

Abstract
Background
Our previous studies have shown that the ciliary beat frequency (CBF) of cultured ciliated airway epithelial cells exposed to chronic ethanol fails to increase in response to β-agonist stimulation. This loss of the ciliary “flight response” correlates with an ethanol-mediated desensitization of adenosine 3’:5’-cyclic monophosphate-dependent protein kinase (PKA), a known regulatory component of CBF stimulation. We hypothesized that a similar ethanol-mediated desensitization of CBF would occur in vivo.
Methods
Sprague Dawley® rats were fed a liquid diet containing various concentrations of ethanol for 1 or 5 weeks. Half were exposed to cigarette smoke for 12 weeks and half were sham exposed. Animals were killed and tracheal epithelial cells analyzed for CBF and PKA activity.
Results
Baseline CBF (~6Hz) was unchanged in tracheal epithelial cells of rats consuming diets containing 0–36% ethanol for 5 weeks. Isoproterenol stimulated CBF to 12 to 13 Hz in the tracheal epithelial cells of control rats not administered ethanol. However, isoproterenol stimulation of CBF was blunted to 7.5 Hz in rats eating a 26% ethanol diet, and there was no stimulation of CBF in rats fed a diet containing 36% ethanol. Similarly, isoproterenol stimulated a 2- to 3-fold increase in PKA activity in control rats, but this PKA response to isoproterenol was blunted in rats fed increasing concentrations of ethanol. No isoproterenol-stimulated PKA response was observed in rats fed 36% ethanol. No ethanol-induced changes in cyclic guanosine monophosphate-dependent protein kinase or protein kinase C were observed in the rats’ tracheal epithelial cells. Cigarette smoke exposure slightly elevated baseline CBF and lowered the ethanol consumption level for isoproterenol-desensitization of CBF and PKA activation to 16%. No isoproterenol desensitization was observed after 1 week of alcohol feeding. Furthermore, 36% ethanol-feeding for 1 week stimulated rat tracheal CBF and PKA.
Conclusion
These data demonstrate that in vivo administration of ethanol to rats results in decreased ciliary beating and the desensitization of PKA. This suggests a mechanism for mucociliary clearance dysfunction in alcoholics.
Keywords: Cilia, Protein Kinase, Rat Model, β-Agonist, Cigarette Smoke
Alcoholism causes increased susceptibility to lung infections and pulmonary disease (Krumpe et al., 1984; Moss and Burnham, 2003). Studies have suggested that an increased risk of pneumonia is associated with high alcohol consumption (Nelson et al., 1991; Reynolds, 1995). However, the role that cigarette smoking plays in alcohol-related lung disease remains understudied. Current population studies report that 23.3% of adults in the United States smoke cigarettes (Cigarette Smoking Among Adults, 2002). Approximately 30% of these smokers are alcoholics (Miller and Gold, 1998). Indeed, reports linking cigarette smoking and alcohol abuse have suggested that as many as 95% of alcoholics are smokers (Patten et al., 1996). Therefore, research into the effects of alcohol on lung function must include a consideration of cigarette smoke as a confounding factor.
Normal lung defense against bacterial inhalation is based on the maintenance of the mucociliary apparatus to provide a first-line barrier of protection. This mucociliary apparatus consists of mucus production and secretion, in concert with orchestrated cilia beating, resulting in lung clearance of aspirated microorganisms and inhaled particles. Mucociliary clearance is highly regulated by pH, temperature, mucus content, viscosity, particle stimulation, and chemical modulators such as β-agonists (Foster et al., 1976). This dynamic organelle regulation results in slow ciliary beating during a resting state and increased ciliary beating to enhance airway clearance at times of stimulation or stress. The increased burden of mucociliary defense during lung infections such as bacterial bronchitis and aspiration pneumonia is a common complication of heavy alcohol ingestion.
We have previously reported the in vitro effects of ethanol on airway ciliary motility (Sisson, 1995). We observed that ethanol acutely elevates ciliary motility in an in vitro cell culture model. In this model, ethanol requires an adenosine 3’:5’-cyclic monophosphate (cAMP) and nitric oxide-dependent signaling pathway (Sisson et al., 1999; Wyatt et al., 2003). This follows the observation that either cAMP or cyclic guanosine monophosphate (cGMP) elevations result in increased cilia beating (Wyatt et al., 1998). However, continued exposure to ethanol eventually results in a return to baseline, unstimulated levels of cilia beating as potentially mediated via the action of a cAMP-phosphodiesterase (Forgèt et al., 2003). Once ciliated cells have been chronically exposed to ethanol in this manner, repeat challenges with ethanol fail to stimulate ciliary motility increase. Furthermore, such chronic ethanol exposure desensitizes the cells even to β-agonist stimulated increases in cilia motility (Wyatt and Sisson, 2001).
Although well characterized in an in vitro tissue culture cell line, the desensitization of ciliary beating in response to chronic ethanol has not yet been demonstrated in vivo. We hypothesized that animals fed a diet containing a significant amount of ethanol would no longer respond to β-agonist challenge with increased ciliary beating compared with the same animals fed a control diet. Furthermore, we hypothesized that cigarette smoke exposure in the same alcohol-fed animals would augment this ethanol-dependent desensitization of ciliary beat. Such findings would suggest that the increased presence of bacterial colonization in the lungs of alcoholics could be in part related to the mechanism of chronic ethanol-induced ciliary desensitization.
METHODS
Animal Model
Adult male Sprague Dawley® rats were obtained from Charles River Labs (Kingston, NY) at an age of 4 to 5 weeks (100–125 g). The rats were maintained in group cages with an ad libitum diet of tap water and rat chow (Purina, St. Louis, MO) for a 1 week adjustment period before initiating any study protocols. Animals were maintained at the Omaha VA Medical Center animal facility approved by the American Association for the Accreditation of Laboratory Animal Care (AAALAC). All animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996).
Rat Exposure to Alcohol and Cigarette Smoke
Rats were treated with passive cigarette smoke using a whole-body smoke exposure system (Teague Enterprises, Davis, CA) as described elsewhere (Gentry-Nielsen, et al., in press). This exposure system involved the simultaneous smoke exposure of 32 cigarettes to 30 rats. The smoke delivery occurred for a period of 1 hr twice a day during the weekdays and once a day on weekends for a total of 12 weeks. The rats were acclimated to this maximum exposure over a 4-day period. Using the Teague device (Teague et al., 1994), both mainstream and sidestream smoke were combined before delivery to the animal exposure chamber. Reference research cigarettes (1R4F) from the University of Kentucky (Lee et al., 1993) were used in this study. Control animals were sham exposed in a similar chamber to room air only. Smoke exposure was monitored by measurement of total suspended particles produced by the machine and blood carboxyhemoglobin and urine cotinine levels in the rats as described (Gentry-Nielsen, et al., in press).
After the smoke exposure period, both smoke-exposed and sham-exposed rats were separated into individual cages and acclimated over a 3 day period to the control Lieber-DeCarli high-fat liquid diet (Lieber and DeCarli, 1994). These rats were further divided into four feeding groups receiving 0, 16, 26, or 36% of their daily caloric intake as ethanol (Fig. 1). Blood ethanol levels were monitored twice for each animal during the treatment using a commercially available kit (Sigma Chemical, St. Louis, MO). These eight distinct groups were then maintained on their respective exposures for an additional 1 or 5 weeks before sacrifice.
Fig. 1.
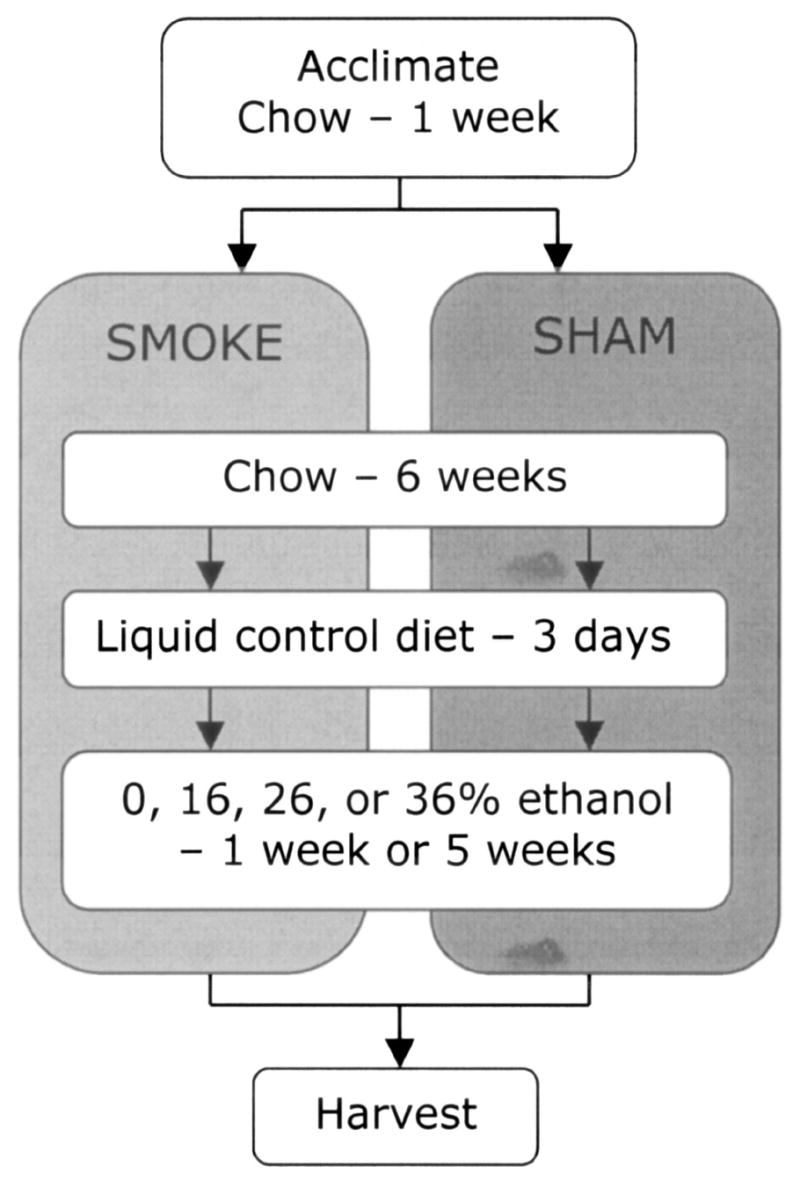
Time line of rat cigarette smoke exposure and dietary-ethanol treatments.
Tissue Preparation
Rats were killed by intraperitoneal injection of 75 mg/kg body weight of Nembutal (Abbott Labs, Chicago, IL). Tracheas were excised and placed in sterile media 199 (M-199; GIBCO, Chagrin Falls, OH) at room temperature. Using a dissection stereo-microscope, thin rings (~1 mm) of trachea were sliced using a sterile scalpel. Tracheal rings were then equilibrated for 30 min at 37°C in humidified 95% air/5% CO2 before treatment in the presence or absence of 100 μM isoproterenol. Tracheal rings were measured for ciliary beat frequency as described subsequently. The remaining trachea were opened by a longitudinal incision and washed in M-199, and epithelial cells were extracted with a sterile cell lifter (Fisher, Springfield, NJ). Extracted tracheal epithelial cells were flash frozen with liquid nitrogen in a cell lysis buffer as previously described (Wyatt et al., 1998).
Determination of Cyclic Nucleotide-Dependent Kinase Activity
PKA activity was determined in crude whole-cell fractions of bronchial epithelial cells. The assay used is a modification of procedures previously described (Jiang et al., 1992) using 130 μM protein kinase (PKA) substrate heptapeptide (LRRASLG), 10 μM cAMP, 0.2 mM isobutylmethyxanthine, 20 mM magnesium-acetate, and 0.2 mM [g-32P] adenosine triphosphate in a 40 mM Tris-HCl buffer (pH 7.5). The cGMP-dependent protein kinase (PKG) activity was assayed in a similar manner to PKA, with the substitution of the peptide RKRSRAE for the heptapeptide substrate, the addition of 10 μM cGMP, and the presence of protein kinase inhibitor peptide (PKI). Samples (20 μl) were added to 50 μl of this reaction mixture and incubated for 15 min at 30°C. Incubations were halted by spotting 50 μl of each sample onto P-81 phosphocellulose papers (Whatman, Hillsboro, OR). Papers were then washed five times for 5 min each in phosphoric acid (75 mM), washed once in ethanol, dried, and counted in nonaqueous scintillant as previously described (Roskoski, 1983). Negative controls consisted of similar assay samples with or without the appropriate substrate peptide or cyclic nucleotide. A positive control of 0.4 ng/ml purified catalytic subunit from type I bovine PKA (Promega, Madison, WI) was included as a sample. Kinase activity was expressed in relationship to total cellular protein assayed and calculated in pmol/min/mg. All samples were assayed in triplicate, and no less than three separate experiments were performed per unique parameter. Data were analyzed for statistical significance using one-way ANOVA, and comparisons between each of the two groups were made using Student’s paired t test (GraphPad Prism, San Diego, CA).
Ciliary Beat Frequency (CBF) Measurements
Actively beating ciliated cells were observed and their motion quantified by measuring CBF using an Olympus IMT-2 inverted phase-contrast microscope with a ×20 objective lens and a ×1.5 tube multiplier (Olympus, Melville, NY). Ciliated cells were digitally analyzed using the Sisson-Ammons Video Analysis (SAVA) system (Sisson et al., 2003). The digital image signal was routed from the camera directly into an IMAQ OCI/PXI-1422 digital image acquisition board (National Instruments, Austin, TX) within a Dell Precision 420 PC. Ciliated cells in culture were maintained at a constant temperature (24 ± 0.5°C) by a thermostatically controlled heated stage because temperature is known to affect CBF (Sanderson et al., 1992). Whole field analysis was performed using software (Ammons Engineering, Mt. Morris, MI) that automatically analyzed the entire captured image of all ciliated cells in a given field. For all experiments, the digital sampling rate was set at 85 frames/sec. The whole field analysis records and labels “motile” zones (having a frequency ≥2 Hz) or “nonmotile” zones (having a frequency <2 Hz). All of the motile whole field zones amplitudes and frequencies were collated, mapped to the screen image, and statistically analyzed to determine the frequency average, standard deviation, and standard error of the entire image. The predominant frequency of a cilium or small group of cilia was viewed and each sampled in at least six separate fields. All frequencies are expressed as mean ± SEM from six separate fields. Baseline CBF was recorded in multiple tracheal rings for each rat treatment condition. Each ring was subsequently stimulated with 100 μM isoproterenol for 30 min and CBF was remeasured. CBF from the same trachea treated with media versus isoproterenol was statistically analyzed using paired t test and post hoc testing (GraphPad Prism, San Diego, CA). Significance was assigned at p ≤ 0.05. All frequencies represent the mean ± one SE of the mean from six separate cell groups or fields.
RESULTS
Ethanol-Feeding Blocks Isoproterenol-Stimulated Cilia Beating in Rat Tracheal Epithelium
Previous results demonstrated that in vitro exposure of airway epithelial cells to chronic ethanol results in the desensitization of β-agonist-stimulated CBF (Wyatt and Sisson, 2001). To determine if chronic ethanol desensitized CBF in vivo, CBF was measured in rats fed various concentrations of ethanol in their diet for a period of 5 weeks. Increasing concentrations of ethanol in the rats’ diet did not significantly alter baseline CBF. Isoproterenol significantly stimulated CBF in tracheal rings obtained from rats fed a liquid diet containing 0% or 16% ethanol, in which mean blood ethanol levels were <5 mg/dL (Fig. 2). However, isoproterenol did not elevate CBF significantly from baseline beating levels in rats fed 26% and 36% ethanol diets. Mean blood alcohol levels from rats eating those diets were elevated >30 mg/dL and >100 mg/dL, respectively. These data indicate that chronic ethanol-mediated desensitization of the β-agonist cilia stimulation response does occur in in vivo ethanol exposure models.
Fig. 2.
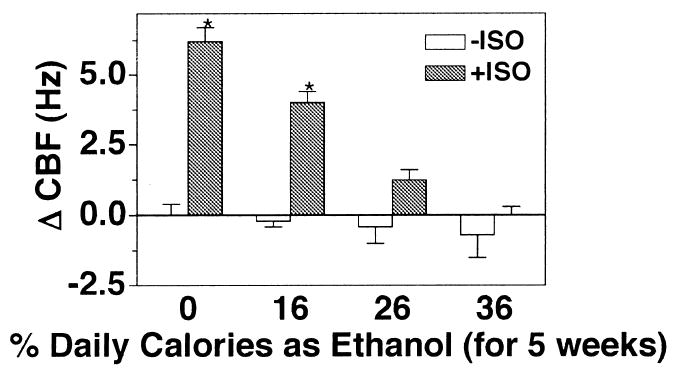
Epithelial cell CBF in tracheal rings of ethanol-fed rats. Rats were fed a liquid diet containing 0, 16, 26, or 36% ethanol for 5 weeks and were killed. Excised tracheal rings were assayed for ciliary beat frequency (CBF) in the absence (white bars) and presence (gray bars) of 100 μM isoproterenol (ISO) for 30 min. Bar caps represent SEM of four different animals. Data from each rat represent the average of 10 or more tracheal rings (*p ≤ 0.05 for tracheal rings treated with isoproterenol for 30 min compared with those treated with media only).
Cigarette Smoke Exposure Augments Ethanol-Mediated Desensitization of PKA
Because acetaldehyde has been shown to decrease CBF (Wyatt et al., 2000), and both cigarette smoke and ethanol metabolism result in the elevation of lung acetaldehyde, half of the rats in this study were exposed to cigarette smoke in combination with ethanol feeding to determine the effects of such exposure on isoproterenol-stimulated CBF. In sham-exposed animals fed a liquid control diet (containing no ethanol), isoproterenol significantly stimulated CBF over baseline (Fig. 3). In smoke-exposed rats fed a liquid control diet, isoproterenol-stimulated CBF was reduced but not significantly inhibited compared with sham-exposed animals. As demonstrated in Fig. 2, increasing ethanol concentrations in the rat diet resulted in decreasing tracheal ring responsiveness to isoproterenol in the sham-exposed animals. In smoke-exposed rats, a significant isoproterenol-stimulated CBF response was inhibited by the 16% ethanol diet, whereas in the sham-exposed rats isoproterenol-desensitization did not occur until they were fed at least 26% ethanol calories. Interestingly, smoke-exposed rats not stimulated with isoproterenol demonstrated an elevated baseline CBF across all dietary ethanol concentrations. These data indicate that the combination of cigarette smoke and ethanol results in a greater inhibition of isoproterenol-stimulated CBF than either exposure alone.
Fig. 3.
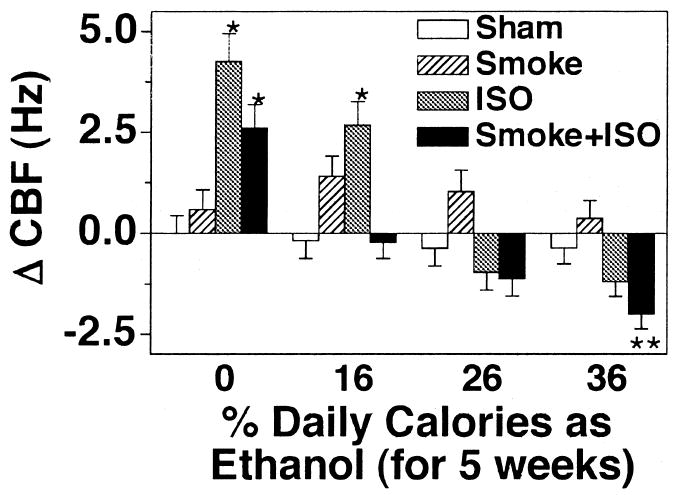
Tracheal epithelial CBF in cigarette smoke-exposed and ethanol-fed rats. Rats fed various concentrations of ethanol in their diets for 5 weeks were either sham exposed (white and gray bars) or cigarette smoke exposed (hatched and black bars) for a total of 12 weeks. Excised tracheal rings were assayed for ciliary beat frequency (CBF) in the absence (white and hatched bars) and presence (gray and black bars) of 100 μM isoproterenol (ISO) for 30 min. Bars represent SEM of six different rats. Data from each rat represent the average of 10 or more tracheal rings (*p ≤ 0.05 for tracheal rings treated with isoproterenol for 30 min compared with those treated with media only).
PKA Activity Decreases With Increasing Ethanol Concentration in Diet
Ethanol stimulates CBF via a dual signaling pathway involving PKG and PKA (Wyatt et al., 2003). Chronic ethanol intake results in the inactivation of PKA in tissue culture models of ethanol exposure (Wyatt and Sisson, 2001). To determine if chronic ethanol ingestion alters PKA in vivo, we assayed tracheal epithelial cell PKA activity from sham-exposed rats fed various concentrations of ethanol in their diet for 5 weeks. Increasing concentrations of ethanol in the rat diet resulted in a general decrease in the baseline activity of PKA (data not shown). A significant reduction in baseline PKA activity was observed at 36% ethanol feeding. No significant differences in PKA activity were observed between sham- and smoke-exposed groups within an alcohol-feeding group. To determine if the expression of PKA was decreased in response to chronic 36% ethanol in the rat diet, total cAMP-stimulatable PKA was assayed in vitro (Fig. 4). An equivalent amount of PKA activity was detected in all animal treatment conditions when the tracheal epithelial cell extracts were assayed in the presence of 10 μM cAMP. This suggests that the amount of PKA contained in the cells remains constant, whereas the ability of the PKA to be activated in situ varies among ethanol exposures. Isoproterenol-stimulated PKA activity paralleled CBF, with decreased isoproterenol-stimulated PKA in response to increasing ethanol in the diet, which was demonstrated at a lower ethanol level in smoke-exposed than in sham-exposed animals (Fig. 5). Consistent with in vitro data, no changes in PKG activity in baseline or isoproterenol-stimulated trachea were observed in response to ethanol or smoke exposure (data not shown).
Fig. 4.
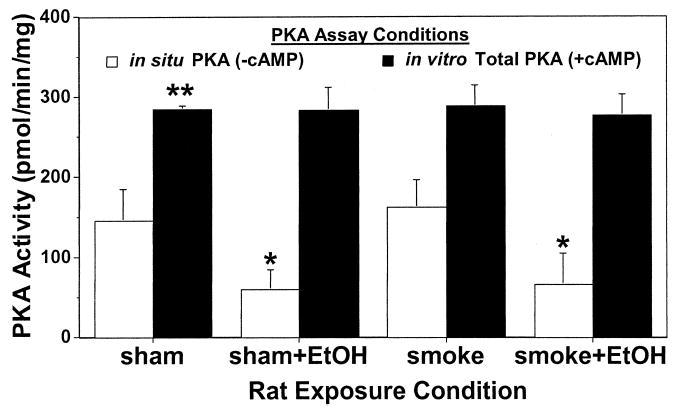
Comparison of total in vitro PKA activity versus in situ PKA activity from rats exposed to cigarette smoke and ethanol feeding. Rats fed 36% ethanol in their diets for 5 weeks were either sham exposed or cigarette smoke exposed for a total of 12 weeks. Tracheal epithelial cells were extracted and assayed for PKA activity in the presence (black bars) or absence (white bars) of 10 μM cAMP. Bars represent SEM of total tracheal epithelial cell PKA from six different rats (*p ≤ 0.05 for cells from ethanol-fed rats compared with control liquid diet-fed rats and **p ≤ 0.01 for cells assayed in the presence of cAMP versus the absence of cAMP).
Fig. 5.
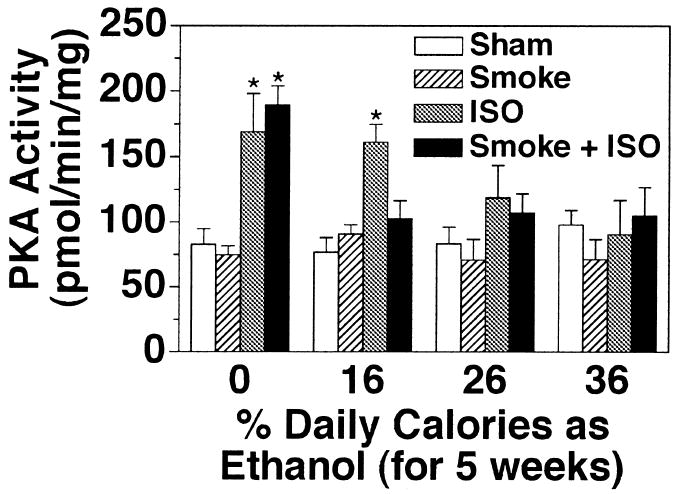
Isoproterenol-stimulated tracheal epithelial PKA activity in cigarette smoke-exposed and ethanol-fed rats. Rats fed various concentrations of ethanol in their diets for 5 weeks were either sham exposed (white and gray bars) or cigarette smoke exposed (hatched and black bars) for a total of 12 weeks. Tracheal epithelial cells were extracted and assayed for PKA activity in the absence (white and hatched bars) and presence (gray and black bars) of 100 μM isoproterenol (ISO) for 30 min. Bars represent SEM of total tracheal epithelial cell PKA from six different rats (*p ≤ 0.05 for trachea treated with isoproterenol for 30 min compared with trachea treated with media only).
Acute Ethanol Exposure Activates PKA
In contrast to chronic ethanol treatment of cells in vitro, short in vitro exposure times of airway epithelial cells to ethanol result in the rapid activation of PKA and increase in CBF (Sisson et al., 1999). To test this in vitro observation in the animal model, rats were sham- or smoke-exposed and fed various concentrations of ethanol for the last 1 week of the total treatment rather than the last 5 weeks. In sham- and smoke-exposed animals fed an ethanol-free liquid control diet, isoproterenol significantly stimulated CBF over baseline (Fig. 6). Increasing ethanol concentrations in the rat diet for 1 week resulted in no significant changes in isoproterenol-stimulated CBF in either the sham- or smoke-exposed animal. In smoke-exposed rats, some reduction of isoproterenol-stimulated CBF occurred at the highest ethanol concentration (36%). However, baseline (without isoproterenol) CBF in sham-exposed rats demonstrated an ethanol dose-dependent increase. As observed previously, baseline CBF in tracheal rings from smoke-exposed rats not stimulated with isoproterenol was elevated across all dietary ethanol concentrations with the highest CBF levels at 36% ethanol. Tracheal ciliated cells of chow-fed control rats demonstrated the same relative baseline and isoproterenol CBF responses as liquid diet control rats. Similar to what was determined after a 5-week ethanol exposure, PKA activity paralleled CBF in the 1-week ethanol-treated animals (Fig. 7). Isoproterenol-stimulated PKA was not blocked by 1 week of ethanol feeding, but 36% ethanol alone activated PKA. These data suggest that a more acute ethanol exposure fails to desensitize rat tracheal CBF to isoproterenol stimulation but rather results in the stimulation of baseline CBF.
Fig. 6.
Isoproterenol-stimulated tracheal epithelial CBF in sham- or cigarette smoke-exposed and ethanol-fed rats. Rats sham exposed (white and gray bars) or cigarette smoke exposed (hatched and black bars) for a total of 12 weeks were fed various concentrations of ethanol in their diets for the final 1 week. Excised tracheal rings were assayed for ciliary beat frequency (CBF) in the absence (white and hatched bars) and presence (gray and black bars) of 100 μM isoproterenol (ISO) for 30 min. Bars represent SEM of six different rats. Data from each rat represent the average of 10 or more tracheal rings (*p ≤ 0.05 for tracheal rings treated with isoproterenol for 30 min compared with those treated with media only; **p ≤ 0.05 for tracheal rings from rats fed diets containing 36% vs. 0% ethanol).
Fig. 7.
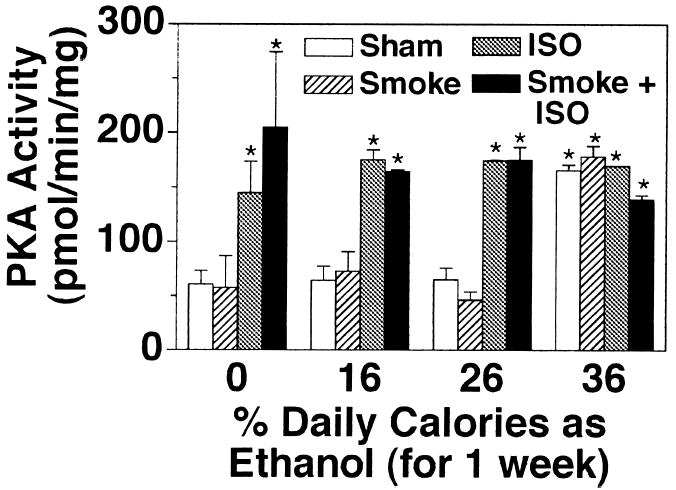
Isoproterenol-stimulated tracheal epithelial PKA activity in cigarette smoke-exposed and ethanol-fed rats. Rats sham exposed (white and gray bars) or cigarette smoke exposed (hatched and black bars) for a total of 12 weeks were fed various concentrations of ethanol in their diets for the final 1 week. Tracheal epithelial cells were extracted and assayed for PKA activity in the absence (white and hatched bars) and presence (gray and black bars) of 100 μM isoproterenol (ISO) for 30 min. Bars represent SEM of total tracheal epithelial cell PKA from six different rats (*p ≤ 0.05 for trachea treated with isoproterenol for 30 min compared with trachea treated with media only and rats fed diets containing 36% vs. 0% ethanol).
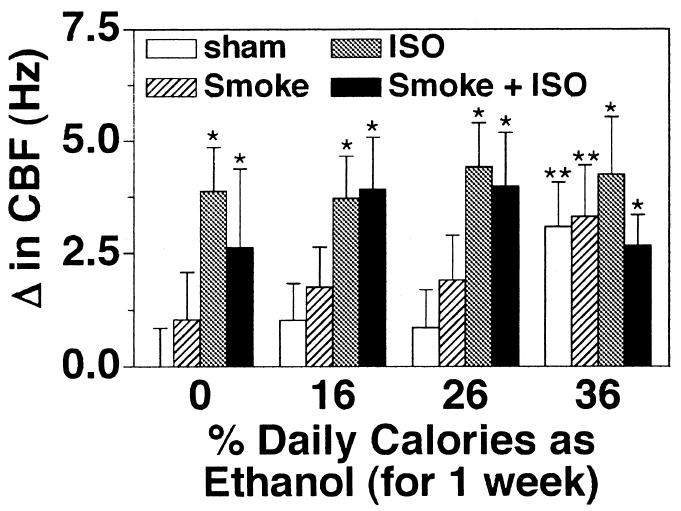
Ethanol-Mediated Desensitization to Isoproterenol Over Time
Our results indicate that 1 week of 36% ethanol calories in a rat diet does not alter the ability of the cilia to be stimulated by β-agonist. In contrast, 5 weeks of 36% ethanol calories in the diet inhibits isoproterenol-stimulated increases in CBF. To determine the time course of the onset of this desensitization response, rats were fed 36% of their daily calories as ethanol for 1, 2, 3, 4, and 5 weeks before they were killed and before measurement of the ciliary beating in the tracheal rings. Isoproterenol stimulated significant increases in CBF in control and ethanol-fed rats after 2 weeks but not in rats fed ethanol for 3 weeks or longer (Fig. 8). The elevation of baseline CBF observed in 1 week ethanol-fed rats was absent after 2 weeks of 36% ethanol in the diet even though the CBF of these animals was still elevated by isoproterenol (data not shown). These data would suggest that the transition from ethanol-stimulated increases in CBF to ethanol-mediated β-agonist desensitization occurs in rats between 1 and 3 weeks of exposure to 36% ethanol in their diet.
Fig. 8.
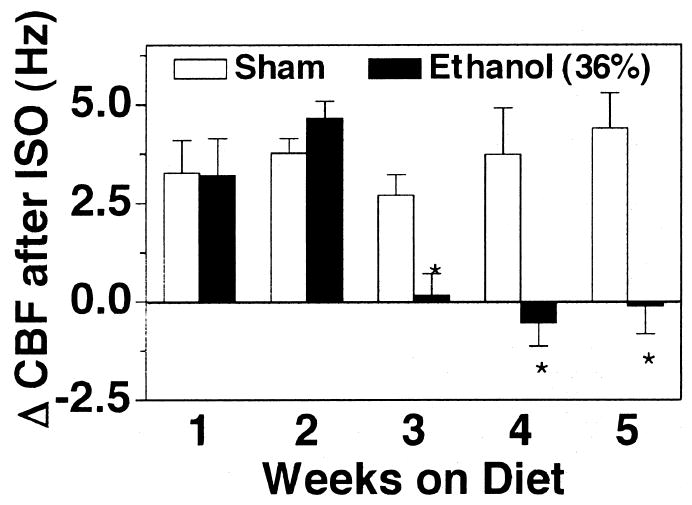
Desensitization of isoproterenol-stimulated CBF in rat tracheal rings exposed to ethanol over time. Rats were fed a liquid diet containing 0 (white bars) or 36% ethanol (black bars) for 1–5 weeks and sacrificed. Excised tracheal rings were assayed for ciliary beat frequency (CBF) in the presence of 100 μM isoproterenol (ISO) for 30 min. Bar caps represent SEM of six different animals. Data from each rat represent the average of 10 or more tracheal rings (*p ≤ 0.05 for rats fed 36% of their daily calories as ethanol compared with those sham-treated with liquid diet only).
DISCUSSION
In these studies, we have described the short-term ethanol stimulation and chronic desensitization of airway epithelial cell CBF and PKA activation in an animal model. This is the first in vivo report of ethanol-mediated desensitization to the effects of isoproterenol on ciliated cells. We observed a significant decline in the ability of isoproterenol to stimulate increased CBF in the tracheal ciliated epithelium of rats fed a diet containing 26% to 36% ethanol calories for 5 weeks. This observation parallels our previous report that chronic ethanol exposure of airway epithelial cells in vitro desensitizes the cells to β-agonist-stimulated increases in CBF (Wyatt and Sisson, 2001). In the present studies, elevated blood alcohol levels corresponded to decreased responsiveness to isoproterenol. Only 27% of the 16% ethanol-fed animals demonstrated measurable blood alcohol levels. CBF in these rats likewise remained highly responsive to isoproterenol (Fig. 1). However, we did observe elevated blood alcohol levels in some rats on a diet of 26% to 36% ethanol. Fifty-five percent of the 26% ethanol-fed rats and 98% of the 36% ethanol-fed rats had measurable blood alcohol levels. These animals also exhibited a decreased CBF response to isoproterenol. Although rats may metabolize ethanol at different rates, the presence of chronic blood ethanol levels equates with cilia desensitization.
Combining cigarette smoke exposure with ethanol feeding in rats further decreases the CBF responsiveness to isoproterenol. In those rats not ingesting alcohol, exposure to cigarette smoke for 12 weeks decreased isoproterenol-stimulated CBF. In smoke- and ethanol-treated animals, the isoproterenol-stimulated CBF also was desensitized at a lower dietary ethanol concentration (16%) compared with sham-exposed animals, in which desensitization occurred only in rats ingesting the 26% to 36% ethanol diets. There appears to be no additional reduction in the CBF response due to smoke exposure at these higher ethanol concentrations. Interestingly, smoke exposure alone elevates baseline CBF compared with sham-exposed rats. This was consistent across all ethanol concentrations in the rat diet. One possible explanation is that elevated particle exposure from the inhalation of cigarette smoke results in CBF elevation as previously reported (Lippmann et al., 1980; Wolff, 1986). It remains unclear why the presence of isoproterenol under these conditions would abrogate smoke-induced elevated baseline beating. These observations suggest that under conditions of cigarette smoke and β-agonist exposure, a combination of CBF signaling mechanisms results in decreased ciliary response to particulate matter. Further studies on cigarette smoke signal transduction pathways in cilia beating mechanisms are required to fully understand this observation.
The loss of isoproterenol-mediated increases in CBF is associated with a loss of isoproterenol-stimulatable PKA activity in the rat tracheal epithelial cells. In the ethanol-fed rat, this appears to be due to an uncoupling of the signal pathway between the β-agonist and PKA rather than any down-regulation of the cAMP-binding enzyme itself. When the same epithelial cell extracts were assayed in vitro in the presence of exogenous cAMP, equal amounts of stimulatable PKA activity were detected across all alcohol and smoke treatment conditions. These data suggest that the intact cell pathway for isoproterenol-stimulated CBF is uncoupled by chronic ethanol exposure. This observation supports our in vitro cellular studies of intact ciliated epithelial CBF desensitization to isoproterenol after chronic ethanol exposure (Wyatt and Sisson, 2001). Results from similar in vitro studies have shown that the ethanol desensitization response is not exclusively regulated at the level of the β-adrenergic receptors (Wyatt and Sisson, 2001), phosphodiesterase activation (Forgèt et al., 2003), or proinflammatory cytokine production (Allen-Gipson et al., 2004). Instead, ethanol appears to acutely stimulate CBF via nitric oxide (Sisson, 1995) and cAMP-dependent (Sisson et al., 1999) pathways. The requirement for this dual pathway regulation suggests that the chronic uncoupling of β-agonist CBF stimulation by ethanol occurs at the level of PKG and PKA substrate phosphorylation on the ciliary axoneme (Wyatt et al., 2003). These observations explain the retention of total cellular PKA, but lack of isoproterenol-responsive CBF, in the ethanol-fed animal model.
Baseline CBF does not appear to be regulated by the PKA activity state. No significant differences existed in the baseline CBF and PKA levels between sham- or smoke-exposed 0% and 36% ethanol-fed rats. These whole animal exposure data support previous results in cellular studies suggesting that a noncyclic nucleotide pathway (Ca2+) also regulates cilia beating (Salathe and Bookman, 1995; Sanderson and Dirksen, 1989). Such a calcium-mediated pathway via PKC might be responsible for the change in baseline beating observed in response to the combination of isoproterenol and ethanol. Cyclic GMP and PKG also have been implicated as regulators of CBF (Wyatt et al., 1998; Li et al., 2000; Zagoory et al., 2002). In the rat model, we observed no significant changes in baseline or isoproterenol-stimulated PKG activity in the tracheal epithelial cells of smoke- or sham- exposed rats ingesting any ethanol concentration. However, the levels of cGMP and PKG activity are significantly lower than cAMP and PKA in airway epithelium (Wyatt et al., 2003), and functionally significant changes at a highly localized region of the cell may not be detectable by the whole-cell assay used.
The duration of ethanol exposure plays an important role in the rat model of ethanol desensitization to isoproterenol. A shorter ethanol-feeding regimen (1 week) resulted in tracheal epithelial cell CBF and PKA activity that remained responsive to isoproterenol-stimulation across all concentrations of ethanol in the diet. In addition, in this short-term feeding model, the highest ethanol concentration (36%) produced a significant baseline elevation in CBF compared with the other rat diets in both the presence and absence of smoke exposure. The combined isoproterenol and smoke-induced decrease in CBF is not observed except at the highest concentration of ethanol (36%) in the diet. This observation in the rat model again parallels the results observed in our in vitro cell treatments. Acute ethanol exposure of airway epithelial cells produces a rapid and transient stimulation of CBF via activation of nitric oxide and PKA (Sisson et al., 1999), whereas longer cell exposures to ethanol produce the β-agonist-desensitized state (Wyatt and Sisson, 2001).
In summary, these studies provide evidence for the existence of ethanol-mediated tracheal epithelial cell desensitization to β-agonists in a whole animal model. Our data indicate that chronic ethanol feeding results in desensitization, whereas short-term ethanol feeding does not. Cigarette smoke exposure augments the chronic ethanol desensitization effect but does not alter the acute ethanol effects observed on CBF and PKA. Additional studies are underway to determine how these ethanol- and smoke-induced alterations in CBF affect susceptibility to bacterial infections in the smoking alcoholic host. These observations support and validate our previous in vitro cultured epithelial cell models of alcohol’s effects on cilia beating and support a specific mechanism by which ethanol impairs airway host defenses.
Acknowledgments
Supported by Grants AA08769 (JHS) and AA10234 (MGN) from NIAAA and a Merit Review Grant from the Department of Veteran Affairs (TAW). TAW is an American Lung Association Career Investigator.
References
- Allen-Gipson D, Romberger DJ, Forget MA, May KL, Sisson JH, Wyatt TA. Il-8 inhibits isoproterenol-stimulated ciliary beat frequency in bovine epithelial cells. J Aerosol Med. 2004;17(2) doi: 10.1089/0894268041457138. [DOI] [PubMed] [Google Scholar]
- Cigarette smoking among adults–United States, 2000. MMWR Morb Mortal Wkly Rep. 2002;51:642–645. [PubMed] [Google Scholar]
- Forgèt M, Sisson JH, Spurzem JR, Wyatt TA. Ethanol increases phosphodiesterase 4 activity in bovine bronchial epithelial cells. Alcohol. 2003;31:31–38. doi: 10.1016/j.alcohol.2003.06.005. [DOI] [PubMed] [Google Scholar]
- Foster WM, Bergofsky EH, Bohning DE, Lippmann M, Albert RE. Effect of adrenergic agents and their mode of action on mucociliary clearance in man. J Appl Physiol. 1976;41:146–152. doi: 10.1152/jappl.1976.41.2.146. [DOI] [PubMed] [Google Scholar]
- Gentry-Nielsen MJ, Vander Top EA, Snitily MU, Casey CA, Preheim LC. Development of a rat model to determine the biomedical consequences of concurrent ethanol ingestion and cigarette smoke exposure. Alcoholism: Clin Exp Res. 2004 doi: 10.1097/01.alc.0000136383.45378.91. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Colbran JL, Francis SH, Corbin JD. Direct evidence for cross-activation of cGMP-dependent protein kinase by cAMP in pig coronary arteries. J Biol Chem. 1992;267:1015–1019. [PubMed] [Google Scholar]
- Krumpe PE, Cummiskey JM, Lillington GA. Alcohol and the respiratory tract. Med Clin North Am. 1984;68:201–219. doi: 10.1016/s0025-7125(16)31250-0. [DOI] [PubMed] [Google Scholar]
- Lee CK, Brown BG, Reed EA, Coggins CR, Doolittle DJ, Hayes AW. Ninety-day inhalation study in rats, using aged and diluted sidestream smoke from a reference cigarette: DNA adducts and alveolar macrophage cytogenetics. Fundam Appl Toxicol. 1993;20:393–401. doi: 10.1006/faat.1993.1051. [DOI] [PubMed] [Google Scholar]
- Li D, Shirakami G, Zhan X, Johns RA. Regulation of ciliary beat frequency by the nitric oxide-cyclic guanosine monophosphate signaling pathway in rat airway epithelial cells. Am J Respir Cell Mol Biol. 2000;23:175–181. doi: 10.1165/ajrcmb.23.2.4022. [DOI] [PubMed] [Google Scholar]
- Lieber CS, DeCarli LM. Animal models of chronic ethanol toxicity. Methods Enzymol. 1994;233:585–594. doi: 10.1016/s0076-6879(94)33061-1. [DOI] [PubMed] [Google Scholar]
- Lippmann M, Yeates DB, Albert RE. Deposition, retention, and clearance of inhaled particles. Br J Ind Med. 1980;37:337–362. doi: 10.1136/oem.37.4.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller NS, Gold MS. Comorbid cigarette and alcohol addiction: epidemiology and treatment. J Addict Dis. 1998;17:55–66. doi: 10.1300/J069v17n01_06. [DOI] [PubMed] [Google Scholar]
- Moss M, Burnham EL. Chronic alcohol abuse, acute respiratory distress syndrome, and multiple organ dysfunction. Crit Care Med. 2003;31(4 Suppl):S207–S12. doi: 10.1097/01.CCM.0000057845.77458.25. [DOI] [PubMed] [Google Scholar]
- National Research Council. Guide for the Care and Use of Laboratory Animals. National Academy Press; Washington DC: 1996. [Google Scholar]
- Nelson S, Bagby G, Andresen J, Nakamura C, Shellito J, Summer W. The effects of ethanol, tumor necrosis factor, and granulocyte colony-stimulating factor on lung antibacterial defenses. Adv Exp Med Biol. 1991;288:245–253. doi: 10.1007/978-1-4684-5925-8_28. [DOI] [PubMed] [Google Scholar]
- Patten CA, Martin JE, Owen N. Can psychiatric and chemical dependency treatment units be smoke free? J Subst Abuse Treat. 1996;13:107–118. doi: 10.1016/0740-5472(96)00040-2. [DOI] [PubMed] [Google Scholar]
- Reynolds HY. Pulmonary host defenses. Alcohol Clin Exp Res. 1995;19:6–10. doi: 10.1111/j.1530-0277.1995.tb01465.x. [DOI] [PubMed] [Google Scholar]
- Roskoski R., Jr Assays of protein kinase. Methods Enzymol. 1983;99:3–6. doi: 10.1016/0076-6879(83)99034-1. [DOI] [PubMed] [Google Scholar]
- Salathe M, Bookman RJ. Coupling of [Ca2+]i and ciliary beating in cultured tracheal epithelial cells. J Cell Sci. 1995;108:431–440. doi: 10.1242/jcs.108.2.431. [DOI] [PubMed] [Google Scholar]
- Sanderson MJ, Dirksen ER. Mechanosensitive and beta-adrenergic control of the ciliary beat frequency of mammalian respiratory tract cells in culture. Am Rev Respir Dis. 1989;139:432–440. doi: 10.1164/ajrccm/139.2.432. [DOI] [PubMed] [Google Scholar]
- Sanderson MJ, Lansley AB, Dirksen ER. Regulation of ciliary beat frequency in respiratory tract cells. Chest. 1992;101(3 Suppl):69S–71S. doi: 10.1378/chest.101.3_supplement.69s. [DOI] [PubMed] [Google Scholar]
- Sisson JH. Ethanol stimulates apparent nitric oxide-dependent ciliary beat frequency in bovine airway epithelial cells. Am J Physiol. 1995;268:L596–L600. doi: 10.1152/ajplung.1995.268.4.L596. [DOI] [PubMed] [Google Scholar]
- Sisson JH, May K, Wyatt TA. Nitric oxide-dependent ethanol stimulation of ciliary motility is linked to cAMP-dependent protein kinase (PKA) activation in bovine bronchial epithelium. Alcohol Clin Exp Res. 1999;23:1528–1533. [PubMed] [Google Scholar]
- Sisson JH, Stoner JA, Ammons BA, Wyatt TA. All-digital image capture and whole-field analysis of ciliary beat frequency. J Microsc. 2003;211:103–111. doi: 10.1046/j.1365-2818.2003.01209.x. [DOI] [PubMed] [Google Scholar]
- Teague S, Pinkerton K, et al. Sidestream cigarette smoke generation and exposure system for environmental tobacco smoke studies. Inhal Toxicol. 1994;6:79–93. [Google Scholar]
- Wolff RK. Effects of airborne pollutants on mucociliary clearance. Environ Health Perspect. 1986;66:223–237. doi: 10.1289/ehp.8666223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt TA, Forget MA, Sisson JH. Ethanol stimulates ciliary beating by dual cyclic nucleotide kinase activation in bovine bronchial epithelial cells. Am J Pathol. 2003;163:1157–1166. doi: 10.1016/S0002-9440(10)63475-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt TA, Schmidt SC, Rennard SI, Tuma DJ, Sisson JH. Acetaldehyde-stimulated PKC activity in airway epithelial cells treated with smoke extract from normal and smokeless cigarettes. Proc Soc Exp Biol Med. 2000;225:91–97. doi: 10.1046/j.1525-1373.2000.22511.x. [DOI] [PubMed] [Google Scholar]
- Wyatt TA, Sisson JH. Chronic ethanol downregulates PKA activation and ciliary beating in bovine bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;281:L575–L581. doi: 10.1152/ajplung.2001.281.3.L575. [DOI] [PubMed] [Google Scholar]
- Wyatt TA, Spurzem JR, May K, Sisson JH. Regulation of ciliary beat frequency by both PKA and PKG in bovine airway epithelial cells. Am J Physiol. 1998;275:L827–L835. doi: 10.1152/ajplung.1998.275.4.L827. [DOI] [PubMed] [Google Scholar]
- Zagoory O, Braiman A, Priel Z. The mechanism of ciliary stimulation by acetylcholine: roles of calcium, PKA, and PKG. J Gen Physiol. 2002;119:329–339. doi: 10.1085/jgp.20028519. [DOI] [PMC free article] [PubMed] [Google Scholar]