using the following thermal cycling profile: 95°C 1 min, followed by 35 cycles of amplification (95°C 15 s, 61°C 30 s, 68°C 30 s).
Northern blot
Forty-eight hours after transfection, RNA samples were prepared from 293T cells using Trizol reagent (Invitrogen). To determine the level of shRNA expression obtained by each individual promoter, 20 µg of total RNA was separated by electrophoresis on a 10% polyacrylamide/7 M urea gel and electroblotted for 4 h at 400 mA. To detect HIV-1 RNA degradation mediated by tat-specific siRNA, 10 µg of RNA was denatured in glyoxal sample loading dye (Ambion), separated on a 1.2% agarose gel and transferred using the Turboblotter system (Schleicher & Schuell, Dassel, Germany). Tubulin mRNA was used as an internal standard. In all cases, RNA was blotted on a Zeta-Probe GT membrane (Bio-Rad) and immobilized by UV crosslinking. Hybridization was carried out at 42°C for siRNA and at 65°C for HIV-1 mRNA detection using Ultrahyb-Oligo Hybridization Buffer (Ambion). HIV-1 specific shRNA was probed with a 32P-labeled tat antisense oligonucleotide. HIV-1 RNA and tubulin mRNA were probed with 32P-labeled gag and tubulin DNA probes, respectively. Membranes were washed twice in 2× SSC, 0.1% SDS and 0.2× SSC, 0.1% SDS at 37 and 60°C, respectively.
Western blot
Supernatant was harvested from 293T cells transfected with HIV-1NL4.3 and passed through a 0.45 µm filter prior to western blot analyses. Protein extracts were prepared by washing cells with phosphate buffered saline (PBS) and lysing in TNT lysis buffer (20 mM Tris/200 mM NaCl/1% Triton X-100) containing protease inhibitors. After 20 min on ice, lysates were spun at 15 000 g for 15 min to remove insoluble material. Protein concentrations were determined by the Bradford procedure. Proteins were resolved on 12% SDS–polyacrylamide gels and transferred onto Hybond PVDF membranes (Amersham, Piscataway, NJ). For sequential probing, blots were stripped in Restore Western Blot Stripping Buffer (Pierce, Rockford, IL). A monoclonal antibody to HIV-1 p24 (183 clone H12-C) was obtained through the NIH AIDS Reagent and Reference program and used at a 1/100 dilution. The monoclonal antibody to α-tubulin (clone DM7 1A) (Sigma, St Louis, MO) was used at a 1/500 dilution. Immunodetection was performed using the peroxidase-based ECL Plus detection system (Amersham) according to the manufacturer’s instructions.
RESULTS
siRNA was designed to target the first exon of the HIV-1 tat gene, which encodes a transcriptional transactivator protein that is essential for viral replication. The siRNA sequence was selected based on systematic alignments of computer-predicted secondary structures (mfold 2.0) of overlapping sequences of the tat exon-1. The five lowest free energy structures were analyzed with respect to secondary structure features such as local free energy (ΔG) of the folded target sequence, the size of single stranded stretches (loops) and the length and stability of stems and helixes. These parameters, among others, have been found to be of predictive value for ribozyme/antisense function (19,20). A 21-nt tat siRNA sequence that was conserved among all energy structures was selected for further use.
293T cells, which are permissive for HIV-1 replication, were co-transfected with synthetic tat siRNA. Viral p24 antigen levels were quantified in cell-free supernatant 48 h post transfection. tat siRNA inhibited virus production by 93% compared to tat specific sense and antisense RNA (Fig. 2).
Figure 2.

Inhibition of HIV-1 expression by tat siRNA. Synthetic tat siRNA was co-transfected with HIV-1NL4.3 in 293T cells and p24 production was assayed 48 h post transfection. Single-stranded sense and antisense tat RNA were used as controls. Values represent averages of three independent experiments, with the range indicated.
To enable intracellular synthesis of shRNA, we created various expression cassettes that consisted of different upstream promoters (MTD, H1, U6, U6+1, U6+27, CMV) followed by a hairpin region containing tat sense and antisense sequences, which were separated by a 6-nt spacer of irrelevant sequence (Fig. 1). A DNA construct expressing luciferase specific shRNA served as a control vector. A five thymidine transcription termination signal was placed downstream of the hairpin sequence in Pol III promoter driven cassettes. Transcription of the CMV driven expression vector was terminated by a polyadenylation signal.
To test the efficacy of the various promoters in expressing tat shRNA, 293T cells were transfected with the indicated expression cassettes (Fig. 1). Expression levels were analyzed by northern blot using an HIV-1 tat-specific oligonucleotide probe. Higher levels of expression were observed with the MTD, U6+1 and U6+27 promoters compared to the U6 and H1 promoters (Fig. 3). There was no detectable expression with the CMV promoter, confirming previous reports that an unmodified CMV promoter is not suitable for shRNA expression (21). The additional 5′ guanosine in the U6+1 transcript proved to be important for efficient priming of transcription, yielding significantly higher expression of tat shRNA when compared to the U6 construct (Fig. 3).
Figure 3.
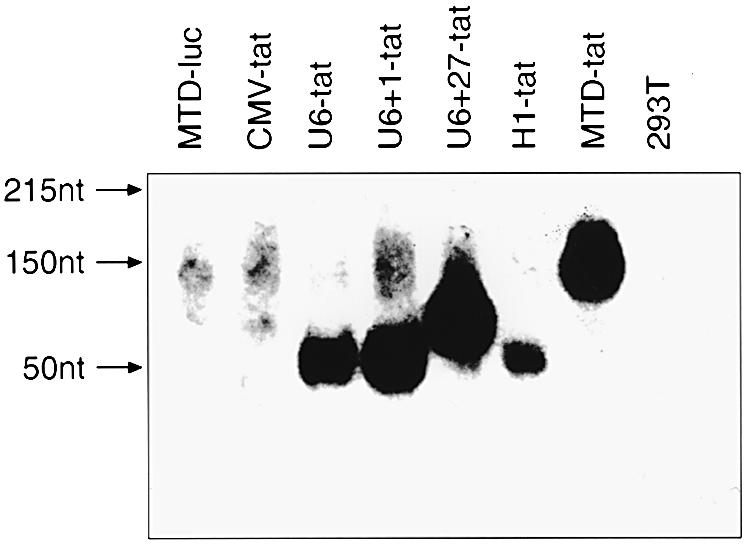
Expression levels of tat shRNA driven by different promoters were analyzed by northern blotting. Total cellular RNA was extracted from 293T cells 48 h after transfection with the indicated DNA constructs. Non-transfected 293T cells served as a negative control. Hybridization was performed using a 32P-labeled DNA oligonucleotide probe complimentary to the sense tat transcript.
Given that all Pol III promoters were able to drive the expression of shRNA, we next determined whether promoter choice affected the antiviral activity of tat shRNA. 293T cells were co-transfected with HIV-1NL4.3 and DNA constructs containing different shRNA expression cassettes. The level of virus production was quantified by measuring viral p24 antigen level in culture supernatant by immunoassay 48 h after transfection. shRNA expressed by the MTD promoter resulted in 93% inhibition of HIV-1 production compared to the MTD-luc control. Significantly, the MTD promoter consistently gave 34–56% greater inhibition of p24 antigen level when compared to the U6+1, U6+27 or H1 promoters (Fig. 4). Western blot analysis of HIV-1 gag protein in cell-free supernatant and cell lysate confirmed the p24 ELISA results. The greatest reduction in gag protein in both fractions was achieved using the vector containing the MTD promoter, followed by the U6+1 promoter expression cassette (Fig. 5). As expected, the CMV promoter did not yield a discernable inhibitory effect on HIV-1 expression.
Figure 4.

Inhibition of HIV-1 virus production by various tat shRNA expression constructs. 293T cells were co-transfected with HIV-1NL4.3 and the indicated DNA vectors. A vector expressing luciferase specific shRNA (MTD-luc) served as a negative control. HIV-1 p24 antigen level in culture supernatant was quantified by ELISA 48 h after transfection. The antiviral effect of the MTD promoter was adjusted to 100% and the potency of the remaining pol III promoters is presented relative to the antiviral effect of the MTD promoter. Values represent the average of duplicate independent experiments, with the range indicated.
Figure 5.

Reduction of HIV-1 p24 antigen in cell-free supernatant and cell lysate determined by western blot analysis. 293T cells were co-transfected with HIV-1NL4.3 and the different shRNA expression vectors. Non- transfected 293T cells served as a negative control. The control plasmid MTD-luc expresses a luciferase shRNA. Tubulin served as a loading control.
Given that RNAi acts on a post-transcriptional level, we next assessed the effect of different expression cassettes on total (spliced and unspliced) intracellular HIV-1 RNA by real-time PCR. The degradation of viral RNA mirrored the decrease in viral proteins with the maximum reduction (88%) observed with the MTD promoter. The MTD promoter consistently gave 34–45% greater reduction in total HIV-1 RNA when compared to the U6+1, U6+27 or H1 promoters (Fig. 6). Finally, the effect of tat shRNA on intracellular unspliced full length HIV-1 RNA expression was determined by northern blot (Fig. 7). The highest level of HIV-1 RNA degradation was achieved using the MTD and U6+1 shRNA expression cassettes.
Figure 6.

Promoter dependent effects of tat shRNA on levels of spliced and unspliced intracellular HIV-1 RNA as quantified by real-time PCR. Variation of RNA input was normalized by GAPDH real-time PCR. To convert threshold cycles to copy number an external standard curve was created with known numbers of HIV-1 in vitro tat transcripts. A vector expressing luciferase specific shRNA (MTD-luc) served as a negative control. The greatest antiviral effect was observed in experiments using the MTD promoter, which was adjusted to 100% to allow comparisons with other promoters. Values represent averages of two independent experiments, with the range indicated.
Figure 7.

Degradation of full length HIV-1 RNA by different tat shRNA expression constructs. Northern blot analysis of total unspliced HIV-1 RNA from 293T cells co-transfected with HIV-1NL4.3 and various shRNA expression vectors 48 h post-transfection. Tubulin expression was used as a loading control.
DISCUSSION
Our results demonstrate that the potency of HIV-1 specific RNAi depends upon the choice of eukaryotic promoter used to drive intracellular expression of shRNA, with the most potent antiviral effects being observed in experiments utilizing the MTD promoter. Our findings confirm the results of Kawasaki and Taira (22) who compared tRNAVal promoter with human and mouse U6 promoters. These investigators found more effective gene silencing when the tRNAVal promoter was used to drive the expression of shRNA targeting K-ras protein (22). Several factors may contribute to these promoter-dependent effects.
It was previously observed that the effect of synthetic siRNA could be abolished if the RNA included non- base-paired nucleotide extensions on the 5′- or 3′-ends of the duplex RNA. Thus, the stable expression of siRNA from DNA vectors appeared to be dependent on promoters with defined transcription initiation and termination sites, such as pol III type III promoters. Of these, the human U6 snRNA and H1, which lie completely upstream of the sequence being transcribed, were successfully introduced to express siRNA and shRNA (1,2). Subsequently, it was shown that the inhibitory effect of shRNA transcripts was not affected by the inclusion of non-specific 5′-end extensions (3). In contrast, it was reasoned that 5′-end extensions of the precursor hairpin siRNA could stabilize and protect the transcript from 5′ nuclease attack. For example, the U6+27 promoter, which contains 27 additional nucleotides upstream of the specific hairpin siRNA, was found to express transcripts to higher levels than the U6+1 promoter (4).
Promoter elements of tRNA promoters (Type II) are intragenic and co-transcribed as the 5′ end of the nascent RNA. The MTD promoter is an optimized human tRNA-derived promoter modified by the inclusion of an additional 5T termination signal directly downstream of the shRNA sequence (5). The resulting transcript consists of a 100-nt extension of promoter elements upstream of the specific hairpin shRNA transcript that is terminated within a stretch of uridine residues. Hairpin precursor RNA must be stabilized against rapid degradation, folded correctly and transported to the cellular compartment where RNAi occurs. The overall secondary structure of the MTD transcript may transfer additional stability to the shRNA and/or improve accessibility of the transcript for the enzyme Dicer to catalyze the initial step of RNAi (23). Human Dicer is localized in the cell cytoplasm and it has been suggested that additional components of the RNAi pathway are likewise cytoplasmic (24–26). Subcellular localization studies have shown that chimeric tRNA transcripts are predominantly found in cytoplasmic fractions, whereas U6 transcripts are more abundant in the nucleus (14,22). Thus, differences in subcellular distribution of precursor transcripts expressed by different promoters could account for variable potencies of shRNA expression units. Therefore, the MTD promoter may be better suited for antiviral RNAi because of its high expression levels of hairpin transcripts, a favorable RNA structure that stabilizes against rapid degradation, and transcripts which are predominantly located in the cytoplasm of the cell.
Interestingly, in our experiments, the expression level of the shRNA transcripts alone did not appear to correlate directly with the magnitude of the antiviral effect. For example, the H1 promoter showed strong inhibition of HIV-1 production, yet had the lowest expression of shRNA in northern blots. Conversely, the U6+27 promoter was associated with strong expression of shRNA, but did not yield concomitantly high suppression of virus production.
RNAi offers investigators a new tool in gene therapy to potentially silence disease-specific genes (27). The effectiveness of permanent transfer of RNAi will depend upon the efficient delivery of molecular constructs and maintenance of gene silencing. Given the rapid replication kinetics of HIV-1, a major barrier to using antiviral RNAi is likely to be the emergence of escape variants that contain nucleotide mutations in regions targeted by siRNA. Analogous to drug-resistance variants associated with HIV-1 therapy, potent inhibition of viral replication will be required to prevent the generation of such escape variants. To achieve this goal, use of the MTD promoter in adenoviral or retroviral siRNA delivery systems merits further study.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by a Clinical Scientist Development Award from the Doris Duke Charitable Foundation, a Daland Fellowship in Clinical Investigation from the American Philosophical Society and a Career Development Grant from NIAID/NIH (B.R.). We thank the Brown/Tufts/Lifespan CFAR Retrovirology Core Laboratory for assay support.
REFERENCES
- 1.Lee N.S., Dohjima,T., Bauer,G., Li,H., Li,M.J., Ehsani,A., Salvaterra,P. and Rossi,J. (2002) Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat. Biotechnol., 20, 500–505. [DOI] [PubMed] [Google Scholar]
- 2.Brummelkamp T.R., Bernards,R. and Agami,R. (2002) Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cells, 2, 243–247. [DOI] [PubMed] [Google Scholar]
- 3.Paddison P.J., Caudy,A.A., Bernstein,E., Hannon,G.J. and Conklin,D.S. (2002) Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev., 16, 948–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paul C.P., Good,P.D., Winer,I. and Engelke,D.R. (2002) Effective expression of small interfering RNA in human cells. Nat. Biotechnol., 20, 505–508. [DOI] [PubMed] [Google Scholar]
- 5.Thompson J.D., Ayers,D.F., Malmstrom,T.A., McKenzie,T.L., Ganousis,L., Chowrira,B.M., Couture,L. and Stinchcomb,D.T. (1995) Effective expression of small interfering RNA in human cells. Nucleic Acids Res., 23, 2259–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brummelkamp T.R., Bernards,R. and Agami,R. (2002) Effective expression of small interfering RNA in human cells. Science, 296, 550–553. [DOI] [PubMed] [Google Scholar]
- 7.Surig D., Bredow,S. and Benecke,B.J. (1993) The seemingly identical 7SK and U6 core promoters depend on different transcription factor complexes. Gene Expr., 3, 175–185. [PMC free article] [PubMed] [Google Scholar]
- 8.Myslinski E., Ame,J.C., Krol,A. and Carbon,P. (2001) An unusually compact external promoter for RNA polymerase III transcription of the human H1RNA gene. Nucleic Acids Res., 29, 2502–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gunnery S., Ma,Y. and Mathews,M.B. (1999) Termination sequence requirements vary among genes transcribed by RNA polymerase III. J. Mol. Biol., 286, 745–757. [DOI] [PubMed] [Google Scholar]
- 10.Elbashir S.M., Harborth,J., Lendeckel,W., Yalcin,A., Weber,K. and Tuschl,T. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- 11.Yu M., Ojwang,J., Yamada,O., Hampel,A., Rapapport,J., Looney,D. and Wong-Staal,F. (1993) A hairpin ribozyme inhibits expression of diverse strains of human immunodeficiency virus type 1. Proc. Natl Acad. Sci. USA, 90, 6340–6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng H., Callison,D., Li,P. and Burrell,C. (1996) Long-term protection against HIV-1 infection conferred by tat or rev antisense RNA was affected by the design of the retroviral vector. Virology, 220, 377–389. [DOI] [PubMed] [Google Scholar]
- 13.Peng H., Callison,D.E., Li,P. and Burrell,C.J. (1997) Long-term protection against HIV-1 infection conferred by tat or rev antisense RNA was affected by the design of the retroviral vector. AIDS, 11, 587–595. [DOI] [PubMed] [Google Scholar]
- 14.Ilves H., Barske,C., Junker,U., Bohnlein,E. and Veres,G. (1996) Retroviral vectors designed for targeted expression of RNA polymerase III-driven transcripts: a comparative study. Gene, 171, 203–208. [DOI] [PubMed] [Google Scholar]
- 15.Sullenger B.A., Lee,T.C., Smith,C.A., Ungers,G.E. and Gilboa,E. (1990) Expression of chimeric tRNA-driven antisense transcripts renders NIH 3T3 cells highly resistant to Moloney murine leukemia virus replication. Mol. Cell. Biol., 10, 6512–6523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bertrand E., Castanotto,D., Zhou,C., Carbonnelle,C., Lee,N.S., Good,P., Chatterjee,S., Grange,T., Pictet,R., Kohn,D. et al. (1997) The expression cassette determines the functional activity of ribozymes in mammalian cells by controlling their intracellular localization. RNA, 3, 75–88. [PMC free article] [PubMed] [Google Scholar]
- 17.Dayton A.I., Sodroski,J.G., Rosen,C.A., Goh,W.C. and Haseltine,W.A. (1986) The trans-activator gene of the human T cell lymphotropic virus type III is required for replication. Cell, 44, 941–947. [DOI] [PubMed] [Google Scholar]
- 18.Ge L. and Rudolph,P. (1997) Simultaneous introduction of multiple mutations using overlap extension PCR. Biotechniques, 22, 28–30. [DOI] [PubMed] [Google Scholar]
- 19.Amarzguioui M., Brede,G., Babaie,E., Grotli,M., Sproat,B. and Prydz,H. (2000) Secondary structure prediction and in vitro accessibility of mRNA as tools in the selection of target sites for ribozymes. Nucleic Acids Res., 28, 4113–4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scherr M., Rossi,J.J., Sczakiel,G. and Patzel,V. (2000) RNA accessibility prediction: a theoretical approach is consistent with experimental studies in cell extracts. Nucleic Acids Res., 28, 2455–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xia H., Mao,Q., Paulson,H.L. and Davidson,B.L. (2002) siRNA-mediated gene silencing in vitro and in vivo. Nat. Biotechnol., 20, 1006–1010. [DOI] [PubMed] [Google Scholar]
- 22.Kawasaki H. and Taira,K. (2003) Short hairpin type of dsRNAs that are controlled by tRNA(Val) promoter significantly induce RNAi-mediated gene silencing in the cytoplasm of human cells. Nucleic Acids Res., 31, 700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernstein E., Caudy,A.A., Hammond,S.M. and Hannon,G.J. (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature, 409, 363–366. [DOI] [PubMed] [Google Scholar]
- 24.Billy E., Brondani,V., Zhang,H., Muller,U. and Filipowicz,W. (2001) Specific interference with gene expression induced by long, double-stranded RNA in mouse embryonal teratocarcinoma cell lines. Proc. Natl Acad. Sci. USA, 98, 14428–14433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hutvagner G. and Zamore,P.D. (2002) A microRNA in a multiple-turnover RNAi enzyme complex. Science, 297, 2056–2060. [DOI] [PubMed] [Google Scholar]
- 26.Zeng Y. and Cullen,B.R. (2002) RNA interference in human cells is restricted to the cytoplasm. RNA, 8, 855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coburn G.A. and Cullen,B.R. (2002) Potent and specific inhibition of human immunodeficiency virus type 1 replication by RNA interference. J. Virol., 76, 9225–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]