

Abstract
Microcephalin (MCPH1) is one of the causative genes for the autosomal recessive disorder, primary microcephaly, characterized by dramatic reduction in brain size and mental retardation. MCPH1 also functions in the DNA damage response, participating in cell cycle checkpoint control. However, how MCPH1 is regulated in the DNA damage response still remains unknown. Here we report that the ability of MCPH1 to localize to the sites of DNA double-strand breaks depends on its C-terminal tandem BRCT domains. Although MCPH1 foci formation depends on H2AX phosphorylation after DNA damage, it can occur independently of MDC1. We also show that MCPH1 binds to a phospho-H2AX peptide in vitro with an affinity similar to that of MDC1, and overexpression of wild type, but not C-BRCT mutants of MCPH1, can interfere with the foci formation of MDC1 and 53BP1. Collectively, our data suggest MCPH1 is recruited to double-strand breaks via its interaction with γH2AX, which is mediated by MCPH1 C-terminal BRCT domains. These observations support that MCPH1 acts early in DNA damage responsive pathways.
Cell cycle checkpoints are critical for detecting damaged DNA and delaying cell cycle progression to allow time for DNA repair. If the damage is too great, they can also trigger cell death. Increasing evidence has shown that defects within these DNA damage checkpoints can lead to genetic instability, a hallmark of cancer cells (1). The DNA damage response is a complex network of proteins that can sense DNA lesions and relay and amplify the signal to downstream effectors to promote accurate and safe transmission of genetic material to the next generation. This pathway is usually classified as having groups of proteins involved in different steps; that is, sensors, mediators, and effectors. Canonically, the DNA damage response is a signal transduction cascade triggered by a series of phosphorylation-dependent events that activate proteins involved in transducing the DNA damage signal (ionizing radiation (IR)3 or stalled replication) to different effector proteins that evoke specific effects, namely halting cell cycle progression, activating DNA repair mechanisms and transcription, and triggering apoptosis. The phosphatidylinositol 3-kinase-like kinase ataxia-telangiectasia-mutated (ATM) becomes active upon IR and relocalizes to the sites of double-strand breaks and phosphorylates H2AX, a histone variant that marks sites of damaged DNA. This then allows for the recruitment of other mediator proteins like MDC1, BRCA1, and 53BP1. These proteins help promote the damage signal to Chk1 and Chk2, which can phosphorylate various substrates to modulate the cell cycle, DNA repair, and apoptosis. However, increasing evidence has shown that the DNA damage response is not a linear pathway; DNA damage proteins can also act both upstream and downstream of their respective functions and maintain cross-talk with other proteins. Instead of a linear pathway as previously thought, there is an intricate network of proteins that function independently and dependently to coordinate the response to DNA damage and maintain genomic stability.
Because of their important roles in maintaining genomic stability, this pathway has been shown to be mutated and deactivated in human cancer, and inherited mutations in several of these proteins cause cancer susceptibility syndromes. Indeed, proteins involved in the DNA damage response and DNA repair, when mutated, cause genetic diseases that predispose individuals to cancer. BRCA1 mutations account for >50% of familial breast cancer cases, and mutations in ATM cause ataxia-telangiectasia syndrome, causing sensitivity to DNA-damaging agents as well as cancer. Recent studies have also provided evidence that the DNA damage response is an anti-cancer barrier, which must be overcome in early neoplastic lesions for tumorigenesis to occur (2, 3).
A new class of mediator proteins including MDC1, BRCA1, 53BP1, and MCPH1 has been shown to be critical in promoting the further activation and transduction of the DNA damage response. MCPH1 function as a mediator is still in debate. Using siRNA targeting MCPH1, several groups have shown that MCPH1 depletion affects BRCA1 and Chk1 protein levels as well as the foci formation of ATM, MDC1, and 53BP1, potentially placing MCPH1 as an early participant in the DNA damage response (4-6). However, cells derived from patients with mutations in MCPH1 do not have differences in BRCA1 and Chk1 protein levels and raise the possibility that MCPH1 may function downstream of ATR and Chk1 in regulating G2/M transition (7). The purpose of our study was to delineate how MCPH1 is regulated in the DNA damage response.
EXPERIMENTAL PROCEDURES
Cell Culture and Plasmids
Human MCPH1 cDNA was graciously provided by Dr. David Stern at Yale University. Full-length and deletions of MCPH1 were generated by PCR and cloned by EcoRI and XmaI sites into a modified pIRES2-EGFP vector that contains an N-terminal S, FLAG, and streptavidin binding peptide tag. MCPH1 BRCT point mutants were generated using the QuikChange site-directed mutagenesis kit and verified by sequencing. All primer sequences used are available upon request. HeLa and 293T cells were purchased from American Tissue Type Culture (Manassas, VA) and maintained in RPMI 1640 supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37 °C in 5% CO2 (v/v). MDC1-/-, 53BP1-/-, H2AX-/-, H2AX-/- reconstituted with WT H2AX and S139A mutant and wild-type mouse embryonic fibroblast (MEF) cells, NBS1-deficient and NBS1-reconstituted cells, ATM-deficient (FT169A) and -reconstituted (YZ5) cells, and HCC1937 and HCC1937-BRCA1 were previously reported (8-11). FANCD2-reconstituted and -deficient cells were a generous gift of Dr. Alan D’Andrea at the Department of Pediatric Oncology at Dana-Farber Cancer Institute (Boston, MA). For irradiation, cells were irradiated using a JL Shepherd Cs137 source at the indicated doses.
Antibodies and Transfection
Rabbit anti-MCPH1 was generated by immunizing rabbits with a peptide containing residues 96-110 of MCPH1 (MNEHLSSLIKKRKC). The antibody was affinity-purified using SulfoLink Plus Immobilization kit (Pierce). BACH1, Claspin, Chk2, 53BP1, MDC1, and H2AX antibodies have been previously described (8, 12). Polyclonal anti-rabbit MCPH1 was purchased from Abcam (ab2612) for Western blotting. Monoclonal ATM clone 2C1 antibody was purchased from GeneTex. Monoclonal anti-FLAG M2 was purchased from Sigma. Transfections were carried out using Lipofectamine per the manufacturer’s instructions. siRNA was performed using Oligofectamine per manufacturer’s instructions and as indicated. Control siRNA, ATM SMARTpool, and MDC1 siRNA were purchased from Dharmacon.
Viral Infection
Full-length MCPH1 was cloned into pEF1A-HA/FLAG retroviral vector using the gateway system. pEF1A-MCPH1 was co-transfected with pCL-Amphobac in BOSC23 packaging cell lines to produce virus. Virus was collected 48 and 72 h after transfection and subsequently used to infect MEF cell lines. 48 h after the last infection, MEFs were irradiated as indicated, and immunofluoresence was performed or selected in puromycin for stable clones.
Immunofluoresence and Western Blotting
Cells grown on coverslips were fixed in 3% paraformaldehyde solution for 20 min, rinsed 1× with PBS ,and then permeabilized using 0.5% Triton X-100 for 5 min. Cells were then washed and incubated with primary antibodies diluted in 5% goat serum at 37 °C for 20 min. Cells were washed again 1× in PBS and incubated in secondary antibodies, either fluorescein isothiocyanate-conjugated goat anti-mouse IgG or rhodamine-conjugated goat anti-rabbit IgG, for 20 min at 37 °C. After washing, cells were counterstained with DAPI, washed with PBS, and then mounted onto slides with anti-Fade. Images were taken using a Nikon ECLIPSE E800 microscope.
Cells were lysed with NETN (20 mm Tris-HCl, pH 8.0, 100 mm NaCl, 1 mm EDTA, 0.5% Nonidet P-40) on ice and then rocked for 10 min at 4 °C. Crude cell lysates were then centrifuged at 14,000 rpm for 10 min, and cleared lysates were collected. Samples were boiled in 2× Laemmli buffer and run on SDS-PAGE. Membranes were blocked in 5% milk, Tris-buffered saline-Tween and then probed with antibodies as indicated.
Fluorescence-activated Cell Sorting
For cell cycle analysis, cells were either trypsinized or collected by mitotic shake-off. Cells were washed in cold PBS 2×, then resuspended in 300 μl of cold PBS and fixed with 700 μl of ice-cold 100% ethanol. Cells were then incubated in RNase A in sodium citrate buffer (100 units/sample) for 30 min, then stained with propidium iodide (50 μg/ml) for 30 min. Cells were then run on a FACScan, and cell cycle analysis was performed.
Preparation of MCPH1 and MDC1 BRCT Domains
The tandem BRCT domains of human MCPH1 (MCPH1-BRCT, residues 640-835) and human MDC1 (MDC1-BRCT, residues 1884-2089) were cloned in an expression plasmid derived from pET15b (Novagen) encoding an N-terminal hexahistidine tag followed by a tobacco etch virus protease cleavage site. For protein production, the plasmid was transformed in Escherichia coli BL21 (DE3) (Novagen), and the cells were grown in LB media at 37 °C until they reach an absorbance at 600 nm of ∼0.8 and then at 18 °C for ∼16 h after induction with 1 mm isopropyl 1-thio-β-d-galactopyranoside. The cells were harvested by centrifugation at 4000 × g for 15 min and resuspended in a bind buffer made up of 50 mm sodium phosphate, pH 7.5, 5 mm imidazole, and 300 mm NaCl. The cells were lysed with a high pressure microfluidizer Emulsiflex C-5 (Avestin Inc.) and centrifuged at 20,000 × g for 30 min. The supernatant fraction was passed through a Ni2+-nitrilotriacetic acid column (Qiagen). Subsequently, the column was washed with a solution of 50 mm sodium phosphate, pH 7.5, 20 mm imidazole, and 300 mm NaCl, and the protein was eluted with a similar solution but with 500 mm imidazole. Tobacco etch virus protease was added to the protein at room temperature overnight to cleave the polyhistidine tag. Further purification was carried out by size exclusion chromatography using a preparative Superdex 75 column (GE Healthcare).
Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) measurements were carried out at 10 °C with a VP-ITC titration calorimeter (MicroCal). The concentrated stocks of MCPH1-BRCT, MDC1-BRCT, and γH2AX peptide (residues 130-142) were in the target buffer (50 mm Tris/HCl, pH 7.5, 40 mm NaCl) and diluted with the same buffer to achieve a protein concentration of 35 μm and 1.3 mm for γH2AX. In a typical experiment the peptide solution was placed in a 298-μl calorimeter syringe and delivered initially as a 3-μl injection and then as a series of 5-μl injections every 5 min to the reaction cell containing 1.4 ml of the protein. Under identical conditions control experiments were also performed to determine the heat of dilution. The initial data point was routinely deleted. Data were analyzed with Levenberg-Marquardt nonlinear regression using a single-binding site model with the software ORIGIN 7.0 (OriginLab).
RESULTS
MCPH1 Localizes to the Sites of DNA Damage
MCPH1 has been shown to relocalize to the sites of DNA breaks generated by IR and ultraviolet (UV) light. We developed an antibody raised against amino acids 96-110 of MCPH1, which readily detects MCPH1 foci in irradiated HeLa cells (Fig. 1A). Damage-induced MCPH1 foci formation was not only detected in cells after IR but was also apparent in cells treated with various DNA-damaging or replication-stressing agents, including hydroxyurea (HU), mitomycin C (MMC), methylmelanosulfate (MMS), and etoposide (Fig. 1A). These induced MCPH1 foci always co-localize with that of γH2AX (Fig. 1A), confirming that MCPH1 relocates to the sites of DNA damage.
FIGURE 1. MCPH1 forms foci in response to various DNA-damaging agents and is expressed throughout the cell cycle.
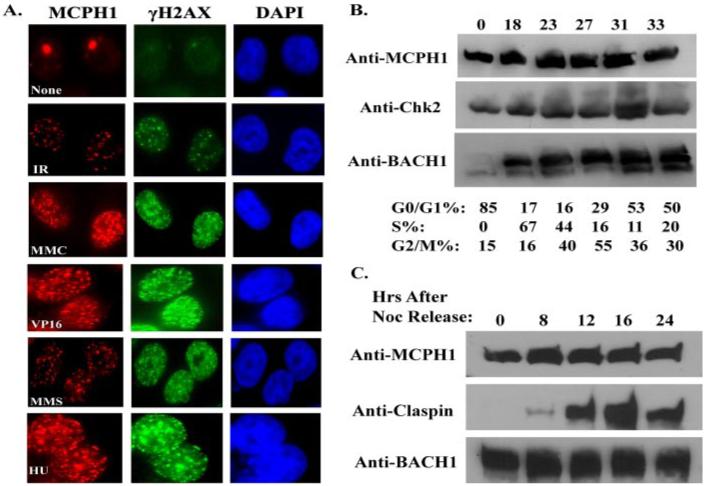
A, DNA damage-induced MCPH1 foci formation. HeLa cells grown on coverslips were treated with 10 Gy IR, 5 mm hydroxyurea (HU), 100 μg/ml mitomycin (MMC), 0.002% methylmelanosulfate (MMS), 2.5 μm etoposide or left untreated for 16 h before fixation and immunostaining with indicated antibodies. B and C, cell cycle expression of MCPH1. T24 cells were allowed to contact inhibit for 96 h and then trypsinized and released into fresh media. Samples were taken at the indicated time points and analyzed by fluorescence-activated cell sorting and Western blotting. C, HeLa cells were arrested overnight with 0.5 μg/ml nocodazole. Mitotic cells were “shaken off” and then released into normal media without nocodazole. Samples were again taken at different time points for Western blotting analysis.
Past studies have shown that several proteins in the DNA damage response are cell cycle-regulated, which impacts on how they function in checkpoint control and in DNA repair (13, 14). We investigated whether MCPH1 expression is cell cycle-regulated using T24 cells that when grown confluent arrest in the G0 or quiescent stage of the cell cycle. After 96 h of contact inhibition these cells were trypsinized and replated to allow re-entry into the cell cycle. We also performed a mitotic shake off experiment using HeLa cells arrested in mitosis with nocodazole. Cells from each were taken at specific time intervals and assayed for MCPH1 protein expression by Western blot and cell cycle analysis using fluorescence-activated cell sorting (Fig. 1, B and C). There is no apparent cell cycle regulation of MCPH1 protein levels, as MCPH1 is detected in all cell cycle phases by immunoblotting. Consistent with this result, we also routinely observe the expression of MCPH1 at all cell cycle stages by immunofluoresence staining (data not shown). Thus, MCPH1 is expressed throughout the cell cycle,; raising the possibility that MCPH1 may function to maintain genomic stability throughout the cell cycle.
C-terminal BRCT Domains of MCPH1 Are Required for Its Foci Formation after DNA Damage
Next, we explored what region of MCPH1 is important for foci formation. Previous reports have shown that the C-terminal BRCT domains of BRCA1 are important for its localization to the sites of DNA damage (15). To determine whether the MCPH1 C-terminal BRCT domains are required for its localization to the sites of DNA damage, we generated epitope-tagged consecutive C-terminal truncation mutants of MCPH1 (Fig. 2A). We expressed wild-type MCPH1 and these MCPH1 deletion mutants in 293T cells and determined their cellular distribution after IR. Indeed, disruption of the C-terminal BRCT domains abolished MCPH1 nuclear foci formation after IR; however, deletion of the N-terminal domain of MCPH1 has no affect on foci formation (Fig. 2B). Interestingly, deletion of residues 301-400 caused abnormal localization of these truncated proteins to the cytoplasm. Within this region spanning amino acids 355-375 there is a canonical nuclear localization signal, RKRVSHGSHSPPKEKCKRKR, that appears to be important for the nuclear localization of MCPH1.
FIGURE 2. The tandem C-terminal BRCT domains of MCPH1 are required for foci formation.
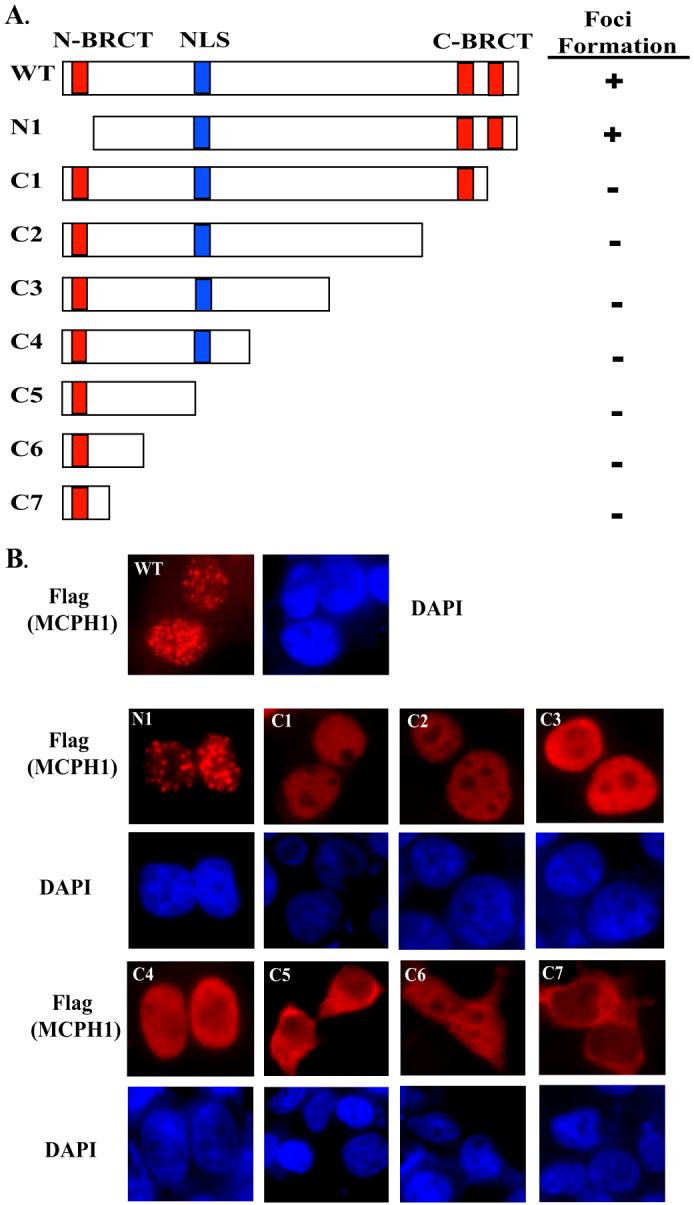
A, diagram of different tagged-MCPH1 (SFB-MCPH1) deletion mutants. 293T cells grown on coverslips were transfected with plasmids encoding wild type or each of these deletion mutants of MCPH1. 24 h after transfection cells were treated with 10 Gy and allowed to recover for 6 h before fixation and immunostaining (B).
Because deletion of the C-terminal BRCT domains abrogated the foci formation of MCPH1, we analyzed if specific mutations within the BRCT domains would affect focus localization of MCPH1. MCPH1 BRCT domains share extended sequence similarity with BRCA1 BRCT domains (Fig. 3A). All of these BRCT domains contain an invariable tryptophan residue, which is important for the three-dimensional structure of the BRCT domain because mutation of this residue abolished the phosphopeptide binding activity of BRCT domains (16). We generated arginine substitution mutations for each of the four conserved tryptophans within MCPH1 BRCT domains (W75R mutation in the N-terminal BRCT domain, W706R, W712R, and W815R mutations in the C-terminal first and second BRCT domains) and evaluated the localization of W75R, W706R, and W815R MCPH1 mutants after IR. Point mutations of the C-terminal BRCT domains abolished foci formation, but mutating the N-terminal BRCT domain had no affect on MCPH1 localization (Fig. 3B). Together with the deletion mutants shown in Fig. 2, these data demonstrate that the C-terminal BRCT domains are crucial for the localization of MCPH1 to the sites of DNA damage.
FIGURE 3. Mutations within the C-terminal BRCT domains abolish MCPH1 foci formation.
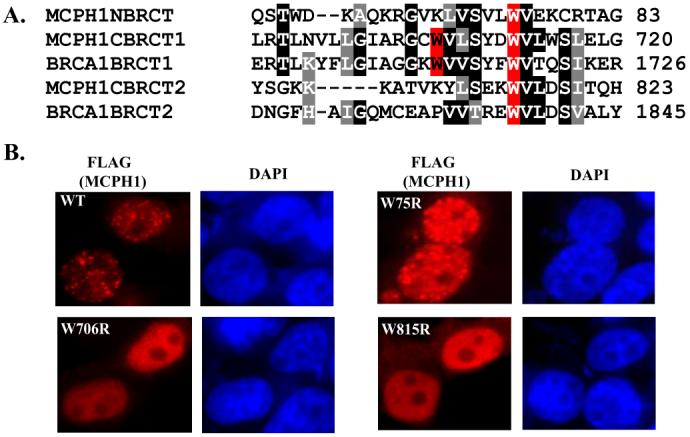
A, ClustalW alignment of the BRCA1 and MCPH1 BRCT domains. Residues shaded in red represent conserved tryptophan residues mutated to arginine. B, mutations of the C-terminal BRCT domains abolished MCPH1 foci formation. Experimental procedure were the same as those described in Fig. 2.
H2AX Phosphorylation Is Critical for MCPH1 Foci Formation after DNA Damage
Several reports have described functional consequences of MCPH1 depletion using siRNA (4-7). These range from defective cell cycle checkpoints and abolishment of foci formation for several important mediator and effector proteins in the DNA damage response to impacting on Cdc25A degradation and normal G2/M transition. However, the results are still controversial, as a MCPH1 knock-out mouse model has not been characterized as of yet, and these studies with siRNA may have off-target effects. Because there are still differing viewpoints on how MCPH1 functions in the DNA damage response, we focused on determining how MCPH1 is regulated upon DNA damage. To answer this question we have evaluated the dependence of MCPH1 foci formation in response to IR using an exhaustive panel of human cells and MEFs that are defective in various DNA damage checkpoint components. As shown in Fig. 4A, MCPH1 forms foci independently of ATM after IR. Similar phenomena have been observed for 53BP1, H2AX, and Pax2 transactivation-interacting protein (PTIP) (8, 17, 18), which are likely due to the compensation of other checkpoint kinases including ATR and DNA protein kinase (DNA-PK) in the absence of ATM. MCPH1 appears to also be independent of the Mre11-Rad50-Nbs1 (MRN) complex and Fanconi anemia (FA) complex, as MCPH1 forms foci in response to IR in Nbs1-deficient and in PD20 (FANCD2-/-) cells (Fig. 4A). We further examined whether MCPH1 foci formation would depend on several mediator proteins, like MDC1, 53BP1, and BRCA1. MCPH1 foci formation was the same in BRCA1-deficient HCC1937 cells as that in HCC1937 cells reconstituted with wild-type BRCA1 (Fig. 4A). Our anti-MCPH1 antibody was generated against human MCPH1 and did not cross-react with mouse MCPH1. To study whether MCPH1 focus localization depends on MDC1, 53BP1, or H2AX using their corresponding knock-out MEFs, we used a retrovirus-based system to express tagged MCPH1 in these MEFs. As shown in Fig. 4B, tagged wild-type MCPH1 readily formed foci after IR in wild-type MEFs. Although MCPH1 foci formation was abolished in H2AX-/- MEFs, the correct localization of MCPH1 was restored when H2AX-/- MEFs were reconstituted with a WT H2AX transgene but not when a H2AX S139A mutant transgene was used (Fig. 4B). Therefore, the focus localization of MCPH1 depends on H2AX phosphorylation in vivo. Interestingly, we observed normal MCPH1 foci formation in MDC1-/- or 53BP1-/- cells, suggesting that MCPH1 acts downstream of H2AX but is independent of MDC1, 53BP1, or BRCA1 after DNA damage. This places MCPH1 at the early steps in the DNA damage induced signal transduction pathways.
FIGURE 4. MCPH1 foci formation depends on H2AX but not on ATM, NBS1, FANCD2, BRCA1, MDC1, and 53BP1.

A, ATM-deficient cells and cells expressing wild-type ATM were evaluated for MCPH1 foci formation before and after ionizing radiation. The same experiments were carried out using Nbs1 wild type and Nbs1-deficient fibroblasts, PD20 wild type and PD20 null fibroblasts, BRCA1-deficient HCC1937 cells and HCC1937 cells reconstituted with wild-type BRCA1. B, Viral transduction of myocyte-specific enhancer binding factor cell lines was performed using retroviral particles expressing hemagglutinin/FLAG-tagged MCPH1. Myocyte-specific enhancer binding factor cell lines were infected twice before they were irradiated with 10 Gy of ionizing radiation. All cells were allowed to recover for 6 h before immunofluoresence staining was performed.
MDC1 has been shown before to be a mediator of the DNA damage checkpoint and is responsible for the recruitment of many checkpoint proteins including NBS1, 53BP1, and BRCA1 (19-21). Our results in MDC1 knock-out mouse embryonic fibroblasts suggest that MCPH1 acts either upstream or parallel of MDC1. To further confirm this result, we used siRNA to down-regulate both ATM and MDC1 and check for MCPH1 foci after irradiation. As a control, we also examined 53BP1 foci in these depleted cells; 53BP1 recruitment to double-strand breaks has been reported to be ATM-independent but MDC1-dependent (8, 17) The siRNA used against ATM and MDC1 worked very efficiently (Fig. 5A), and 48 h after the first transfection, cells were irradiated, allowed to recover, and immunostained with 53BP1 and MCPH1 antibodies. As expected, 53BP1 foci formation is not reduced in cells treated with ATM siRNA; however, 53BP1 foci are decreased in MDC1 knock-down cells (Fig. 5B). On the other hand, MCPH1 foci formation is not dependent on either ATM or MDC1 (Fig. 5B), which support our observations using ATM- or MDC1-deficient cells (Fig. 4A). There is a slight decrease of MCPH1 foci formation in the MDC1 siRNA-treated cells (Fig. 5B). We have shown previously that MDC1 functions to amplify the DNA damage signaling cascade by promoting further phosphorylation of H2AX near the sites of DNA damage (8). It is, therefore, possible that with the depletion of MDC1, the amplification step of H2AX phosphorylation is absent and may cause the slight defect in MCPH1 foci formation observed in these experiments.
FIGURE 5. 53BP1, but not MCPH1, recruitment to double-strand breaks is dependent on MDC1.
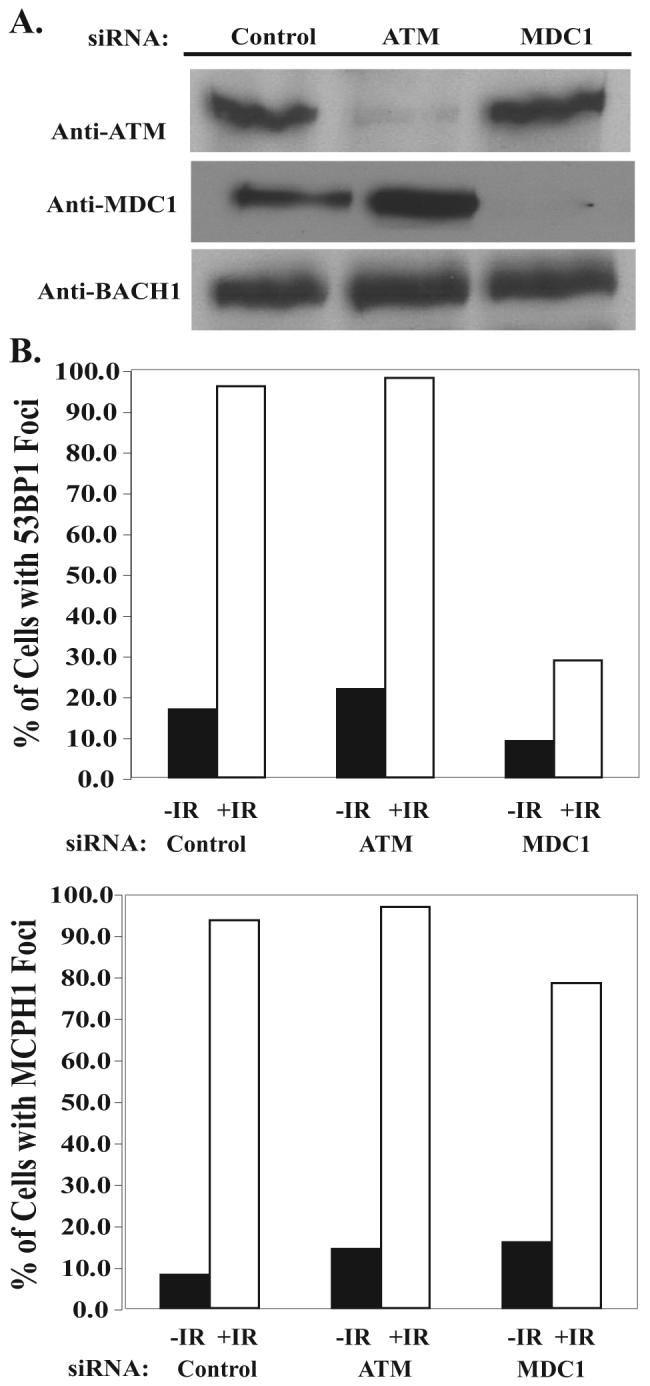
A, HeLa cells were transfected with Control, ATM, or MDC1 siRNA. 48 h after the first transfection cells were collected for Western analysis. B, HeLa cells on coverslips from A were subjected to 10 Gy and allowed to recover for 6 h, and then immunofluoresence was performed. Approximately 250 cells were counted for each treatment.
MCPH1 Binds Directly to Phosphorylated H2AX
Our data indicate that MCPH1 acts downstream of H2AX and may function either upstream or parallel of MDC1. This also made us inquire as to whether, like MDC1, whose C BRCT domains have been shown to bind efficiently to phosphorylated H2AX (22), MCPH1 also interacts with H2AX through its BRCT domains. As determined by ITC assay, MCPH1 tandem BRCT domains (MCPH1-BRCT, residues 640-835) bind tightly to a γH2AX peptide (residues 130-142) that is phosphorylated at Ser-139 (Fig. 6). The dissociation constant (KD)is 0.26 ± 0.01 μm. The interaction is driven by favorable enthalpic (ΔH° = -12.4 kcal/mol) and entropic (-TΔS° = 3.9 kcal/mol) contributions. For comparison, under similar conditions the KD of the tandem BRCT domains of MDC1 (MDC1-BRCT, residues 1884-2089) for the same γH2AX peptide is 0.57 ± 0.02 μm with ΔH° = -9.6 kcal/mol and -TΔS° = 1.5 kcal/mol. The observation that the thermodynamic parameters for the interactions of MCPH1-BRCT and MDC1-BRCT with γH2AX are similar strongly suggests that MCPH1-BRCT is a bona fide binding partner of γH2AX.
FIGURE 6. ITC titration of MCPH1 tandem BRCT domains with a γH2AX peptide.
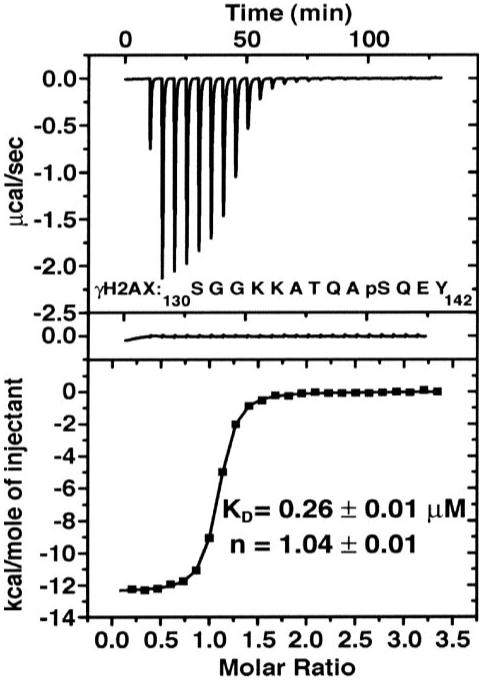
Shown are the integrated heat measurements from raw titration data (upper plot), control experiment with peptide injected into buffer solution (middle plot), and curve fitting to a standard one-site model (lower plot). The dissociation constant (KD) and stoichiometry number (n) are reported with the associated errors determined by nonlinear least squares analysis. The γH2AX peptide sequence (residues 130-142) is shown in the upper plot (pS stands for phosphoserine).
Previously, it has been shown that the BRCT domain of MDC1 when overexpressed can interfere with the foci formation of 53BP1 and BRCA1 (22). Because MCPH1 has a similar binding affinity as MDC1 for H2AX, we performed a similar set of experiments with MCPH1 WT and point mutants that abolish MCPH1 foci formation (W706R, W712R). As shown in Fig. 7, A-C, overexpression of WT MCPH1 can interfere with the foci formation of MDC1 and 53BP1. To some extent some cells with overexpression of wild-type MCPH1 had bright uniform staining of γH2AX instead of discrete foci seen in the second WT panel. This is potentially due to the ability of MCPH1 to bind and protect γH2AX from dephosphorylation, a phenomena similar to that observed in cells with overexpression of MDC1 BRCT domains (22). In contrast to WT MCPH1, the expression of MCPH1 BRCT mutants does not affect either MDC1 or 53BP1 foci formation (Fig. 7, A-C). All of these data are quantified and summarized in Fig. 7, D-F.
FIGURE 7. Overexpression of WT MCPH1 effects MDC1 and 53BP1 foci formation in response to IR.

293T cells were either untransfected or transfected with WT, W706R, or W712R MCPH1. 24 h later cells were irradiated with 10 Gy and allowed to recover for 6 h, and immunofluoresence staining were performed with H2AX (A), MDC1 (B), and 53BP1 (C) antibodies. Quantifications of the results in A-C are shown in D-F. 100 transfected cells were counted and scored for the staining of different proteins analyzed.
DISCUSSION
Our results demonstrate that MCPH1 is a stable protein expressed throughout the cell cycle and localizes to sites of DNA lesions induced by a variety of DNA-damaging agents. The data confirm the critical role of the C-terminal BRCT domains in mediating the ability of MCPH1 to localize to the sites of double-strand breaks. We have also demonstrated that MCPH1 foci formation in response to IR depends on H2AX but is independent of ATM, NBS1, FANCD2, MDC1, 53BP1, and BRCA1. Using ITC, we have shown that MCPH1 C BRCT domains bind tightly to a phospho-H2AX peptide in vitro and that the affinity is similar to that of MDC1. Finally, like MDC1, overexpression of WT but not mutant MCPH1 can affect the foci formation of downstream proteins in response to IR. Collectively, our data present evidence to support MCPH1’s lofty position at the top of the DNA damage response hierarchy.
MCPH1 relocates to the sites of damage in response to diverse genotoxic agents, similar to γH2AX. MCPH1, like BRCA1 and MDC1, contains tandem BRCT domains in its C terminus that are crucial for its ability to localize to the sites of damaged DNA. MCPH1 does appear to be a mediator in the pathway, but unlike MDC1, 53BP1, and BRCA1, it is farther upstream, only depending upon H2AX for foci formation. This is an intriguing result that the ability of MCPH1 to localize to the sites of DNA breaks is MDC1-independent because MDC1 has been shown to be required for the stable recruitment of many checkpoint proteins to the sites of damage (8, 21). This raises the possibility that MCPH1 somehow functions with MDC1 in an overlapping or parallel pathway. One possibility is that MCPH1, like MDC1, promotes the amplification of the DNA damage response by binding to H2AX at the sites of breaks and recruiting other mediator proteins.
MCPH1 is also similar to MDC1 in that depends on H2AX in vivo for foci formation. Our ITC data support this hypothesis as the MCPH1 C-BRCT domains bind to a phospho-H2AX peptide with an affinity similar to that of MDC1. Characterizing how MDC1 and MCPH1 both interact with H2AX to modulate checkpoint activation and protein recruitment will help to resolve questions as to how MCPH1 and MDC1 function. Two models supported by our data are that MCPH1 functions upstream of MDC1 or in a parallel pathway with MDC1. We are currently attempting to purify a MCPH1 protein complex in the hope of discovering MCPH1-interacting proteins that can help elucidate MCPH1-dependent checkpoint responses.
It has been proposed that MCPH1 functions in the ATR/Chk1 pathway (7). Cells derived from patients with mutations in MCPH1 appear to have a defect in Cdc25A degradation, which has been shown to be mediated by Chk1. However, a recent report has shown that there is a family with a genomic deletion spanning the promoter and half (exons 1-6) of the MCPH1 gene (23). Notably, ATR-Seckel cells still express full-length ATR at low levels (24). ATR and other proteins intimately involved in DNA replication are lethal if they are completely deleted (25). It is unknown if these patients express a truncated MCPH1 protein product. Genetic and biochemical studies with these patient cell lines will help to investigate the role of MCPH1 in ATR-dependent pathways.
We have demonstrated the importance of the C-BRCT domains of MCPH1 for foci formation in response to IR. Little is known, however, of the possible function of the N-BRCT domain of MCPH1. Mutation of the conserved tryptophan residue in the N-BRCT domain did not affect foci formation after IR (Fig. 3B). A recent report describes a missense mutation in the N-BRCT domain that changes a conserved Thr to Arg (T27R) in the BRCT domain (26). What is interesting is that the patient had a mild clinical and cellular phenotype compared with known mutant alleles of MCPH1 that are truncating mutations. It is, therefore, possible that both the N- and C-terminal BRCT domains are implicated in the clinical and PCC-like phenotype that is a hallmark of these cells.
MCPH1 is important for normal brain development and possibly tumor suppression (6). It is important to note that the majority of mutations found in MCPH1 patients harbor premature stop codons, deleting the C-terminal BRCT domains. Thus, the patients harbor mutations that would affect their ability to go to the sites of DNA damage and exert their checkpoint functions. Currently, there is no report of primary microcephaly patients developing tumors, similar to ATR-Seckel syndrome. Because of both the recent identification of these patients and with a small number of cases, future genetic studies will be crucial in determining if these patients are predisposed toward tumorigenesis. However, a recent study has reported loss of heterozygosity at the MCPH1 locus in breast cancer and deregulation of MCPH1 expression levels in some breast cancer cell lines (6). Future studies of MCPH1 knock-out mice will help to determine whether MCPH1 is a potential tumor suppressor in vivo.
Acknowledgments
We thank all past and present members of the Chen laboratory, especially Ja-Eun Kim, Zhenkun Lou, and Michael Huen for helpful discussions and reagents.
Footnotes
- IR
- ionizing radiation
- ATM
- ataxia-telangiectasia-mutated
- PTIP
- Pax2 transactivation-interacting protein
- MCPH1
- microcephalin
- MDC1
- mediator of DNA damage checkpoint protein 1
- Gy
- gray
- DAPI
- 4′,6-diamidino-2-phenylindole
- siRNA
- small interfering RNA
- WT
- wild type
- MEF
- mouse embryonic fibroblast
- PBS
- phosphate-buffered saline
- ITC
- isothermal titration calorimetry
This work was supported in part by National Institute of Health Grants CA092312 and CA100109 (to J. C.) and CA109449 (to G. M.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Bartek J, Lukas J. Cancer Cells. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 2.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr., Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 3.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 4.Xu X, Lee J, Stern DF. J. Biol. Chem. 2004;279:34091–34094. doi: 10.1074/jbc.C400139200. [DOI] [PubMed] [Google Scholar]
- 5.Lin SY, Rai R, Li K, Xu ZX, Elledge SJ. Proc. Natl. Acad. Sci. U. S. A. 2005;102:15105–15109. doi: 10.1073/pnas.0507722102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rai R, Dai H, Multani AS, Li K, Chin K, Gray J, Lahad JP, Liang J, Mills GB, Meric-Bernstam F, Lin SY. Cancer Cell. 2006;10:145–157. doi: 10.1016/j.ccr.2006.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alderton GK, Galbiati L, Griffith E, Surinya KH, Neitzel H, Jackson AP, Jeggo PA, O’Driscoll M. Nat. Cell Biol. 2006;8:725–733. doi: 10.1038/ncb1431. [DOI] [PubMed] [Google Scholar]
- 8.Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT, Alt FW, Chen J. Mol. Cell. 2006;21:187–200. doi: 10.1016/j.molcel.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 9.Maser RS, Zinkel R, Petrini JH. Nat. Genet. 2001;27:417–421. doi: 10.1038/86920. [DOI] [PubMed] [Google Scholar]
- 10.Yu X, Fu S, Lai M, Baer R, Chen J. Genes Dev. 2006;20:1721–1726. doi: 10.1101/gad.1431006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rappold I, Iwabuchi K, Date T, Chen J. J. Cell Biol. 2001;153:613–620. doi: 10.1083/jcb.153.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chini CC, Wood J, Chen J. Oncogene. 2006;25:4165–4171. doi: 10.1038/sj.onc.1209447. [DOI] [PubMed] [Google Scholar]
- 13.Peschiaroli A, Dorrello NV, Guardavaccaro D, Venere M, Halazonetis T, Sherman NE, Pagano M. Mol. Cell. 2006;23:319–329. doi: 10.1016/j.molcel.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 14.Yu X, Chen J. Mol. Cell. Biol. 2004;24:9478–9486. doi: 10.1128/MCB.24.21.9478-9486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Au WW, Henderson BR. J. Biol. Chem. 2005;280:6993–7001. doi: 10.1074/jbc.M408879200. [DOI] [PubMed] [Google Scholar]
- 16.Yu X, Chini CC, He M, Mer G, Chen J. Science. 2003;302:639–642. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 17.DiTullio RA, Jr., Mochan TA, Venere M, Bartkova J, Sehested M, Bartek J, Halazonetis TD. Nat. Cell Biol. 2002;4:998–1002. doi: 10.1038/ncb892. [DOI] [PubMed] [Google Scholar]
- 18.Jowsey PA, Doherty AJ, Rouse J. J. Biol. Chem. 2004;279:55562–55569. doi: 10.1074/jbc.M411021200. [DOI] [PubMed] [Google Scholar]
- 19.Goldberg M, Stucki M, Falck J, D’Amours D, Rahman D, Pappin D, Bartek J, Jackson SP. Nature. 2003;421:952–956. doi: 10.1038/nature01445. [DOI] [PubMed] [Google Scholar]
- 20.Lou Z, Minter-Dykhouse K, Wu X, Chen J. Nature. 2003;421:957–961. doi: 10.1038/nature01447. [DOI] [PubMed] [Google Scholar]
- 21.Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- 22.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. Cell. 2005;123:1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 23.Garshasbi M, Motazacker MM, Kahrizi K, Behjati F, Abedini SS, Nieh SE, Firouzabadi SG, Becker C, Ruschendorf F, Nurnberg P, Tzschach A, Vazifehmand R, Erdogan F, Ullmann R, Lenzner S, Kuss AW, Ropers HH, Najmabadi H. Hum. Genet. 2006;118:708–715. doi: 10.1007/s00439-005-0104-y. [DOI] [PubMed] [Google Scholar]
- 24.O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. Nat. Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- 25.Brown EJ, Baltimore D. Genes Dev. 2000;14:397–402. [PMC free article] [PubMed] [Google Scholar]
- 26.Trimborn M, Richter R, Sternberg N, Gavvovidis I, Schindler D, Jackson AP, Prott EC, Sperling K, Gillessen-Kaesbach G, Neitzel H. Hum. Mutat. 2005;26:496. doi: 10.1002/humu.9382. [DOI] [PubMed] [Google Scholar]