

Abstract
PURPOSE
The aim of the present study was to investigate effects of thrombin and thrombin in combination with other proangiogenic factors on VEGF expression in hRPE cells.
METHODS
hRPE cells were stimulated with thrombin TNF-α, monocytes and TGF-β2. Following stimulation, conditioned medium and lysed cells were subjected to ELISA, Western blotting, immunocytochemistry and RT-PCR analyses. Inhibitors specific for various signal transduction pathways were employed to determine the signal pathways involved.
RESULTS
Treatment of RPE cells with thrombin resulted in dose- and time-dependent increases in VEGF mRNA levels and protein production. hRPE VEGF expression is predominantly PAR-1 dependent. About 80% of thrombin-induced VEGF secretion was abrogated by inhibitors of MAPK/ERK kinase (MEK), p38, c-Jun NH2-terminal kinase (JNK), protein tyrosine kinase (PTK), phosphatidylinositol 3-kinase (PI3K), protein kinase C (PKC), nuclear factor-κB (NF-κB), and reactive oxygen species (ROS). Analyses of VEGF protein production and mRNA synthesis revealed that VEGF induction by thrombin plus TNF-α or co-culture with monocytes was additive, while that by co-incubation with TGF-β2 was synergistic. The co-stimulated VEGF production by TGF-β2 plus thrombin averaged threefold higher than the sum of that induced by each agent alone. Furthermore, BAPTA, a calcium chelator, blocked the VEGF secretion induced by thrombin and thrombin plus TGF-β2 by 65% and 20%, respectively, but had no effect on that by TGF-β2 alone.
CONCLUSIONS
Thrombin alone and in combination with TNF-α, monocytes and TGF-β2 potently stimulated VEGF expression in hRPE cells via multiple signal pathways. The thrombin-induced calcium mobilization may play an important permissive role in maximizing TGF-β2–induced VEGF expression in RPE cells.
Introduction
Retinal pigment epithelial (RPE) cells, strategically located between neuroretinal photoreceptors and choriocapillaris of choroidal tissue, form the outer blood-retina barrier. In addition to supporting normal photoreceptor function, RPE cells secrete a variety of cytokines, growth factors, and extracellular matrix components that enable RPE cells to actively participate in retinal and choroidal neovascularization.1,2 Neovascularization is a pathological process common to many ocular diseases of diverse etiologies such as proliferative diabetic retinopathy (PDR), proliferative vitreoretinopathy (PVR) and age-related macular degeneration (AMD). Retinal and choroidal neovascularization is the most common and important complication of these diseases and often leads to blindness.3
Vascular endothelial growth factor (VEGF) is a multifunctional cytokine strongly implicated in angiogenesis. Like pericytes, endothelial cells, Müller cells, and astrocytes, hRPE cells also secrete VEGF.4 There is considerable evidence that VEGF secretion by RPE cells leads to neovascularization in the posterior segment of the eye. Several therapies to control angiogenesis involve antagonizing VEGF or its receptors.5 Thus, identifying precise molecular mechanisms, upstream of VEGF gene expression in RPE cells, may prove useful for developing treatment that is even more effective at controlling ocular neovascularization.
Thrombin stimulates signaling pathways via interactions with G-protein-coupled protease-activated receptors (PARs). Human thrombin receptor is a protein with seven transmembrane domains and a large extracellular amino-terminal extension. Thrombin cleaves within this extension, thereby creating a new receptor amino-terminus that functions as a tethered ligand and activates the receptor. At least 3 subtypes of PARs, PAR-1, PAR-3 and PAR-4, are activated by thrombin. PAR-1 and PAR-3 are constitutively expressed in RPE cells, but PAR-1 is likely the major form of PAR mediating thrombin activation in RPE cells.6,7
Classically, thrombin generation is triggered when disruption of vascular integrity allows coagulation factors in the plasma to contact extravascular tissue factor (tissue thromboplastin; coagulation factor III). Prothrombin in 1 ml of human plasma, when fully converted, forms 15-38 U thrombin which is concentrated in the resulting clot.8, 9 Thrombin is formed from prothrombin in areas of increased vascular permeability and hemorrhage at sites of blood-retina barrier breakdown in many retinal diseases, such as ocular trauma, PVR, PDR, and AMD. Thrombosis is also enhanced by localized increases in thrombin concentration in the retinal circulation during retinal ischemia that results in increased leakage of serum components, including prothrombin.10 Thrombin itself may induce gap formation between endothelial cells, further enhancing its own formation.11 Thrombin stimulates VEGF in many cell types12–14 and interplays with VEGF synergistically in promoting angiogenesis.15 Despite the likely exposure of RPE cells to thrombin in retinal diseases, no study has shown that thrombin induces RPE cells to produce VEGF. Therefore, it is of pathophysiological importance to assess the role of thrombin in stimulating VEGF expression in RPE cells. Moreover, thrombin is also likely to co-exist with other pro-angiogenic factors such as TNF-α and TGF-β that have been detected in diseased ocular tissues.16–19 Histopathological studies of choroidal neovascular membranes from patients with AMD have demonstrated the presence of various growth factors, including TGF-β.20 Nevertheless, interactions of thrombin with other pro-angiogenic factors in stimulating VEGF secretion have not been reported. Apart from its central role in blood coagulation, thrombin also regulates other cellular functions, including those involved in wound healing and inflammation. One example is thrombin’s mitogenic effects on RPE cells.21 The latter finding has led to thrombin being used as a therapeutic agent.22,23 As a mitogen of retinal cells, thrombin has been used in intraocular surgery for diabetic retinopathy and ocular trauma to control bleeding and to close macular holes. However, thrombin treatment frequently causes significant inflammation. This clinical observation is consistent with our previous observations showing the potential pro-inflammatory role of thrombin, TNF-α, and monocytes by stimulating RPE cells to secrete chemokines.24–26 These observations underscore the potential importance of investigating whether thrombin at sites of blood-retinal barrier breakdown works in concert with leulocytes to stimulate VEGF gene expression in ocular tissues. In this study we demonstrated that thrombin, working additively with TNF-α and monocytes and synergistically with TGF-β2, triggers multiple signal pathways and lead to enhanced VEGF gene expression in human RPE cells.
Materials and Methods
Materials
Human thrombin was purchased from ICN Pharmaceuticals, Inc (Costa Mesa, CA). TNF-α and TGF-β2 were purchased from R&D System (Minneapolis, MN). U0126 was obtained from Promega (Madison, WI), SB202190 from Calbiochem (San Diego, CA), CAPE (caffeic acid phenethyl ester) from BIOMOL (Plymouth Meeting, PA), Nac (N-acetyl-cysteine), DPI (diphenyleneiodonium chloride) and BAPTA [1,2-bis (o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid] and all other reagents were obtained from Sigma-Aldrich Company (St. Louis, MO). Human thrombin receptor 1 (PAR-1)-activating peptide (SFLLRNPNDKYEPF, H-Ser-Phe-Leu-Leu-Arg-Asn-Pro-Asn-Asp-Lys-Tyr-Glu-Pro-Phe-OH) and human thrombin receptor 3 (PAR-3)-activating peptide (TPRGAP, H-Thr-Phe-Arg-Gly-Ala-Pro-OH) were obtained from Bachem Americas (Torrence, CA). SCH79797 was obtained from Tocris Cookson Inc (Ellisville, Mo).
The signal transduction inhibitors used in this study and their molecular targets are summarized in Table 1.
Table 1.
Inhibitors used in this study*
| Inhibitor | Protein Target |
|---|---|
| U0126 | MEK |
| SB202190 | P38 |
| Sp600125 | JNK |
| AG490 | Jak2 |
| Genistein | PTK |
| Ro318220 | PKC |
| CAPE | NF-κB |
| Ly294002 | PI3K |
| Nac | ROS |
| DPI | ROS |
| BAPTA/AM | Calcium chelator |
MEK, mitogen-activated protein kinase; JNK, c-Jun NH2-terminal kinase; jak2, Janus kinase 2; PTK, protein tyrosine kinase; PKC, protein kinase C; NF-κB, nuclear factor-κB; PI3K, phosphatidylinositol 3-kinase; Nac, N-acetyl-cysteine; DPI, diphenyleneiodonium chloride; ROS, reactive oxygen species; BAPTA/AM, 1,2-bis-(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid/acetoxy-methyl ester.
Cell isolation and culture
Human RPE cells were isolated within 24 hr of death from donor eyes that were obtained from the Midwest Eye Bank as previously described. 27 In brief, the sensory retina tissue was separated gently from the RPE monolayer, and the RPE cells were removed from Bruch’s membrane with papain (5 U/ml). The RPE cells were cultured in Dulbecco’s modified essential medium (DMEM) containing 15% fetal bovine serum, penicillin G (100 u/ml), streptomycin sulfate (100 μg/ml), and amphotericin B (0.25 μg/ml) in Falcon Primaria culture plates to inhibit fibroblast growth. The human RPE monolayers exhibited typical hexagonal arrays with uniform immunohistochemical staining for fibronectin, laminin, cytokeratin 8/18, and type IV collagen in the chicken-wire distribution characteristic of these epithelial cells. The identity of RPE cells in the culture was also confirmed by apical immunohistochemical staining of Na+-K+-ATPase.28 Cells were subcultured, grown to reach near confluence, and used for experiments. The cells were in culture for up to four to six passages.
Human monocytes were freshly isolated from the peripheral blood of healthy volunteers using modification of the methods by Yoshida et al.24. Mononuclear cells were separated by density gradient centrifugation. The cells were washed and then layered onto density gradient (Fico-Lite monocytes, 1.068 g/ml) for the enrichment of monocytes. The purity of the cell was greater than 97 percent.
ELISA
The levels of antigenic VEGF in the serial dilutions of human RPE supernatants were quantitated by modification of a double ligand ELISA method as previously described.28. Standards included 0.5 log dilutions of recombinant VEGF (R&D Systems) from 5pg to 100ng/well.
Quantifying mRNA of VEGF
Nearly confluent human RPE cells seeded in 60 mm dishes were treated with thrombin, TGF-β2, or thrombin plus TGF-β2 with or without the thrombin inhibiter, hirudin, for 6 hr, respectively. Quantikine mRNA colorimetric quantification kits (R&D Systems, Minneapolis, MN) were used to quantitate RPE VEGF mRNA. In brief, following removal of excess media, the cell lysates were obtained by adding cell lysis diluent to the cell monolayers. Then 150 μl diluent was hybridized in hybridization plate with 50 μl of VEGF gene-specific biotin-labeled capture oligonucleotide probes in a 65 °C water bath for 1 hr. Next, 150 μl of hybridization solution was transferred to a streptavidin-coated microplate and incubated for 60 min at room temperature on a shaker. After wash, 200 μl of anti-digoxigenin conjugate was added to each well and incubated for 60 min. After the unbound conjugate solution had been washed away, 50 μl of substrate solution and 50 μl of substrate were in turn added and incubated for 60 or 30 min. The chromogen was generally developed for 30 min, but was monitored depending on the amount of VEGF present in the experimental samples and the samples used for standard calibration. The absorbance at 490 nm with correction of 690 nm was determined following the addition of 50 μl of stop solution. The concentration of VEGF mRNA was calculated by interpolation of a standard calibration curve. All samples and calibrators were in duplicate.
Semi-quantitative reverse transcription polymerase chain reaction (RT-PCR)
The total cellular RNA was isolated from nearly confluent cultures of human RPE cells using TRIZOL reagent according to the manufacturer’s procedure. The cDNA synthesis reaction was set up according to the protocol for a reverse transcription system (Invitrogen, Carsbad, CA). Briefly, 5 μg of RNA was added to the reaction mixture with M-MLV reverse transcriptase (100 U/μl) and 1μl random primers for a total volume of 20 μl. PCR for each product was performed with three different cycles (15, 25 and 35). The PCR reactions were accepted as semi-quantitative when individual amplificates were carried out in the mid-linear portion of the response curve. Specific cDNA was amplified by 28 (1 μl cDNA) and 20 cycles (1 μl cDNA) for VEGF, and β-actin, respectively. The following conditions were used in PCR reaction for VEGF and β-actin: denaturation at 94°C for 1 min, annealing at 62°C for 1 min, and extension at 72°C for 2 min. The reaction was initiated by adding 0.15 μl of Taq DNA polymerase (5 U/μl) to a final volume of 20 μl. The synthetic proprietary oligonucleotide primers for human VEGF were obtained from R&D (Catalog number RDP-33-025). To ensure that an equal amount of templates was used in each amplification reaction, human β-actin sense (5′-GTGGGGCGCCCCAGGCACCA-3′) and antisense (5′-GCTCGGCCGTGGTGGT GAAGC-3′) primers were used in parallel. Each PCR product was analyzed by electrophoresis on a 2% agarose gel and stained with ethidium bromide.
Immunocytochemistry
Immunochemical staining was performed according to manufacturer’s protocol (ABC kit; Vector, Burlingame, CA). Nearly confluent human RPE cells were fixed with 4% paraformaldehyde for 15 min at room temperature. The cells were incubated with primary rabbit polyclonal antibody (1:200; Santa Cruz Biotechnology, Inc, Santa Cruz, CA) to VEGF at 37°C for 1 hr. The cell-bound antibody complexes were then visualized by development in the substrate solution containing 3-amino-9-ethylcarbazole (AEC) to yield a red reaction product as previously described.27 The same concentration of nonspecific rabbit IgG replaced primary anti-VEGF antibody and immunostaining of unstimulated RPE cells served as controls.
Statistic Analysis
The mean VEGF concentration ± SEM was determined for each assay condition. Various assay conditions were compared by Two-way ANOVA using software STATVIEW (Cary, NC), and p values<0.05 was considered to be statistically significant, p<0.05 is indicated as *, p<0.01 **, P<0.001***.
Results
Thrombin induces human RPE VEGF protein production and mRNA expression
To determine stimulation of VEGF secretion, human RPE cells were challenged by 10 U/ml of thrombin. The growth medium was harvested after 8, 24, and 48 hr of stimulation. As shown in Fig. 1A, thrombin at 10 U/ml induced time-dependent increases in VEGF secretion. The induction of VEGF was noted within 8 hr of stimulation, was more evident at 24 hr and showed sustained increases from 24 to 48 hr, reaching 3.5- and 4.8-fold higher than non-stimulated RPE cells. This stimulation by thrombin was dose-dependent. Thrombin at 10 U/ml resulted in about 40% more VEGF secretion as compared to that at 1 U/ml concentration. To ensure that stimulation of VEGF by thrombin was specific, two approaches were employed. We first used hirudin, a potent and specific thrombin inhibitor to examine if co-incubation with hirudin could inhibit the thrombin action. As shown in Fig. 1A, the enhanced VEGF secretion was 50% reduced by co-incubation with hirudin (40 U/ml). Second, we used specific agonist peptides to determine which forms of PARs mediated thrombin induced VEGF expression. Specific agonist peptides have been developed as useful tools to validate which forms of PARs are present in various cell types.29 To find out whether the observed thrombin-mediated effects on RPE VEGF secretion may be due to specific activation of PAR-1 and PAR-3, the two PAR forms previously shown on human RPE cells, we used specific thrombin receptor agonist peptides for RPE treatment. As shown in Fig. 1B, thrombin receptor 1 agonist peptide induced dose-dependent RPE VEGF secretion. At 100 μM the PAR-1 specific thrombin receptor activated peptide (TRAP) produced the levels of VEGF secretion nearly identical to that by 10 U/ml of thrombin, while PAR-3 (50, 100 μM) specific TRAP did not induce measurable VEGF production. In addition, SCH79797, a potent non-peptide antagonist for PAR-1,30 induced dose-dependent reduction in hRPE VEGF expression. SCH79797 inhibited hRPE VEGF reduction by 66% at 30 μM and 37% at 3 μM (Fig. 1C). These results suggest that thrombin-induced human RPE VEGF secretion is predominantly mediated by PAR-1.
Figure 1.

Stimulation of human RPE VEGF secretion. A. Time- and dose-dependent stimulation of RPE VEGF secretion by thrombin. Human RPE cells were stimulated by thrombin (0, 1 or 10 U/ml) with or without hirudin and SCH79797 for 8, 24, and 48 hs. B. Stimulation of RPE VEGF secretion by thrombin receptor-activating peptides (TRAPs). The RPE cells were stimulated with thrombin or TRAPs for thrombin receptor PAR-1 and PAR-3. C. I nhibition of thrombin-stimulated human RPE VEGF production by SCH79797. Unstimulated RPE cells were used as control. The conditioned media were assayed for VEGF by ELISA. The values represent means ± SEM (n=3). R1, PAR-1; R3, PAR-3; SCH30, SCH79797 at 30 μM; SCH3, SCH79797 at 3 μM.
To determine if the stimulation of VEGF secretion by thrombin occurs at transcriptional and/or translational levels, steady-state VEGF mRNA was quantified using semi-quantitative RT-PCR. As seen in Fig. 2A, induction of VEGF mRNA expression by thrombin was evident, as early as 1 hr after stimulation, peaked at 2 hr, and sustained for at least 20 hr. Consistent with the results by ELISA, induction of VEGF mRNA expression by thrombin was greater than around 1.5 fold of that found in untreated RPE cells (p<0.01) and was significantly abolished by 40 U/ml of hirudin (Fig. 2B).
Figure 2.
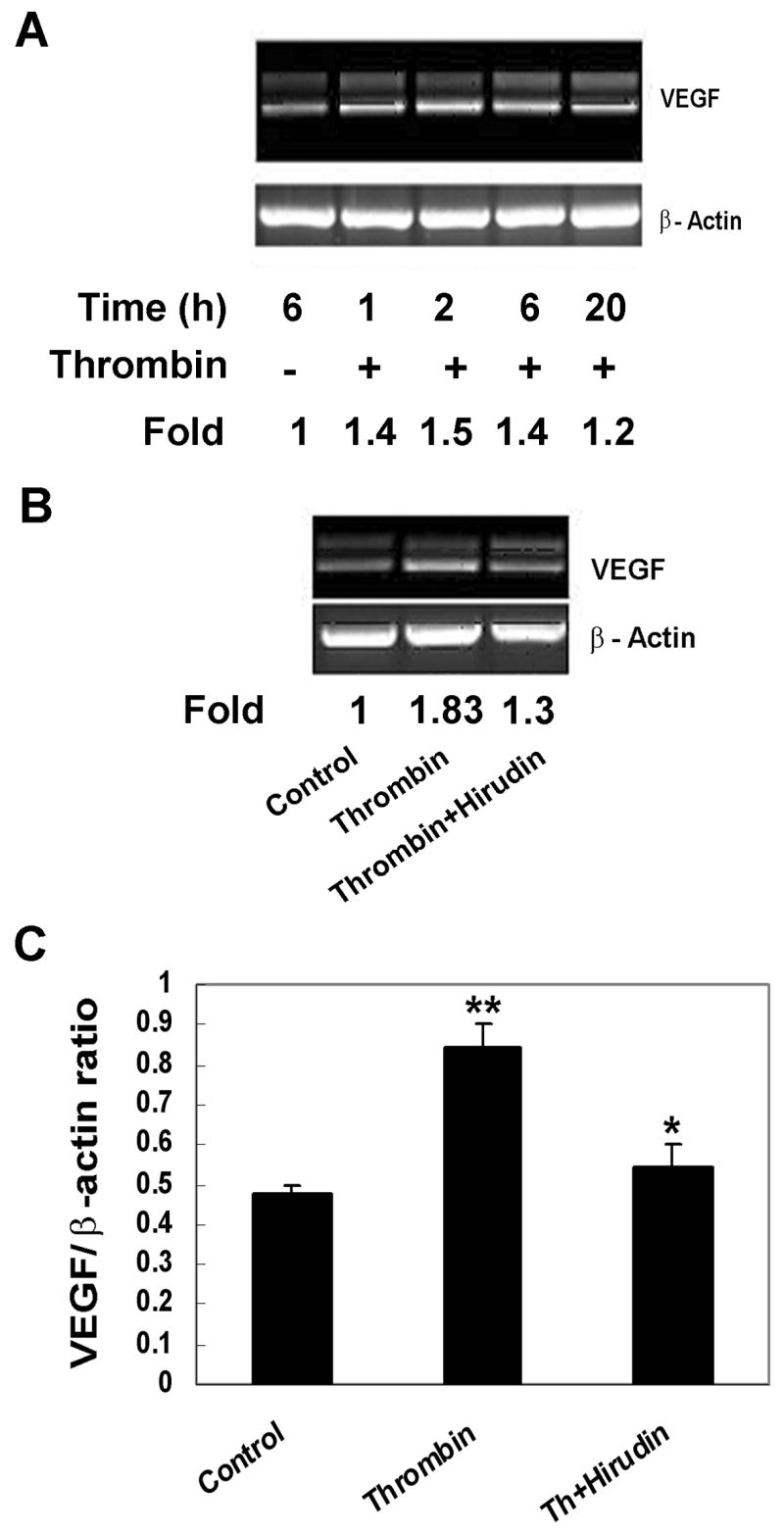
Stimulation of human RPE VEGF mRNA expression by thrombin. The cells were treated with thrombin (0 or10U/ml) for 1, 2, 6, and 20 hr (A) and with thrombin (10 U/ml) with or without hirudin (40 U/ml) for 6 hr (B). Total mRNA was isolated and subjected to RT-PCR analysis as described in Materials and Methods. The data shown in A and B represent results from a typical experiment. The fold changes were ratios of the normalized densities of VEGF PCR products by β-actin between treated and untreated samples (A and B). Statistical analysis of thrombin-induced VEGF mRNA expression in parallel samples is shown in lower panel of B. ** p<0.01; * p<0.05.
Multiple signal transduction pathways involved in thrombin-induced human RPE VEGF expression
Although the signal pathways for thrombin have been well documented in many cell types,31 the signal pathways for thrombin-induced VEGF production in human RPE cells remain unknown. To assess the signal transduction pathways involved in thrombin-induced VEGF gene expression in RPE cells, various inhibitors specific for signal mediators were employed (Table 1). The inhibitors included Ly294002 (100 μM), a PI3K inhibitor; U0126 (20 μM), an inhibitor of MEK, the signal molecule upstream from ERK; SB202190 (30 μM), a p38 inhibitor; Sp600125 (20 μM), a JNK inhibitor; CAPE (25 μg/ml), a NF-κB inhibitor; AG490 (50 μM), a Jak2 inhibitor; genistein (25 μg/ml), an inhibitor of tyrosine kinase; Ro31820 (10 μM), a PKC inhibitor; and Nac (25, 10, 1 mM) and DPI (25 μM), blockers of reactive oxygen species. As shown in Fig. 3, more than 90% of the thrombin-induced VEGF protein production was abolished by CAPE, Nac (25mM), and DPI (92, 100, and 93%). Nac inhibited thrombin-induced VEGF by 92% and 87% at 25 and 10 mM, respectively, while Nac at 1 mM resulted in only slight inhibition (Fig. 3B). The stimulated VEGF expression was also highly sensitive to Ro318220, U0126, SB202190, Ly294002, genistein, and Sp600125 with inhibition rates of 88, 80, 80, 75, 73 and 70%, respectively. By contrast, thrombin-induced VEGF production was resistant to AG490. To compare the signal pathways induced by thrombin to those stimulated by TGF-β2, results of similar experiments were obtained (data not shown) with one exception.
Figure 3.
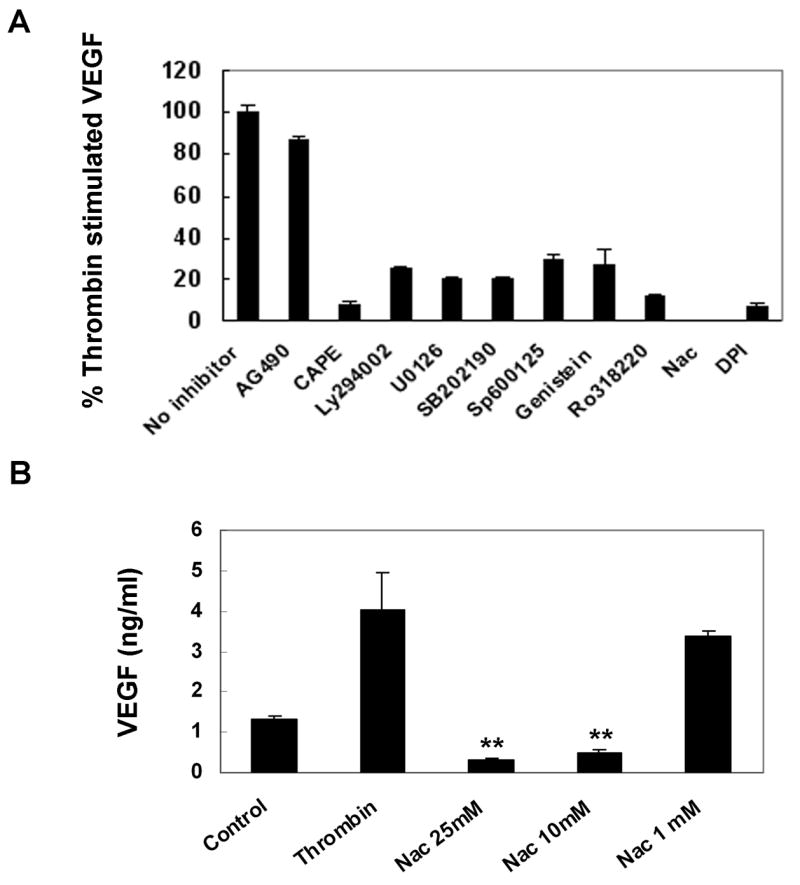
Inhibition of thrombin-stimulated human RPE VEGF production by inhibitors specific for various signal pathways. RPE cells were pretreated with AG490 (50 μM), CAPE (25 μg/ml), Ly294002 (100μM), U0126 (20 μM), SB202190 (30 μM), Sp600125 (20 μM), genistein (25 μg/ml), Ro31822 (10 μM), Nac (25, 10 and 1 mM), DPI (25 μM), and then co-stimulated with thrombin (10U/ml) for 24 hr. The thrombin-stimulated RPE cells were used as the positive control. Values represent are means ± SEM (n=3). Statistic analyses: all values versus the corresponding control, except AG490 had p<0.001.
Additive increase in thrombin-induced human RPE VEGF production by TNF-α and human monocyte co-culture
The study of thrombin in combinations with other pro-angiogenic stimuli is also important for fully understanding the role of thrombin in vivo. TNF-α was previously shown to induce human RPE VEGF.32 We previously showed that thrombin-induced TNF-α is responsible for human RPE/monocyte co-culture-induced chemokine expression.24 These interactions led us to examine induction of RPE VEGF secretion by thrombin plus TNF-α and by thrombin plus co-culture with monocytes. Incubation of human RPE cells with TNF-α (20ng/ml) induced VEGF protein secretion above basal levels (Fig. 4). As compared to induction by TNF-α and by thrombin alone (0.48± 0.04 and 1.96 ± 0.07ng/ml, respectively), combined stimulation with both agents resulted in enhanced VEGF secretion very close to the sum of VEGF induced by the two factors alone (2.71 vs. 2.44ng/ml), suggesting that TNF-α and thrombin are additive in stimulating VEGF production in RPE cells.
Figure 4.
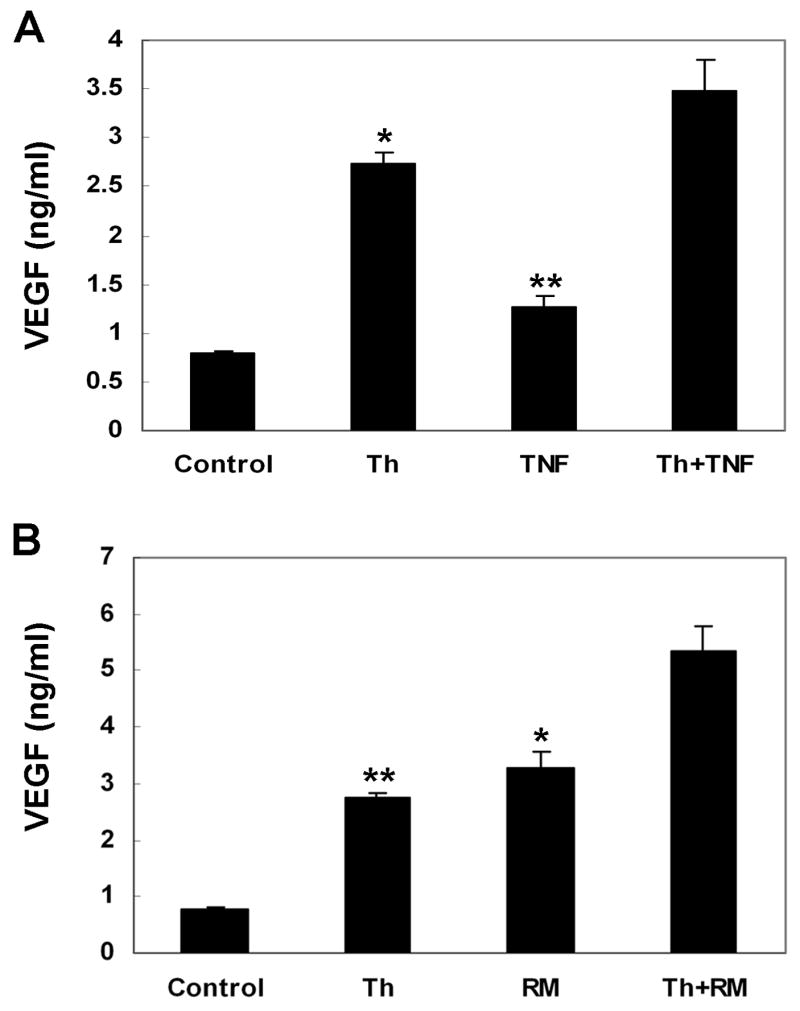
Effects of TNF-α and human RPE/monocyte co-culture on thrombin-stimulated RPE VEGF secretion. The RPE cells were stimulated by thrombin (10 U/ml) alone and in combination with TNF-α(20 ng/ml) (A), and co-culture with human monocytes (ratio, 1: 1.5) (B) for 24 hr. Unstimulated RPE cells were used as control. The conditioned media were assayed for VEGF using ELISA. Values represent means ± SEM (n=3) in ELISA assay.
We then tested the effect of thrombin on monocyte-induced RPE VEGF secretion. Similar additive induction was observed when monocytes were overlaid onto human PRE cells in the presence of thrombin. Monocytes co-cultured with PRE cells for 24 hr induced VEGF secretion (3.28 ± 0.27ng/ml) more than that obtained with 10 U/ml thrombin (2.74 ± 0.1 ng/ml). The combined induction by monocyte co-culture with thrombin was approximately equal to the sum of VEGF induced by each of these stimulants alone (4.58 vs. 4.46ng/ml).
Synergistic effect of thrombin plus TGF-β2 on stimulating RPE VEGF expression
As TGF-β2 is the predominant form of TGF-β found in ocular tissues, 33 we next focused on the effect of TGF-β2 on VEGF expression induced by thrombin, to determine if TGF-β2 alone or in combination with thrombin alters VEGF mRNA and protein production in human RPE cells. We also performed experiments to dermine whether thrombin induces TGF beta2 production in hRPE cells and found that it does not do so (unpublished data).
Incubation of RPE cells with TGF-β2 (10ng/ml) for 24 hr potently induced VEGF secretion. The level of induction by TGF-β2 was about 4-fold of that stimulated by 10 U/ml thrombin (4.18 VS 0.94ng/ml; p<0.001) (Fig. 5A). The same concentration of TGF-β2 markedly synergized thrombin-induced VEGF protein secretion. The amount of VEGF jointly induced by TGF-β2 plus thrombin was about 3.5-fold of the level anticipated if both factors were simply additive in stimulating VEGF secretion (18.1 vs. 5.12ng/ml; p<0.001). To further characterize this synergistic effect, the treated cells were lysed and subjected to ELISA to determine cell associated VEGF. In unstimulated RPE cells, VEGF was barely detectable (0.11 ± 0.03 ng/ml). Upon stimulation by thrombin (10 U/ml) for 24 hr, intracellular VEGF was enhanced, albeit at very low level (0.1ng/ml), while in TGF-β2 (10 ng/ml) treated RPE cells the induced VEGF protein was about 4–fold higher than that resulting by thrombin stimulation (0.42 VS 0.1 ng/ml). Consistent with the synergistic effect of RPE VEGF secretion observed in the conditioned medium, RPE cells exposed to TGF-β2 and thrombin contained VEGF at levels 3.4-fold more than that expected simply from an additive effect (1.78 vs. 0.52ng/ml; p<0.01).
Figure 5.

Effects of TGF-β2 on thrombin-stimulated human RPE VEGF protein secretion (A), cell-associated VEGF protein production (B), and VEGF mRNA expression (C). RPE cells were stimulated with thrombin (10 U/ml) (Th) alone or in combination with of TGF-β2 (10 ng/ml) (TGF) for 6 (C) or 24 hr (A, B). The media overlying the cells were then collected and the cells were lysed for VEGF determination by ELISA. For assessing VEGF mRNA expression in RPE cells, total RNA was extracted from RPE cells, and VEGF mRNA levels were determined with a Quantikine mRNA coloriometric quantification kit. Unstimulated RPE cells and RPE cells incubated with hirudin were used as controls.
To illustrate synergistic increases in VEGF mRNA production by co-administration of thrombin and TGF-β2, we examined VEGF mRNA by colorimetric quantification of PCR products. As seen in Fig. 5C, combined stimulation by thrombin and TGF-β2 (146.5 ± 2.5 amole/μg total RNA) resulted in a 3-fold increase in VEGF mRNA over the sum of the VEGF mRNA induced by TGF-β2 and thrombin separately (29.6 ± 1.4 and 19.0 ± 1.9 amole/μg total RNA, respectively; p<0.001). This synergistic increase (3-fold) at the mRNA level was close to the protein increases (3.5-fold) found by ELISA, suggesting that the synergistic effect at the protein level is due to increases in both VEGF mRNA expression and translation. Thrombin induced VEGF mRNA synthesis was also proved to be sensitive to inhibition of thrombin enzymatic activity by hirudin, consistent with the ELISA (Fig. 1) and RT-PCR findings (Fig. 2).
In agreement with ELISA data depicted in Fig 5A and 5B, immunocytochemical staining of intracellular RPE VEGF protein by anti-VEGF antibodies showed similar qualitative trends (Fig. 6). Human RPE cells stimulated by thrombin plus TGF-β2 (Fig. 6E) resulted in the most intense immunoreactivity when compared to cells stimulated by thrombin and TGF-β2 alone (Fig. 6C and D). By contrast, RPE cells treated with pre-immune serum (Fig. 6A) were completely negative and unstimulated quiescent cells showed only weak immunostaining for VEGF (Fig. 6B).
Figure 6.
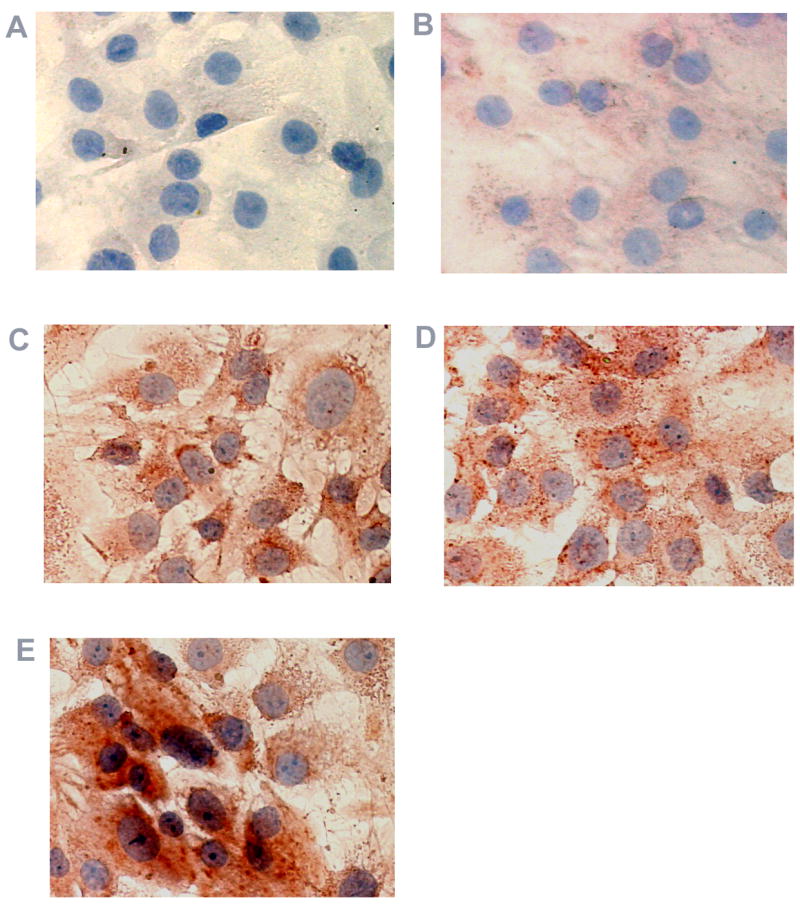
Immunohistochemical analysis of VEGF protein expression in cultured human RPE cells. RPE cells were exposed to thrombin, TGF- β2, or thrombin plus TGF- β2 for 16 hr. Cells in (A) were stained with normal rabbit serum serving as non-specific control. Untreated RPE cells (B) and cells treated with thrombin (C), TGF- β2 (D), and thrombin plus TGF- β2 (E) were stained using rabbit anti-human VEGF antibody and the red chromogen, AEC.
Thrombin is a potent mediator of calcium mobilization in endothelial cells. This effect has been suggested to be the major metabolic link between thrombin and TNF-α in synergistically stimulating tissue factor (thromboplastin; factor III) expression in endothelial cells.34 Thus, the role which thrombin-stimulated calcium mobilization plays in VEGF induction is of interest. To investigate this possibility, we pretreated human RPE cells with BAPTA/AM, a cell-permeable calcium chelator for 1 hr prior to challenge with TGF-β2, thrombin, or thrombin plus TGF-β2. ELISA data showed that BAPTA/AM did not affect TGF-β2-induced VEGF secretion (Fig. 7). In contrast, more than 65% of thrombin-induced VEGF expression was blocked by BAPTA/AM. VEGF stimulation due to thrombin plus TGF-β2 was only reduced by 20%, but the actual reduction in VEGF protein (2.1ng/ml) was about 5-fold more than the amount of VEGF inhibited due to thrombin stimulation alone (0.44 ng/ml). These data suggest that thrombin-mediated calcium mobilization, at least in part, accounts for the synergistic increases in VEGF expression caused by thrombin plus TGF-β2.
Figure 7.

Effects of intracellular calcium chelation on VEGF expression induced by thrombin and TGFβ-2. RPE cells were stimulated after preincubation with 5 μM BAPTA [1, 2-bis(o-aminophenoxy)ethane-N,N,N′, N′-tetraacetic acid] for 1 hr, then stimulated with thrombin (10 U/ml), TGFβ-2 (10 ng/ml) or both for 24 hr. The conditioned media were assayed for VEGF using ELISA. The values represent means ± SEM (n=3). Ctl, control; BA, BAPTA; TGFβ, TGF-β2; Th, thrombin.
Discussion
VEGF protein exists as a homodimer of four alternatively spliced variants of a single gene.35 Upon binding to its tyrosine kinase receptors (KDR/VEGF-R1 and Flt-1/VEGF-R2), VEGF triggers a cascade of downstream signaling events, which leads to physiological and pathological angiogenesis.36 While VEGF is expressed in various cell types, expression of KDR and flt-1 is primarily restricted to vascular endothelial cells.37
There is ample immunohistochemical, molecular, genetic, and pharmacologic evidence that VEGF is one of the most important factors leading to ocular neovascularization.38, 39 For example, intraocular VEGF levels are elevated in patients with PDR, and may reach 15 to 30 times higher than those present in nondiabetic control patients. VEGF is a pro-angiogenic mitogen that also promotes vascular permeability. It is implicated as a cause of blood-retina barrier breakdown within the neurosensory retina in diabetic retinopathy. In addition, RPE-derived VEGF may also contribute to dysfunction of neurosensory and outer retinal blood barrier, and induce choroidal neovascularization. In experimental animals, injection of adeno-associated virus-VEGF into the subretinal space of rats causes extensive subretinal neovascularization, degeneration of photoreceptors, and proliferation of RPE cells. In addition, silencing VEGF by small interfering RNA (siRNA) effectively inhibits choroidal neovascularization in a mouse model.
On the other hand, overexpression of VEGFs and their receptors increases microvascular permeability and angiogenesis of the outer retina in AMD.40 VEGF protein has been known expressed in human RPE cells of surgically excised choroidal neovascular membranes of AMD and other ocular diseases.32 VEGF is induced by various stimuli, including hypoxia, increased levels of reactive oxygen species, vasopressor hormones (angiotensin II and vasopressin, advanced glycation end products), cytokines (TGF-β, IL-1), and growth factors such as basic fibroblast growth factor (bFGF). The induction of VEGF is thought to be the consequence of local inflammatory response.41 In this study we demonstrated for the first time that thrombin is a potent stimulus for VEGF expression by RPE cells. This action is mediated by thrombin receptor PAR-1, not PAR-3. The thrombin-specific action was evidenced by inhibition with hirudin, a thrombin antagonist, and by mimicking the stimulation with a PAR-1 specific agonist. These results suggest that thrombin is a potential angiogenic factor in RPE-mediated neovascularization and that RPE-derived VEGF may then perpetrate its own production by enhancing thrombin accumulation at sites of blood-retina barrier breakdown.
The angiogenic action of thrombin has been demonstrated both in vivo and in vitro. 42, 43 Many thrombin-mediated cellular effects on endothelial cells may contribute to angiogenesis. It has been reported that thrombin causes time- and dose-dependent decreases in endothelial attachment to basement membrane components, permits endothelial cell migration, and increases DNA synthesis in endothelial cells, thus promoting endothelial cell proliferation.15 Thrombin also up-regulates expression of integrin αvβ3, a marker for angiogenic endothelial cells, and activates matrix metalloproteinase 2.15 Furthermore, our laboratory and others have shown that thrombin stimulates pro-angiogenic IL-8 and MCP-1 secretion by RPE cells.24, 44
In the present study we also investigated the signal pathways leading to enhanced VEGF gene expression by thrombin and TGF-β2. The mechanisms underlying regulation of VEGF gene expression and protein production have been the subject of intensive investigation.31,45 Expression of VEGF is controlled at both transcriptional and post-transcriptional (mRNA stabilization) levels via complicated signaling pathways31,45–47 that lead transcription factors, including HIF-1, Smads, Ap1, Ap-2, Sp-1, and NFkB, to bind VEGF promoter at specific, functional DNA-binding sequences.46–48 VEGF expression is further tuned by stabilization of VEGF mRNA14, 49, 50 and through crosstalk between different pathways.51 In this study we used a pharmacological approach, demonstrating that the stimulation of VEGF expression by thrombin was mediated by multiple signal pathways post PAR-1, including MAPK/ERK, p38, JNK MAPK pathways, PI3-K and NF-kB pathways, as well as by activation of PTK, PKC, and ROS. Those signal pathways are also shared by TGF-β2 in stimulation of VEGF expression in RPE cells, although thrombin-induced RPE VEGF expression was more sensitive to the inhibition by Sp600125 than that induced by TGF-β2 (data not shown), suggesting that the JNK pathway is more important for thrombin induction of RPE VEGF than for TGF-β2 induction.
Depending on cell types and stimuli, all these signal molecules have been reported for VEGF gene expression.31,45, 52 Although STAT3, the downstream target of jak2, is required for transcriptional control of VEGF gene expression and blockade of Jak2 by AG490 abrogates the induced VEGF production in many cell types,53 the present studies do not support an involvement of Jak2 pathway in thrombin- and TGF-β2-induced VEGF gene expression in RPE cells.
Up-regulation of VEGF has been observed in various cell types by thrombin, 13, 14, 54 TNF-α,44 TGF-β2,55 and monocyte contact.56 As demonstrated in the present study, thrombin, monocyte cell-to-cell contact, TGF-β2, and TNF-α all independently stimulate human RPE to produce VEGF. Our results with the latter two pro-angiogenic factors were consistent with previous reports.44,55 In addition to these angiogenic effects by thrombin alone, thrombin may amplify its effects through interplays with other pro-angiogenic agents such as TNF-α and TGF-β. For example, thrombin works in synergy with TNF-α, causing large increases in endothelial permeability,57,58 and simultaneous treatment of HepG2 cells with thrombin and TGF-β synergistically enhances the levels of plasminogen activator inhibitor type-1.59 As shown in this study, stimulation of human RPE VEGF by thrombin and TGF-β2 appears to be mediated by highly overlapped signaling pathways. However, the mechanism underlying the synergistic effect of thrombin and TGF-β2 in stimulating RPE VEGF is not fully understood. The synergy between thrombin and TGF-β2 is likely to result from the combination of the signaling pathways unique to each stimulus. The Smad pathway by TGF-β51 and the Ca+2 pathway by thrombin60 are the likely candidates for the synergistic action of the two factors. Consistent with this hypothesis is our finding that 36% of thrombin-induced and 19% of thrombin plus TGF-β2-induced, but not TGF-β2-induced RPE VEGF stimulation was sensitive to the calcium chelator, BAPTA. The remaining BAPTA-insensitive synergy between the two factors remains to be investigated. The TGF-β2-specific Smad pathway is likely to be the major contributor.
Our findings of the independent and interactive up-regulation of VEGF production by thrombin, TNF-α, TGF-β2 and monocytes in hRPE cells are likely to be pathologically relevant. Thrombin is generated in areas of hemorrhage and in diseased retinal tissue.61, 62 Thrombin has been thought to be a potent stimulus for inflammation, which aggravates disease processes.21,63 The role of thrombin in ocular neovascularizaiton is reminiscent of its role in angiogenesis and tumor progression.64 On the one hand, thrombin is able to induce VEGF expression in tumor cells. On the other hand, thrombin up-regulates thrombin receptors (KDR and Flt-1) on endothelial cell. The dual role of thrombin results in a synergistic activation of angiogenesis. Likewise, it is possible that plasma-derived thrombin potentiates other pro-angiogenic cytokines such as TGF-β2 and TNF-α, as well as mononuclear phagocytes to stimulate RPE VEGF secretion. The secreted VEGF then binds to VEGF receptors in endothelial cells that are up regulated by thrombin, exacerbating ocular angiogenesis. Further studies of the interplay among those pro-angiogenic factors in ocular neovascularization under diseased conditions will shed more insight into the important role of these factors in PDR, PVR, and AMD, and suggest potential approaches for pharmacological interventions. One such intervention may be the specific inhibition of PAR-1 mediated, thrombin of RPE VEGF.
Acknowledgments
The authors wish to thank Margarete G Ribianszky for excellent technical assistance.
* This study was supported by NIH Grants EY-09441, EY007003, and Research to Prevent Blindness-Senior Scientific Investigator Award (VME).
References
- 1.Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005;85:845–881. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- 2.Elner VM, Burnstine MA, Strieter RM, Kunkel SL, Elner SG. Cell-associated human retinal pigment epithelium interleukin-8 and monocyte chemotactic protein-1: immunochemical and in-situ hybridization analyses. Exp Eye Res. 1997;65:781–789. doi: 10.1006/exer.1997.0380. [DOI] [PubMed] [Google Scholar]
- 3.Marano RJ, Rakoczy PE. Treatments for choroidal and retinal neovascularization: a focus on oligonucleotide therapy and delivery for the regulation of gene function. Clin Experiment Ophthalmol. 2005;33:81–89. doi: 10.1111/j.1442-9071.2005.00952.x. [DOI] [PubMed] [Google Scholar]
- 4.Adamis AP, Shima DT, Yeo KT, et al. Synthesis and secretion of vascular permeability factor/vascular endothelial growth factor by human retinal pigment epithelial cells. Biochem Biophys Res Commun. 1993;193:631–638. doi: 10.1006/bbrc.1993.1671. [DOI] [PubMed] [Google Scholar]
- 5.Adamis AP, Shima DT, Tolentino MJ, et al. Inhibition of vascular endothelial growth factor prevents retinal ischemia-associated iris neovascularization in a nonhuman primate. Arch Ophthalmol. 1996;114:66–71. doi: 10.1001/archopht.1996.01100130062010. [DOI] [PubMed] [Google Scholar]
- 6.Scholz M, Vogel JU, Hover G, et al. Thrombin stimulates IL-6 and IL-8 expression in cytomegalovirus-infected human retinal pigment epithelial cells. Int J Mol Med. 2004;13:327–331. [PubMed] [Google Scholar]
- 7.Yang Z, Cohen RL, Lui GM, Lawrence DA, Shuman MA. Thrombin increases expression of urokinase receptor by activation of the thrombin receptor. Invest Ophthalmol Vis Sci. 1995;36:2254–2261. [PubMed] [Google Scholar]
- 8.Fenton JW, Fasco MJ, Stackrow AB. Human thrombins. Production, evaluation, and properties of alpha-thrombin. J Biol Chem. 1977;252:3587–3598. [PubMed] [Google Scholar]
- 9.Shuman MA, Majerus PW. The measurement of thrombin in clotting blood by radioimmunoassay. J Clin Invest. 1976;58:1249–1258. doi: 10.1172/JCI108579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sagripanti A, Romani A, Ferretti A, et al. Blood coagulation parameters in retinal arterial occlusion. Graefes Arch Clin Exp Ophthalmol. 1999;237:480–483. doi: 10.1007/s004170050265. [DOI] [PubMed] [Google Scholar]
- 11.Sakamoto T, Sakamoto H, Sheu SJ, Gabrielian K, Ryan SJ, Hinton DR. Intercellular gap formation induced by thrombin in confluent cultured bovine retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1994;35:720–729. [PubMed] [Google Scholar]
- 12.Dupuy E, Habib A, Lebret M, Yang R, Levy-Toledano S, Tobelem G. Thrombin induces angiogenesis and vascular endothelial growth factor expression in human endothelial cells: possible relevance to HIF-1alpha. J Thromb Haemost. 2003;1:1096–1102. doi: 10.1046/j.1538-7836.2003.00208.x. [DOI] [PubMed] [Google Scholar]
- 13.Bassus S, Herkert O, Kronemann N, Gorlach A, Bremerich D, Kirchmaier CM, Busse R, Schini-Kerth VB. Thrombin causes vascular endothelial growth factor expression in vascular smooth muscle cells: role of reactive oxygen species. Arterioscler Thromb Vasc Biol. 2001;21:1550–1555. doi: 10.1161/hq0901.095148. [DOI] [PubMed] [Google Scholar]
- 14.Huang YQ, Li JJ, Hu L, Lee M, Karpatkin S. Thrombin induces increased expression and secretion of VEGF from human FS4 fibroblasts, DU145 prostate cells and CHRF megakaryocytes. Thromb Haemost. 2001;86:1094–1098. [PubMed] [Google Scholar]
- 15.Maragoudakis ME, Tsopanoglou NE, Andriopoulou P. Mechanism of thrombin-induced angiogenesis. Biochem Soc Trans. 2002;30:173–177. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 16.Hirase K, Sugiyama T, Ikeda T, et al. Transforming growth factor beta(2) increases in subretinal fluid in rhegmatogenous retinal detachment with subretinal strands. Ophthalmologica. 2005;219:222–225. doi: 10.1159/000085731. [DOI] [PubMed] [Google Scholar]
- 17.Markomichelakis NN, Theodossiadis PG, Sfikakis PP. Regression of neovascular age-related macular degeneration following infliximab therapy. Am J Ophthalmol. 2005;139:537–540. doi: 10.1016/j.ajo.2004.09.058. [DOI] [PubMed] [Google Scholar]
- 18.Limb GA, Little BC, Meager A, et al. Cytokines in proliferative vitreoretinopathy. Eye. 1991;5 :686–693. doi: 10.1038/eye.1991.126. [DOI] [PubMed] [Google Scholar]
- 19.Doganay S, Evereklioglu C, Er H, et al. Comparison of serum NO, TNF-alpha, IL-1beta, sIL-2R, IL-6 and IL-8 levels with grades of retinopathy in patients with diabetes mellitus. Eye. 2002;16:163–170. doi: 10.1038/sj.eye.6700095. [DOI] [PubMed] [Google Scholar]
- 20.Amin R, Puklin JE, Frank RN. Growth factor localization in choroidal neovascular membranes of age-related macular degeneration. Invest Ophthalmol Vis Sci. 1994;35:3178–3188. [PubMed] [Google Scholar]
- 21.Hackett SF, Singer JH, Leschey KH, Campochiaro PA. Thrombin is a stimulator of retinal pigment epithelial cell proliferation. Exp Eye Res. 1991;53:95–100. doi: 10.1016/0014-4835(91)90150-d. [DOI] [PubMed] [Google Scholar]
- 22.Olsen TW, Sternberg P, Martin DF, Capone A, Jr, Lim JI, Aaberg TM. Postoperative hypopyon after intravitreal bovine thrombin for macular hole surgery. Am J Ophthalmol. 1996;121:575–577. doi: 10.1016/s0002-9394(14)75437-6. [DOI] [PubMed] [Google Scholar]
- 23.Kim SH, Cho YS, Choi YJ. Intraocular hemocoagulase in human vitrectomy. Jpn J Ophthalmol. 1994;38:49–55. [PubMed] [Google Scholar]
- 24.Yoshida A, Elner SG, Bian ZM, Kunkel SL, Lukacs NW, Elner VM. Thrombin regulates chemokine induction during human retinal pigment epithelial cell/monocyte interaction. Am J Pathol. 2001;159:1171–1180. doi: 10.1016/S0002-9440(10)61793-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elner VM, Strieter RM, Elner SG, Baggiolini M, Lindley I, Kunkel SL. Neutrophil chemotactic factor (IL-8) gene expression by cytokine-treated retinal pigment epithelial cells. Am J Pathol. 1990;136:745–750. [PMC free article] [PubMed] [Google Scholar]
- 26.Bian ZM, Elner SG, Yoshida A, Elner VM. Human RPE-monocyte co-culture induces chemokine gene expression through activation of MAPK and NIK cascade. Exp Eye Res. 2003;76:573–583. doi: 10.1016/s0014-4835(03)00029-0. [DOI] [PubMed] [Google Scholar]
- 27.Elner SG, Elner VM, Pavilack MA, et al. Huber AR: Modulation and function of intercellular adhesion molecule-1 (CD54) on human retinal pigment epithelial cells. Lab Invest. 1992;66:200–211. [PubMed] [Google Scholar]
- 28.Bian ZM, Elner SG, Yoshida A, Elner VM. Differential involvement of phosphoinositide 3-kinase/Akt in in human RPE MCP-1 and IL-8 expression. Invest Ophthalmol Vis Sci. 2004;45:1887–1896. doi: 10.1167/iovs.03-0608. [DOI] [PubMed] [Google Scholar]
- 29.Vassallo RR, Jr, Kieber-Emmons T, Cichowski K, Brass LF. Structure-function relationships in the activation of platelet thrombin receptors by receptor-derived peptides. J Biol Chem. 1992;267:6081–6085. [PubMed] [Google Scholar]
- 30.Ahn HS, Foster C, Boykow G, Stamford A, Graziano M. Inhibition of cellular action of thrombin by N3-cyclopropyl-7-{[4-(1-methylethyl)phenyl]methyl}-7H-pyrrolo[3,2-f]quinazoline-1,3-diamine ( SCH79797), a nonpeptide thrombin receptor antagonist. Biochem Pharmacol. 2000;60:1425–1434. doi: 10.1016/s0006-2952(00)00460-3. [DOI] [PubMed] [Google Scholar]
- 31.Mazure NM, Brahimi-Horn MC, Pouyssegur J. Protein kinases and the hypoxia-inducible factor-1, two switches in angiogenesis. Curr Pharm Des. 2003;9:531–541. doi: 10.2174/1381612033391469. [DOI] [PubMed] [Google Scholar]
- 32.Oh H, Takagi H, Takagi C, et al. The potential angiogenic role of macrophages in the formation of choroidal neovascular membranes. Invest Ophthalmol Vis Sci. 1999;40:1891–1898. [PubMed] [Google Scholar]
- 33.Pfeffer BA, Flanders KC, Guerin CJ, Danielpour D, Anderson DH. Transforming growth factor beta 2 is the predominant isoform in the neural retina, retinal pigment epithelium-choroid and vitreous of the monkey eye. Exp Eye Res. 1994;59:323–333. doi: 10.1006/exer.1994.1114. [DOI] [PubMed] [Google Scholar]
- 34.Liu N, Makino T, Nogaki F, et al. Coagulation in the mesangial area promotes ECM accumulation through factor V expression in MsPGN in rats. Am J Physiol Renal Physiol. 2004;287:F612–620. doi: 10.1152/ajprenal.00322.2003. [DOI] [PubMed] [Google Scholar]
- 35.Houck KA, Leung DW, Rowland AM, Winer J, Ferrara N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J Biol Chem. 1992;267:26031–26037. [PubMed] [Google Scholar]
- 36.Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- 37.Terman BI, Dougher-Vermazen M, Carrion ME. Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochem Biophys Res Commun. 1992;187:1579–1586. doi: 10.1016/0006-291x(92)90483-2. [DOI] [PubMed] [Google Scholar]
- 38.Miller JW. Vascular endothelial growth factor and ocular neovascularization. Am J Pathol. 1997;151:13–23. [PMC free article] [PubMed] [Google Scholar]
- 39.Takahashi H, Shibuya M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin Sci. 2005;109:227–241. doi: 10.1042/CS20040370. [DOI] [PubMed] [Google Scholar]
- 40.Kliffen M, Sharma HS, Mooy CM, Kerkvliet S, de Jong PT. Increased expression of angiogenic growth factors in age-related maculopathy. Br J Ophthalmol. 1997;81:154–162. doi: 10.1136/bjo.81.2.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kvanta A, Algvere PV, Berglin L, Seregard S. Subfoveal fibrovascular membranes in age-related macular degeneration express vascular endothelial growth factor. Invest Ophthalmol Vis Sci. 1996;37:1929–1934. [PubMed] [Google Scholar]
- 42.Tsopanoglou NE, Pipili-Synetos E, Maragoudakis ME. Thrombin promotes angiogenesis by a mechanism independent of fibrin formation. Am J Physiol. 1993;264:C1302–1307. doi: 10.1152/ajpcell.1993.264.5.C1302. [DOI] [PubMed] [Google Scholar]
- 43.Haralabopoulos GC, Grant DS, Kleinman HK, Maragoudakis ME. Thrombin promotes endothelial cell alignment in Matrigel in vitro and angiogenesis in vivo. Am J Physiol. 1997;273:C239–245. doi: 10.1152/ajpcell.1997.273.1.C239. [DOI] [PubMed] [Google Scholar]
- 44.Yoshida S, Ono M, Shono T, et al. Involvement of interleukin-8, vascular endothelial growth factor, and basic fibroblast growth factor in tumor necrosis factor alpha-dependent angiogenesis. Mol Cell Biol. 1997;17:4015–4023. doi: 10.1128/mcb.17.7.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arisato T, Sarker KP, Kawahara K, et al. The agonist of the protease-activated receptor-1 (PAR) but not PAR3 mimics thrombin-induced vascular endothelial growth factor release in human smooth muscle cells. Cell Mol Life Sci. 2003;60:1716–1724. doi: 10.1007/s00018-003-3140-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Josko J, Mazurek M. Transcription factors having impact on vascular endothelial growth factor (VEGF) gene expression in angiogenesis. Med Sci Monit. 2004;10:RA89–98. [PubMed] [Google Scholar]
- 47.Shima DT, Kuroki M, Deutsch U, Ng YS, Adamis AP, D'Amore PA. The mouse gene for vascular endothelial growth factor. Genomic structure, definition of the transcriptional unit, and characterization of transcriptional and post-transcriptional regulatory sequences. J Biol Chem. 1996;271:3877–3883. doi: 10.1074/jbc.271.7.3877. [DOI] [PubMed] [Google Scholar]
- 48.Sanchez-Elsner T, Botella LM, Velasco B, Corbi A, Attisano L, Bernabeu C. Synergistic cooperation between hypoxia and transforming growth factor-beta pathways on human vascular endothelial growth factor gene expression. J Biol Chem. 2001;276:38527–38535. doi: 10.1074/jbc.M104536200. [DOI] [PubMed] [Google Scholar]
- 49.Pages G, Berra E, Milanini J, Levy AP, Pouyssegur J. Stress-activated protein kinases (JNK and p38/HOG) are essential for vascular endothelial growth factor mRNA stability. J Biol Chem. 2000;275:26484–26491. doi: 10.1074/jbc.M002104200. [DOI] [PubMed] [Google Scholar]
- 50.Berra E, Pages G, Pouyssegur J. MAP kinases and hypoxia in the control of VEGF expression. Cancer Metastasis Rev. 2000;19:139–145. doi: 10.1023/a:1026506011458. [DOI] [PubMed] [Google Scholar]
- 51.Javelaud D, Mauviel A. Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: implications for carcinogenesis. Oncogene. 2005;24:5742–5750. doi: 10.1038/sj.onc.1208928. [DOI] [PubMed] [Google Scholar]
- 52.Grand RJ, Turnell AS, Grabham PW. Cellular consequences of thrombin-receptor activation. Biochem J. 1996;313:353–368. doi: 10.1042/bj3130353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chang KT, Tsai CM, Chiou YC, Chiu CH, Jeng KS, Huang CY. IL-6 induces neuroendocrine dedifferentiation and cell proliferation in non-small cell lung cancer cells. Am J Physiol Lung Cell Mol Physiol. 2005;289:L446–453. doi: 10.1152/ajplung.00089.2005. [DOI] [PubMed] [Google Scholar]
- 54.Lockwood CJ, Krikun G, Koo AB, Kadner S, Schatz F. Differential effects of thrombin and hypoxia on endometrial stromal and glandular epithelial cell vascular endothelial growth factor expression. J Clin Endocrinol Metab. 2002;87:4280–4286. doi: 10.1210/jc.2001-011969. [DOI] [PubMed] [Google Scholar]
- 55.Nagineni CN, Samuel W, Nagineni S, et al. Transforming growth factor-beta induces expression of vascular endothelial growth factor in human retinal pigment epithelial cells: involvement of mitogen-activated protein kinases. J Cell Physiol. 2003;197:453–462. doi: 10.1002/jcp.10378. [DOI] [PubMed] [Google Scholar]
- 56.Hojo Y, Ikeda U, Ohya K, et al. Interaction between monocytes and vascular endothelial cells induces adrenomedullin production. Atherosclerosis. 2001;155:381–387. doi: 10.1016/s0021-9150(00)00607-9. [DOI] [PubMed] [Google Scholar]
- 57.Liu Y, Pelekanakis K, Woolkalis MJ. Thrombin and tumor necrosis factor alpha synergistically stimulate tissue factor expression in human endothelial cells: regulation through c-Fos and c-Jun. J Biol Chem. 2004;279:36142–36147. doi: 10.1074/jbc.M405039200. [DOI] [PubMed] [Google Scholar]
- 58.Tiruppathi C, Naqvi T, Sandoval R, Mehta D, Malik AB. Synergistic effects of tumor necrosis factor-alpha and thrombin in increasing endothelial permeability. Am J Physiol Lung Cell Mol Physiol. 2001;281:L958–968. doi: 10.1152/ajplung.2001.281.4.L958. [DOI] [PubMed] [Google Scholar]
- 59.Hopkins WE, Fujii S, Sobel BE. Synergistic induction of plasminogen activator inhibitor type-1 in HEP G2 cells by thrombin and transforming growth factor-beta. Blood. 1992;79:75–81. [PubMed] [Google Scholar]
- 60.Sarker KP, Biswas KK, Yamaji K, et al. Inhibition of thrombin-induced vascular endothelial growth factor production in human neuroblastoma (NB-1) cells by argatroban. Pathophysiol Haemost Thromb. 2005;34:41–47. doi: 10.1159/000088547. [DOI] [PubMed] [Google Scholar]
- 61.Imokawa Y, Brockes JP. Selective activation of thrombin is a critical determinant for vertebrate lens regeneration. Curr Biol. 2003;13:877–881. doi: 10.1016/s0960-9822(03)00294-x. [DOI] [PubMed] [Google Scholar]
- 62.Malukiewicz-Wisniewska G, Kotschy M. Thrombin-antithrombin III complexes in plasma and subretinal fluid in patients with perforated retinal detachment. Klin Oczna. 2000;102:267–270. [PubMed] [Google Scholar]
- 63.Chen LB, Buchanan JM. Mitogenic activity of blood components. I. Thrombin and prothrombin. Proc Natl Acad Sci U S A. 1975;72:131–135. doi: 10.1073/pnas.72.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maragoudakis ME, Tsopanoglou NE, Andriopoulou P, Maragoudakis MM. Effects of thrombin/thrombosis in angiogenesis and tumour progression. Matrix Biol. 2000;19:345–351. doi: 10.1016/s0945-053x(00)00079-2. [DOI] [PubMed] [Google Scholar]