

Abstract
In this work, we search for coordination as an organizing principle in a complex signaling system using a multilevel hierarchical paradigm. The objective is to explain the underlying mechanism of Interferon (IFNγ) induced JAK-STAT (specifically JAK1/JAK2-STAT1) pathway behavior. Starting with a mathematical model of the pathway from the literature, we modularize the system using biological knowledge via principles of biochemical cohesion, biological significance, and functionality. The modularized system is then used as a basis for in silico inhibition, knockdown/deletion and perturbation experiments to discover a coordination mechanism. Our analysis shows that a module representing the SOCS1 complex can be identified as the coordinator. Analysis of the coordinator can then be used for the selection of biological experiments for the discovery of ’soft’ molecular drug targets, that could lead to the development of improved therapeutics. The coordinator identified is also being investigated to determine its relationship to pathological conditions.
Keywords: JAK-STAT, multilevel, hierarchical, coordination, systems biology, mathematical modeling
1 Introduction
Janus Kinase - Signal Transducer and Activator of Transcription (JAK-STAT) signaling, and specifically JAK1/JAK2-STAT1 considered in this work, is a highly conserved pathway implicated in numerous cellular processes from inflammatory response to hematopoiesis. Consequently, aberrant activation of this pathway can lead to pathological conditions. In particular, activation of STAT has been observed in various cancer cell lines and human tumor tissues including multiple myeloma, lymphomas, leukemia, and breast cancer (Buettner et al., 2002; Yu and Jove, 2004). The activation of STAT is usually followed by various inactivation mechanisms that further prevent undesirable gene transcription by STAT. Such activation/inactivation mechanisms are not intuitive and are poorly understood because the interacting proteins form a complex system.
In this study, we employ a complex systems biology approach based on a multilevel hierarchical paradigm to search for organizing principles. In particular we focus on coordination to explain the underlying mechanism of the Interferon γ (IFNγ) induced JAK-STAT pathway behavior. Results of our study establish a bridge from the general concept of coordination introduced in our earlier work (Mesarovic et al., 2004), particularized here to the JAK-STAT pathway behavior, and to the logical sequel of in vitro and in vivo experimentation. In our analysis we use mathematical modeling as a tool to gain an understanding of the pathway, but we do not use this for prediction.
Starting with a biochemical model of the pathway, we use knowledge of biological functionality to modularize the system. This forms the basis for in silico inhibition, knockdown/deletion, and perturbation experiments aimed at discovering a coordination mechanism. In addition to these in silico experimental results agreeing with biological data in the literature, we also show that a subsystem involving a multi-component Suppressors of Cytokine Signaling (SOCS1) complex is a coordinator for the pathway.
The identification of the SOCS1 complexes as a coordinator is used to guide the selection of biological experiments for the discovery of ’soft’ molecular drug targets. The hypothesis is that interruption and/or modification of these targets will lead to the development of improved therapeutics. It is expected that this will result in a future design of therapeutic strategies. Biological experiments are suggested to confirm the existence of the coordinator discovered in this work and to develop insight into the signaling and phenotype level behavior. The new behavior, identified from the in silico experiments, is also being investigated in its relationship to pathological conditions.
1.1 JAK-STAT Pathway Mechanism
In this study we focus our attention on the IFNγ-induced JAK1/JAK2-STAT1 pathway. In the absence of stimulus, STAT1 remains latent in the cytoplasm. Upon receptor activation by IFNγ stimulus, STAT1 is recruited to the receptor cytoplasmic domain and subsequently phosphorylated before forming a dimer through a signal cascade in the JAK-STAT pathway. As a dimer, STAT1 translocates to the nucleus so as to initiate transcription. The duration of STAT activation typically ranges from a few minutes to a couple of hours in normal physiological conditions, and it is found to be constitutively active in human tumor cell lines especially for STAT1, STAT3, and STAT5 (Buettner et al., 2002).
The STAT inactivation mechanism is more intricate and not yet fully elucidated. Identified negative regulatory mechanisms include: JAK-receptor complex endocytosis; dominant-negative STATs; down regulation through Protein Inhibitor of Activated STAT (PIAS), the Cytokine-Inducible SH2 containing protein (CIS)/SOCS/STAT-induced STAT Inhibitor (SSI) families of proteins, and the Tyrosine phosphatases (Ward et al., 2000). SOCS1 acts as a negative feedback with its gene expression being regulated by STAT1. Proposed mechanisms of SOCS1 mediated JAK-STAT inactivation include inhibiting JAK kinase activity and targeting signaling components for degradation (Lehmann et al., 2003). The Src-Homology2-containing phosphatase (SHP2) interacts with the pathway through dephosphorylation of activated JAK (Ward et al., 2000), or STAT (Chen et al., 2004; Yu et al., 2000; Chughtai et al., 2002; Chen et al., 2003). Depending on the ligands or ligand-binding receptors, SHP2 inhibits or promotes activation of different JAK kinases (You et al., 1999; Yu et al., 2003).
Ali et al. (2003) demonstrate that SHP2 binding to JAK prevents SOCS from targeting JAK for degradation, providing more active JAK to instigate the pathway. Other tyrosine phosphatases such as Phosphotyrosine Phosphatase 1B (PT1B) and T Cell Protein Tyrosine Phosphatase (TC-PTP) are also involved in deactivating STAT dimers in the nucleus and the cytoplasm (Aaronson and Horvath, 2002; Valentino and Pierre, 2006).
Though the negative regulators of the pathway are simultaneously acting on various JAK-STAT signaling components, the mechanism seems to fail in inflammation and cancer. In this study we attempt to understand constitutive activation of STAT by analyzing the dynamical behavior of the JAK-STAT pathway through the use of mathematical modeling and a systems science approach, as a complement to in vitro/in vivo studies. Although STAT1 is more often identified as a tumor suppressor (proapoptosis), its activation has been observed in multiple myeloma, erythroleukemia, and Acute Myeloid Leukemia (AML) (Buettner et al., 2002; Yu and Jove, 2004).
2 Materials and Methods
A commonly used concept in systems biology is feedback, although there are other systems science concepts that can provide additional insight and improve our understanding of complex biological phenomena. Through the use of a mathematical model, Ashtagiri and Lauffenburger (2001) show that negative feedback in the Mitogen-Activated Protein Kinase (MAPK) pathway generates signal adaptation, while Swameye et al. (2003) show that nucle-ocytoplasmic cycling of STAT5 acts as a remote sensor between the nucleus and cytoplasm. In this study we introduce the concept of “coordination” and demonstrate its applicability to JAK-STAT pathway using an existing mathematical model. The results of this study provide valuable information that can be used to guide biological experiments.
While Swameye et al. (2003) have developed a simplified EPO-induced JAK-STAT5 pathway model, in this work we are interested in the roles of SOCS1 and SHP2 as negative regulators, and therefore we employed an IFNγ-induced STAT1 pathway mathematical model (Section 2.1) developed by Yamada et al. (2003). This model has been calibrated and validated with published biological data, refer to Yamada et al. (2003) for details. We modularized the pathway (Section 2.2), and carried out analysis to identify coordination in the model as described in Section 2.3.
2.1 JAK-STAT mathematical model
The mathematical model is constructed from a set of biochemical reactions that describe the pathway (Yamada et al., 2003; Zi et al., 2005). These biochemical reactions (Figure 1) are translated into first order nonlinear Ordinary Differential Equations (ODE) using mass action kinetics with the exception of the transcription rate which is approximated using Michaelis-Menten kinetics (see Yamada et al. (2003)). The resulting model consists of 31 equations and 51 rate parameters, with IFNγ as the input. Each state variable represents the concentration of a signaling molecule. Nuclear active STAT1 dimer concentration, denoted as [P-STAT1n2], is the output of the system as it is a proxy for transcriptional activity as well as an indicator for pathological conditions, characterized by constitutive activation of STAT1. The set of nonlinear ODE was coded into MATLAB and numerically solved using ode15s.
Fig. 1.

Biochemical reactions in JAK1/JAK2-STAT1 pathway model (adapted from (Zi et al., 2005)). Here ⊘ represents degradation. Numbers in bold (1–5) represent module.
2.2 Modularization and Block Diagram
Signal transduction in biochemical pathways are known to exhibit complex system behavior (Ashtagiri and Lauffenburger, 2001). Modularization is a strategy to deal with such complexity with the objective of improving our understanding of the system behavior. It also facilitates categorizing the results of inhibition, knockdown, and perturbation experiments. Though there is no single preferred method for modularization (Saez-Rodriguez et al., 2005) we can group the approaches according to: (i) species of interacting molecules (biochemical cohesion) (Hartwell et al., 1999); (ii) functionality such as control modules (Lauffenburger, 2000); (iii) network-theoretic systems concepts (Saez-Rodriguez et al., 2005); and (iv) analogous electrical circuit behavior (Deckard and Sauro, 2004).
Here, we employ a hybrid method based on the principles of biochemical cohesion, biological significance, and functionality. Each module or subsystem is then modeled by a set of biochemical reactions that are similar in function, representing biochemical contiguity and functional cohesiveness. Non-parameter variables in the subsystem model that are not state variables define the inputs, while the outputs are variables that are required as inputs to other subsystems.
Using the above principles, modularization was achieved with five subsystems (Figure 1), by rationally combining reactions associated with activities that potentially can have a critical effect on the overall system (biological significance and functionality) and which could function as a group (biochemical cohesion). These include a 1st module that includes receptor/JAK activation essential for initiation of the signaling pathway. The receptor activation requires JAK and IFNγ association, followed by receptor complex dimerization. The three variables (R, R-JAK, IFN-R-JAK) belong to this subsystem that activates the receptor. Once activated, the receptor complex can be inactivated by the effects of either SHP2 in the 2nd module or SOCS1 in the 4th module. If the signaling proceeds past phosphorylation, then STAT1 dimerization can occur in the third module leading to release from the receptor. Additional regulation by cytosolic phosphatase is also possible in this 3rd module. Finally nuclear shuttling and a nuclear phosphatase can regulate the transcriptional output resulting from activated STAT1 in the 5th module. Compartmentalization of the system into 4 sub-systems in the cytoplasm and one representing the activity in the nucleus, facilitates not only the conceptual understanding of signal transduction, but also provides a framework within which modeling equations can be analyzed.
The modularized system (Figure 1) is shown in the form of a block diagram representing the causality signal flow in Figure 2 and this representation is useful in the formulation of the in silico experiments discussed in Section 3. In the block diagram, the modules are labeled based on the dominant signaling component within each module (Module 1: Receptor and JAK, Module 2: SHP2, Module 3: STAT1, Module 4: SOCS1, Module 5: Nucleus).
Fig. 2.

Block diagram of JAK-STAT pathway.
2.3 A Multilevel Hierarchical Systems Framework
In this study, we searched for a coordinating process as an organizing principle for pathway behavior using a multilevel hierarchical system framework. Multilevel hierarchical systems consist of interacting subsystems in multiple levels, where each subsystem has its own distinct goal (Mesarovic et al., 1970, 2004). On the highest level is the coordinator or coordinating process, whose role is to regulate the subsystems at the lower level to achieve the overall objective of the system. To qualify as a coordinator the following conditions must be satisfied: (i) Subsystems in the lower level must be functionally independent. (ii) The coordinator can change the behavior of the lower level subsystems. (iii) The coordinator has the ability to change the behavior of the subsystems in the lower level such that the overall system objective is advanced. Condition (ii) implies that not all behavior of the lower level subsystems can be changed by the coordinator due to the constraints that may be natural to the system. Whereas condition (iii) specifically identifies the behavior that the coordinator can effect and consequently changes the overall system in an improved direction towards achieving its goal.
The search for a subsystem that satisfies the above coordination conditions was conducted through the following systematic, biologically plausible in silico experiments: (a) knockdown - by setting rates and initial conditions of the targeted component to zero, (b) inhibition - excising the connection between subsystems in the block diagram in Figure 2 that represent cause and effect, (c) targeted parametric studies - by varying the rates of binding constants.
3 Results
Starting with the Wild-Type (WT) system - considered the nominal system, we conducted experiments with a pulsed input (various amplitude and durations), and observed the resulting concentration of one or more signaling components in which the phosphorylated nuclear STAT1 dimer is the main focus, as it is a proxy for pathological conditions and only phosphorylated STAT1 can accumulate in nucleus (Meyer et al., 2003). Note that our interest is not in the prediction of individual time-course experiments and hence not in the exact concentration value of the output, since this varies between biological experiments and cell lines. Rather, we analyze the general qualitative behavior of the output and characterize the in silico results based on its temporal profile, and then group these into different behavior response categories or simply behavior categories. A behavior category is defined as a set of responses that have similar temporal characteristics. Such a characterization is useful in labeling simulation results as nominal or pathological in subsequent analysis. Before analyzing the simulation results, we first define the different behavior categories found in this study below.
A nominal behavior category is defined as a set of pathway response under nominal parameter values (as listed in Yamada et al. (2003)). Under this condition, STAT1 is activated only for a couple of hours. This response is characterized by a first peak that is immediately followed by a trough nearly reaching initial condition, and a second peak before it settles to its initial condition. This nominal behavior category is illustrated in Figure 3A. Any responses that exhibit such characteristics, regardless of the peak values, will be categorized as nominal behavior category.
Fig. 3.

Behavior categories for the JAK-STAT system response: (A) nominal behavior category, (B) pathological behavior category, (C) extreme behavior category, (D) high-level activation behavior category, (E) medium-level activation behavior category and (F) low-level activation behavior category.
A pathological behavior category is characterized by constitutively active STAT1. This means that phosphorylated STAT1 concentration remains at a high level even when the stimulus is withdrawn as illustrated in Figure 3B. Under this behavior the system response still exhibits a peak, but instead of returning to its initial condition it oscillates before settling to a value that is higher than its initial condition. In an extreme case of pathological behavior category, the response stays at its peak even after stimulus is withdrawn (Figure 3C). Such a response is labeled as extreme behavior category.
A high-level activation behavior category includes system response in which the first peak that exists under nominal behavior category disappears and the high-level STAT1 concentration is retained for the duration of the stimulus (Figure 3D). In the medium-level activation behavior category the output exhibits a first peak and the concentration is retained at medium-level (with regards to the peak) for the duration of input stimulus. Once the stimulus is withdrawn, the output concentration is slowly returned to its initial condition. Similarly, in a low-level behavior category the output continues to show a first peak, followed by persistent low-level activation that lasts only for the duration of the stimulus (Figure 3E). Under this category, immediately after the stimulus is withdrawn, the output concentration returns to the initial condition.
3.1 Wild-Type System Behavior
We stimulated WT cells in silico with 1nM IFNγ for 8 hours and observed the dynamical behavior of different signaling components (Figure 4) consistent with the results in the literature (Yamada et al., 2003; Zi et al., 2005). Cytoplasmic STAT1 activation reached its peak within one hour and immediately translocated to the nucleus (Figure 4A). Once activated STAT1 reaches its peak, it is instantly brought down to low-level concentration, indicating a normal physiological condition. The IFNγ concentration of 1nM for 8 hours is the nominal input and its corresponding response (Figure 4) is considered to belong to the nominal behavior category.
Fig. 4. Wild-Type Simulation.

Input is set to 1nM IFNγ(nominal input). (A) Various STAT1 concentration, (B) SOCS1 concentration, and (C) SOCS1 mRNA concentration.
We investigated the system response to various pulse input conditions (amplitude and duration) to identify its membership in an appropriate behavior category. In silico experiment results show that the JAK-STAT1 pathway is highly robust to varied input duration and continues to exhibit nominal behavior (Figure 5). Higher input duration only increased the first peak amplitude of STAT1 activation. These simulations suggest that the negative regulators of the pathway are capable of stabilizing STAT1 activation regardless of the input concentration and duration, leading to further analysis of the negative regulation mechanisms.
Fig. 5. Nuclear active STAT dimer concentration while varying input duration and amplitude.

(A) Input amplitude is set to 0.5nM and input duration is varied from 1 to 15 hrs. (B) Input duration is set to 8 hrs while input amplitude is varied from 0.1 to 50 nM.
3.2 Knockdown Experiments
Four negative regulators are represented in this model: SOCS1, SHP2, and two protein phosphatases for nuclear and cytoplasmic STAT1 (PPN and PPX, respectively). Knocking down SOCS1 results in high-level activation behavior (Figure 6A).
Fig. 6. Negative Regulators Knockdown Simulation.

Input is set to 1nM IFNγ and various STAT1 concentration is shown. (A) and (B) SOCS1, SHP2, and both SOCS1 and SHP2 knockdown.
In SHP2 knockdown, the first peak amplitude of [P-STAT1n2] is higher than the nominal response. Under this condition the system exhibits a pathological behavior where the [P-STAT1n2] concentration subsequently oscillates and settles at about 50 nM even after the input is removed (Figure 6A, B). Taken together, it appears that [P-STAT1n2] oscillations are governed by SOCS1 while constitutive activation is determined by SHP2. Furthermore, since with SOCS1 knockdown, STAT1 activation remains high in the presence of the input, SHP2 must be responsible for down-regulating STAT1 at a later time period. Consequently, the first peak still exists in the SHP2 knockdown but STAT1 is constitutively activated, illustrating that SOCS1 is regulating STAT1 deactivation early in the stimulus period.
When SOCS1 and SHP2 are simultaneously knocked down, with the nominal stimulus of 1nM IFNγ for 8 hours, [P-STAT1n2] response exhibits an extreme behavior. This suggests that PPN and PPX alone cannot regulate the system response back to its initial state. In the absence of PPN, active STAT1 accumulates in the nucleus because STAT1 cannot translocate to the cytoplasm without dephosphorylation.
3.3 Parametric Studies
In WT cells with nominal input, changing parameters in either the SHP2 or nuclear subsystems resulted in low-level activation behavior (Figures 7A and 7C). Such activation provides a window for the transcriptional activity, although whether it is sufficient to facilitate pathological conditions remains an open question. Varying STAT1 subsystem parameter does not change the nominal response (Figure 7).
Fig. 7. Parameter Variation in WT.

Input is set to 1nM IFNγand changing parameter in: (A) the SHP2 subsystem, (B) the STAT1 subsystem, (C) the nuclear subsystem, and (D) the SOCS1 subsystem.
Varying SOCS1 subsystem parameter (Figure 7D) leads to a response that is similar to those in pathological behavior category. However, instead of settling to a high-level concentration after stimulus withdrawal, STAT1 activation very slowly decreases. Twelve hours after the stimulus was removed, STAT1 activation had not reached its initial state. Thus, the SOCS1 subsystem parameter is capable of bringing STAT1 to near constitutive activation.
When SOCS1 was knocked down (Figure 6A), STAT1 remained active only in the presence of the input, but changing the SHP2 subsystem parameter (rate constant for SHP2 association with receptor complex) could change this behavior. Setting SHP2 parameter to a high value leads to extreme behavior, whereas low value results in high-activation behavior (Figure 8A). When both SOCS1 and SHP2 are knocked down, no parameter change can alter the system’s behavior (data not shown).
Fig. 8.

Parameter Variation in Knockdown Cells. Input is set to 1nM IFN. (A) With SOCS1 knockdown, SHP2 subsystem parameter is varied - nominal value is 0.2s−1. SHP2 is knockdown and varying (B) the STAT1 subsystem parameter - nominal value is 0.003s−1.
A SHP2 knockdown results in pathological behavior (Figure 6A, B). Decreasing the dephosphorylation rate constant of active STAT1 by PPX in the cytoplasm (STAT1 subsystem parameter, v12) kept [P-STAT1n2] at a higher concentration and dampened the oscillations (Figure 8B). Increasing the aforementioned rate constant lowers the concentration of the [P-STAT1n2], with STAT1 constitutively active and more pronounced oscillations (Figure 8B).
In exploring the parameter perturbations we observed bounded autonomy (tolerance to parameter variations) in the subsystem behavior (Mesarovic et al., 2004). The pathway can exhibit responses in different behavior categories under different conditions including parameter changes. Figure 9 illustrates bounded autonomy in the SOCS1 subsystem. Here the parameters in reaction 31 (refer to Figure 1) are varied. For each different input amplitude and duration, the minimum value of the parameter for which the nominal condition response (Figure 1) is preserved is shown. The parameter space above the bounded autonomy surface maintains the nominal behavior.
Fig. 9. Bounded autonomy surface.

The surface indicates threshold for parameter in SOCS1 subsystem (kf31) for nearly constitutive activation behavior. Setting parameter above the surface will result in a normal physiological behavior. 10B) where the response is retained at about 180 nM for the rest of the input duration. Removing the input causes [P-STAT1n2] concentration to gradually decrease to zero.
3.4 Inhibition Experiments
The block diagram in Figure 2 shows that the SHP2 and SOCS1 subsystems are interacting. Inhibiting the SHP2 and SOCS1 connection is tantamount to setting the reaction rates to zero: (i) the binding of SHP2 to SOCS1-IFNRJ2*-STAT1c ,and (ii) the dissociation of SOCS1-IFNRJ2*-STAT1c-SHP2. The result (Figure 10A) is similar to setting reaction 31 in the SOCS1 subsystem (SHP2 and SOCS1-IFNRJ2*-STAT1c-SHP2 binding) to 0.001 in Figure 7D. This suggests that the overall effects of SHP2 and SOCS1 are tightly coupled.
Fig. 10. Inhibit connection between subsystems.
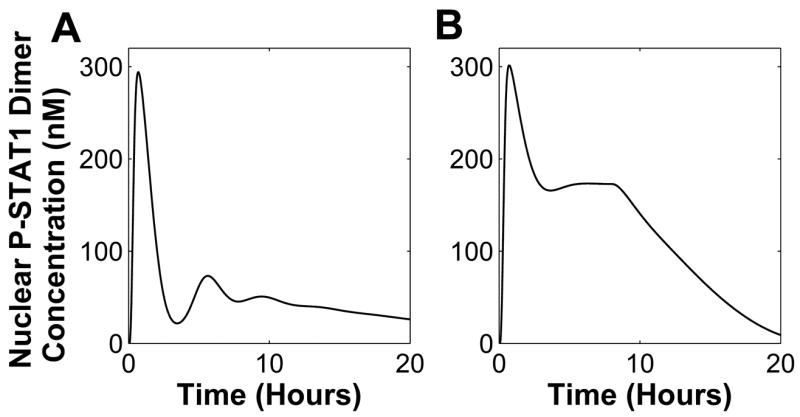
Input is set to 1nM IFN. (A) Cutting connection between SOCS1 and SHP2 subsystems. (B) Inhibiting connection between STAT1 and SOCS1 subsystems
We further examined the interaction between the STAT1 and SOCS1 subsystems, through STAT1 binding to the SOCS1-IFNRJ2* complex (reaction 30) and the SOCS1-IFNRJ2*-STAT1c-SHP2 dissociation reaction (reaction 32). Inhibiting these reactions results in medium-level activation behavior (Figure
3.5 SOCS1 as a Candidate Coordinator
In the absence of either SHP2 or SOCS1 the system exhibits a response that falls under a different behavior category than the nominal, suggesting that both SHP2 and SOCS1 subsystem can alter the behavior of STAT1 subsystem. Hence both are candidate coordinators, satisfying the second condition for characterizing a coordinator (Section 2.3). The first coordination condition requires that the coordinated subsystems are independent of each other. With a knockdown of the SHP2 subsystem, the lower level consists of SOCS1 and STAT1 subsystems with SOCS1 production dependent on STAT1. This eliminates the SHP2 subsystem as a viable candidate coordinator. Furthermore varying parameters in the SHP2 subsystem does not significantly change the behavior of the pathway (Figure 7A), failing the third condition.
On the other hand, the SHP2 and STAT1 subsystems are independent, suggesting SOCS1 as a candidate coordinator. We note that the SOCS1 subsystem is capable of changing the behavior of the lower level subsystems. In silico studies in Section 3.3 and Figure 7 show that by varying only the SOCS1 subsystem parameter the nuclear STAT1 activation changed significantly to near constitutive activation (Figure 7B). Changing only the SHP2 subsystem parameter results in responses that are in the same nominal behavior category. This evidence strongly suggests SOCS1 as the potential coordinator.
The third condition for a coordinator requires that the coordinator is able to change the lower level subsystem behavior to achieve the overall objective of the system. We define the overall goal of the pathway as having nominal STAT1 activation. Based on this condition if the system results in a pathological behavior, parameters in the SOCS1 subsystem should be capable of bringing the system back to its nominal behavior. The pathological behavior was created by the SHP2 knockdown or by alternatively changing one or more parameter in the SHP2 subsystem (Figure 11) such that SHP2 is not fully capable of deactivating the receptor complex (mutation). Three parameters in the SOCS1 subsystem were varied to restore the system back to its nominal behavior.
Fig. 11. SOCS1 subsystem coordination action.

(A) SHP2 knockdown and parameter in SOCS1 subsystem is varied in order to reach WT behavior. (B) SHP2 is nearly knockdown or mutated (i..e. not performing its function at its full capability), simulated by setting kf9=0.5μM−1 s−1, kb9=20s−1, kf10=0.0003s−1, and SHP2 initial condition to 5 nM. To restore the system, SOCS1 subsystem parameters are changed to kb21=0.008s−1 and kb31=0.03s−1.
From the simulation results above, we conclude that the SOCS1 subsystem is a candidate coordinator, and this result needs to be further confirmed by biological experiments. The SOCS1 subsystem has the capability of changing the independently functioning lower-level subsystem behaviors such that WT response is achieved even with mutation. The hierarchical representation of the system with SOCS1 as a coordinator is illustrated in Figure 12.
Fig. 12.
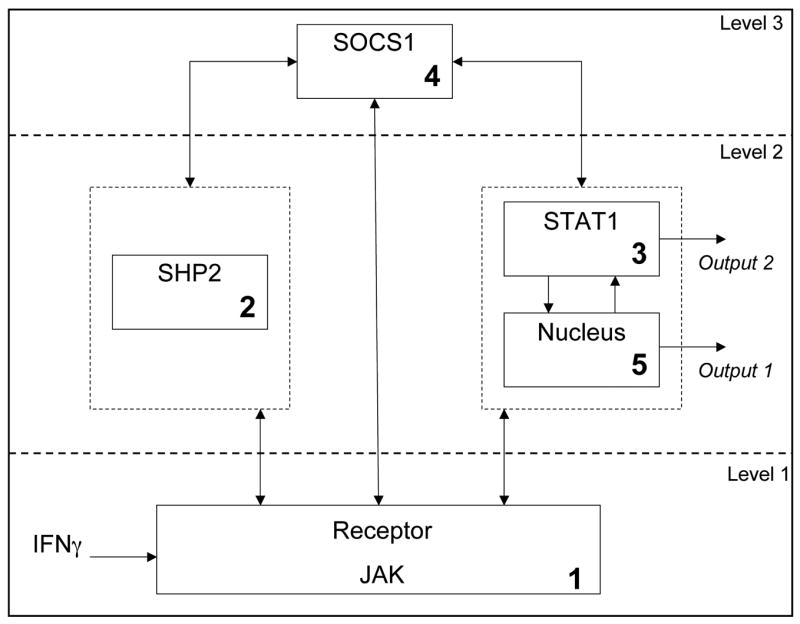
Hierarchical representation of the JAK-STAT pathway with SOCS1 subsystem as a coordinator
4 Discussion
Through the use of a complex systems biology approach and a hierarchical system viewpoint we explored the behavior of the JAK-STAT pathway under WT and mutant conditions to discover an organizing principle, more specifically coordination. In silico experiments demonstrated that the pathway in WT exhibits responses belonging to the same behavior category. It was observed that the WT system response is invariant to input amplitude and duration changes, and we further showed that a SOCS1 knockdown leads to high-level activation behavior, while SHP2 knockdown results in constitutive activation (pathological behavior). Cytokine, growth factors, and autocrine signaling commonly occur in the progression of cancer and inflammation. This suggests that with abundant cytokines/growth factors, STAT1 can be persistently activated if a SOCS1 mutation exists. Without the high availability of cytokines/growth factors, it is SHP2 that is regulating the constitutive activation of STAT1.
Biological data in Wormald et al. (2006) shows STAT1 phosphorylation in the SOCS1 knockout for bone marrow-derived macrophage cells is retained for only up to 100 minutes. We believe this discrepancy between our in silico experiments and the biological data in (Wormald et al., 2006) is due to the different cell lines. The parameters that we are using (Yamada et al., 2003) were fitted to data in the literature including Brysha et al. (2001) in which phosphorylation of STAT1 in knockdown hepatocytes is prolonged for at least up to 3 hours, given a 10 minute stimulation. In addition, the in silico experimental results of STAT1 phosphorylation with SOCS1 knockdown shown by Yamada et al. (2003), agrees with our results and is consistent with Brysha et al. (2001).
We identified SOCS1 as a candidate coordinator that is regulating the lower-level subsystems (SHP2 and STAT1) such that the overall objective of the pathway is achieved. As a coordinator, the SOCS1 subsystem can be a potential target for therapy since changes in SOCS1 can alter the behavior of the STAT1 subsystem itself. Our discovery of SOCS1 as a coordinator is in line with the recent publication (Hanada et al., 2006) that shows biological data supporting SOCS1 as a crucial signaling component regulator. We suggest that specific biological experiments need to be conducted to confirm that SOCS1 is a coordinator. Further it will be interesting to examine the resulting in vivo phenotype of the various perturbation experiments that were conducted here in silico.
We have only considered the JAK-STAT pathway whereas in an actual living cell crosstalk often exists. In addition to acting as a phosphatase for JAK-STAT, SHP2 can also act as a docking protein for the MAP Kinase pathway. Such crosstalk needs to be further investigated in silico.
Acknowledgments
We thank Mihajlo Mesarovic for helping us understand multilevel hierarchical systems and in particular coordination theory. Ashish Desai and Michael Weis conducted many of the simulations and we are thankful to them. We gratefully acknowledge the support of US NIH grant K25 CA 113133 for SNS and RPS, and US NIH grant U56 CA 112963 for SNS, KAL, and RPS.
Abbreviations
- IFN
Interferon
- JAK
Janus Kinase
- STAT
Signal Transducer and Activator of Transcription
- PIAS
Protein Inhibitors of Activated STATs
- CIS
Cytokine-Inducible SH2 containing protein
- SOCS
Suppressors of Cytokine Signaling
- SSI
STAT-induced STAT Inhibitor
- SHP2
Src-homology 2 (SH2)-containing phosphatase
- PTP1B
Phosphotyrosine Phosphatase 1B
- TC-PTP
T Cell Protein Tyrosine Phosphatase
- AML
Acute Myeloid Leukemia
- ODE
Ordinary Differential Equation
- PPN
Nuclear Phosphatase
- PPX
cytoplasmic phosphatase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aaronson DA, Horvath CM. A road map for those who don’t know JAK-STAT. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- Ali S, Nouhi Z, Chughtai N, Ali S. SHP-2 regulates SOCS-1-mediated Janus Kinase-2 ubiquitination/degradation downstream of the prolactin receptor. Journal of Biological Chemistry. 2003;278:52021–31. doi: 10.1074/jbc.M306758200. [DOI] [PubMed] [Google Scholar]
- Ashtagiri AR, Lauffenburger DA. A computational study of feedback effects on signal dynamics in a Mitogen-Activated Protein Kinase (MAPK) pathway model. Biotech Prog. 2001;17:227–239. doi: 10.1021/bp010009k. [DOI] [PubMed] [Google Scholar]
- Brysha M, Zhang J, Bertolino P, Corgin J, Alexander W, Nicola N, Hilton D, Starr R. Suppressor of Cytokine Signaling-1 attenuates the duration of Interferonγ signal transduction in vitro and in vivo. J. Biol. Chem. 2001;276 (25):22086–22089. doi: 10.1074/jbc.M102737200. [DOI] [PubMed] [Google Scholar]
- Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clinical Cancer Research. 2002;8:945–954. [PubMed] [Google Scholar]
- Chen J, Yu W, Bunting KD, Qu CK. A negative role of SHP-2 tyrosine phosphatase in growth factor-dependent hematopoietic cell survival. Oncogene. 2004;23 (20):3659–69. doi: 10.1038/sj.onc.1207471. [DOI] [PubMed] [Google Scholar]
- Chen Y, Wen R, Yang S, Schuman J, Zhang E, Yi T, Feng GS, Wang D. Identification of SHP-2 as a STAT5A phosphatase. J Biol Chem. 2003;278:16520–16527. doi: 10.1074/jbc.M210572200. [DOI] [PubMed] [Google Scholar]
- Chughtai N, Schimchowitsch S, Lebrun JJ, Ali S. Prolactin induces SHP-2 association with STAT5, nuclear translocation, and binding to the beta-casein gene promoter in mammary cells. J Biol Chem. 2002;277:31107–31114. doi: 10.1074/jbc.M200156200. [DOI] [PubMed] [Google Scholar]
- Deckard A, Sauro HM. Preliminary studies on the in silico evolution of biochemical networks. ChemBiochem. 2004;5 (10):1423–1431. doi: 10.1002/cbic.200400178. [DOI] [PubMed] [Google Scholar]
- Hanada T, Kobayashi T, Chinen T, Saeki K, Takaki H, Koga K, Minoda Y, Sanada T, Yoshioka T, Mimata H, Kato S, Yoshimura A. IFNγ-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J Exp Med. 2006;203(6):1391–1397. doi: 10.1084/jem.20060436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Hopfield JJ, Leibler S, Murray A. From molecular to modular cell biology. Nature. 1999;402:C47–52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- Lauffenburger D. Cell signaling pathways as control modules: complexity for simplicity. PNAS. 2000;57 (10):5031–5033. doi: 10.1073/pnas.97.10.5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann U, Schmitz J, Weissenbach M, Sobota RM, Hörtner M, Friederichs K, Behrmann I, Tsiaris W, Sasaki A, Schneider-Mergener J, Yoshimura A, Neel BG, Heinrich PC, Schaper F. SHP2 and SOCS3 contribute to Tyr-759-dependent attenuation of interleukin-6 signaling through gp130. Journal of Biological Chemistry. 2003;278 (1):661–671. doi: 10.1074/jbc.M210552200. [DOI] [PubMed] [Google Scholar]
- Mesarovic M, Macko D, Takahara Y. Theory of Hierarchical Multilevel System. Academic Press; USA: 1970. [Google Scholar]
- Mesarovic M, Sreenath S, Keene J. Search for organising principles: understanding in systems biology. IEE Systems Biology. 2004;1 (1):19 – 21. doi: 10.1049/sb:20045010. [DOI] [PubMed] [Google Scholar]
- Meyer T, Marg A, Lemke P, Wiesner B, Vinkemeier U. DNA binding controls inactivation and nuclear accumulation of the transcription factor Stat1. Genes and Developement. 2003;17:1992–2005. doi: 10.1101/gad.268003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez-Rodriguez J, Kremling A, Gilles E. Dissecting the puzzle of life: modularization of signal transduction networks. Computers and Chemical Engineering. 2005;29:619–629. [Google Scholar]
- Swameye I, Müller T, Timmer J, Sandra O, Klingmüller U. Identification of nucleocytoplasmic cycling as a remote sensor in cellular signaling by data-based modeling. PNAS. 2003;100:1028–33. doi: 10.1073/pnas.0237333100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentino L, Pierre J. JAK/STAT signal transduction: Regulators and implication in hematological malignancies. Biochemical Pharmacology. 2006;71 (6):713–721. doi: 10.1016/j.bcp.2005.12.017. [DOI] [PubMed] [Google Scholar]
- Ward AC, Touw I, Yoshimura A. The JAK-STAT pathway in normal and perturbed hematopoiesis. Blood. 2000;95 (1):19–29. [PubMed] [Google Scholar]
- Wormald S, Zhang JG, Krebs DL, Mielke LA, Silver J, Alexander WS, Speed TP, Nicola NA, Hilton DJ. The comparative roles of Suppressor of Cytokine Signaling-1 and -3 in the inhibition and desensitization of cytokine signaling. J Biol Chem. 2006;281 (16):11135–11143. doi: 10.1074/jbc.M509595200. [DOI] [PubMed] [Google Scholar]
- Yamada S, Shiono S, Joo A, Yoshimura A. Control mechanism of JAK/STAT signal transduction pathway. FEBS Letters. 2003;534:190–196. doi: 10.1016/s0014-5793(02)03842-5. [DOI] [PubMed] [Google Scholar]
- You M, Yu D, Feng G-S. SHP-2 tyrosine phosphatase functions as a negative regulator of the interferon-stimulated JAK/STAT pathway. Mol Cell Biol. 1999;19:2416–2424. doi: 10.1128/mcb.19.3.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CL, Jin YJ, Burakoff S. Cytosolic tyrosine dephosphorylation of STAT5. Potential role of SHP2 in STAT5 regulation. J Biol Chem. 2000;275:599–604. doi: 10.1074/jbc.275.1.599. [DOI] [PubMed] [Google Scholar]
- Yu H, Jove R. The STATs of cancer - new molecular targets come of age. Nature Reviews Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- Yu WM, Hawley T, Qu CK. Catalytic-dependent and -independent roles of SHP-2 tyrosine phosphatase in interleukin-3 signaling. Oncogene. 2003;22 (38):5995–6004. doi: 10.1038/sj.onc.1206846. [DOI] [PubMed] [Google Scholar]
- Zi Z, Cho KH, Sung MH, Xia X, Zheng J, Sun Z. In silico identification of the key components and steps in IFNγ induced JAK-STAT signaling pathway. FEBS Letters. 2005;579:1101–1108. doi: 10.1016/j.febslet.2005.01.009. [DOI] [PubMed] [Google Scholar]