


Abstract
Parallel, highly specific analysis methods are required to take advantage of the extensive information about DNA sequence variation and of expressed sequences. We present a scalable laboratory technique suitable to analyze numerous target sequences in multiplexed assays. Sets of padlock probes were applied to analyze single nucleotide variation directly in total genomic DNA or cDNA for parallel genotyping or gene expression analysis. All reacted probes were then co-amplified and identified by hybridization to a standard tag oligonucleotide array. The technique was illustrated by analyzing normal and pathogenic variation within the Wilson disease-related ATP7B gene, both at the level of DNA and RNA, using allele-specific padlock probes.
INTRODUCTION
Over the last few years millions of single nucleotide polymorphisms (SNPs) have been identified that represent a large proportion of common genetic variation among the humans. It is an important objective of future genetic studies to correlate these molecular genetic variations with human phenotypic variation in health and disease. It remains an analytical challenge, however, to comprehensively access information about the genetic constitution of individual patients. A great number of genotyping methods have been developed to analyze large sets of SNPs. Most methods are based on analysis of individual markers, typically using PCR to amplify the segments harboring the variable nucleotide positions (1). In the interest of greater throughput, a number of approaches have recently been established for parallel analyses of large sets of markers in individual reactions. Amplification by PCR is inherently difficult to perform in a highly multiplexed format, but multiple individual or modestly multiplexed PCR reactions can be pooled and analyzed in parallel on dense resequencing microarrays, revealing thousands of genotypes per hybridization in a serial–parallel process (2,3). A parallel–parallel process was used to genotype and analyze the expression of up to 1000 markers by performing multiplexed target-dependent ligation of pairs of oligonucleotides, followed by amplification and sorting by hybridization to fiber optic bead arrays with DNA tag sequences (4,5).
We have recently presented an approach for parallel gene analysis using large pools of sequence-tagged padlock probes. These were added to individual DNA samples and subjected to gap-fill ligation followed by exonuclease treatment to remove both unreacted and any cross-reactive products that might arise through intermolecular ligation. Next, the circularized probes were molecularly inverted by linearization at a position remote from the site of ligation and amplified by PCR. Finally, the probes were analyzed on commercially available tag sequence arrays (6). Using this strategy individual DNA samples were analyzed for more than 1500 SNPs in parallel, and more recently the technique has been used to analyze sets of 13 000 markers (T.Willis, ParAllele BioScience, personal communication). The approach exploits some useful properties of padlock probes, oligonucleotides that circularize in target sequence-dependent ligation reactions (7). The requirement for recognition of adjacent target sequences by the two ends of these linear probes provide sufficient specificity to analyze SNPs in total genomic DNA without prior target amplification (6,8–10). Moreover, as shown by Hardenbol et al. (6), large numbers of padlock probes can be combined in single reactions since amplification of unreacted and cross-reacted probes can be counteracted by exonuclease treatment.
In this paper we report an alternative protocol for padlock-based analyses of sequence variants in genomic DNA or transcribed sequences. Sets of allele-specific padlock probes were combined in a single ligation reaction, followed by exonucleolytic removal of unreacted and cross-reacted probes. The remaining circularized probes were amplified and the products identified in an array-of-arrays, prepared using a standard printing robot. Twenty-six allele-specific padlock probes were used to genotype 75 individuals at 100% call rate and accuracy, and with sufficient signal strength for further scale-up. The method was also used to evaluate allele-specific gene expression by comparing genomic DNA samples from liver necropsies with cDNA prepared from the same samples. We discuss differences between the present approach and that used in Hardenbol et al. (6).
MATERIALS AND METHODS
DNA and RNA samples
DNA samples were from patients with Wilson’s disease, cared for at Uppsala University hospital (11), and from anonymous blood donors. Liver necropsies were kindly provided by Dr Fredrik Pontén. DNA and RNA were extracted using a Trizol protocol (Gibco BRL). Second strand cDNA was prepared by incubating 0.1 µg/µl total RNA in 50 µl of 50 mM Tris–HCl pH 8.3, 3 mM MgCl2, 75 mM KCl, 10 mM DTT, 1 µM random hexamer primers (Gibco BRL), 0.8 U/µl HPRI ribonuclease inhibitor (Amersham Bioscience) and 3 U/µl M-MLV reverse transcriptase (USB) for 30 s at 50°C, followed by 1 h at 37°C and finally 80°C for 10 min. An aliquot of 5 µl of second strand reaction mix, containing 50 mM Tris–HCl pH 7.5, 10 mM MgCl2, 1 mM DTT, 0.25 µg BSA, 5 µM random primers and 5 U Klenow fragment (Amersham Bioscience) was added to 20 µl of the first strand reaction and incubated at 37°C for 1 h, followed by 65°C for 15 min. The reactions were finally diluted 10-fold in water.
Oligonucleotides
All padlock probes were evaluated using Oligo 6.6 software (Molecular Biology Insights Inc.) to avoid secondary structure that might interfere with probe function. The 104 nt long probes were synthesized, chemically 5′-phosphorylated and gel purified by Thermo Electron (Ulm, Germany). Tag oligonucleotide sequences were selected from the GeneFlex™ Tag Array collection (Affymetrix, Santa Clara, CA) that contains sequence information for 2000 oligonucleotides with minimal tendency for cross-hybridization. Padlock probe sequences and amplification primers are presented in Table 1.
Table 1. Oligonucleotide sequences used for parallel gene analyses (5′→3′).
| Padlock probe | 5′-End segment | Common sequence | Allele tag | Locus tag | 3′-End segment |
|---|---|---|---|---|---|
| 1216(G/T) | gggattataaagaactgttgc | CTCGACCGTTAGCAGCATGAtt | (A1/A2) | GTTGATGTCATGTGTCGCAC | ttcttctgggctaattacag(c/a) |
| 1366(C/G) | agatgtaggtgtaccatcAgtag | CTCGACCGTTAGCAGCATGAtt | (A1/A2) | TCGTGCCTTGTCATTCGGGA | gggagccacttcctgca(g/c) |
| 1877(C/G) | caatttccttgtcattaaaaa | CTCGACCGTTAGCAGCATGAtt | (A1/A2) | CGTGCAAGTTACCGAGCTGA | gggccagggaagcatgaaag(g/c) |
| 2128(A/G) | gaggagctgaaagacaaggaA | CTCGACCGTTAGCAGCATGAtt | (A1/A2) | TAGATCAGTTGGACTCGATG | Cacgtagaagtaccacccac(t/c) |
| 2495(G/A) | tgacgatatcgccccgctTca | CTCGACCGTTAGCAGCATGAtt | (A1/A2) | GCAGGGAATTGCCGACCATA | acttAcccccagggaccacc(c/t) |
| 2855(G/A) | tctgaacaacaccaaaatcga | CTCGACCGTTAGCAGCATGAtt | (A1/A2) | ACGTTCGTCAAGAGTCGCAT | Atcaacttacaggaaagtat(c/t) |
| 2930(C/T) | tgatggacgtctggaaagcaa | CTCGACCGTTAGCAGCATGAuu | (A1/A2) | CGTTCCTAAAGCTGAGTCTG | ggcaggcaatgcacagcacc(g/a) |
| 2973(G/A) | gtggccagccccagggagcag | CTCGACCGTTAGCAGCATGAtt | (A1/A2) | GAGAGGCCGTCGCTATACAT | ccTaccatgacagccgtggg(c/t) |
| 3207(C/A) | tgttcactgctggcctccgca | CTCGACCGTTAGCAGCATGAtt | (A1/A2) | AAGCCAGATCGACCATCGTA | gtgactgccacgcccaaggg(g/t) |
| 3317(A/T) | ctttgcacccaattccacagc | CTCGACCGTTAGCAGCATGAtt | (A1/A2) | GACGCCGTTATGAGAGTCCA | ggatgccttccacgttgctg(t/a) |
| 3419(T/C) | ctgcatctattcaaaagaggc | CTCGACCGTTAGCAGCATGAtt | (A1/A2) | ATATCGTGTCACAGGTCGTT | tCacagagaaggtctggggg(a/g) |
| 3457(C/T) | ctcacggtttccaatcagcac | CTCGACCGTTAGCAGCATGAtt | (A1/A2) | ATGATGTGCAAAGTGCCGTC | taaTccgttgcgcctcagcc(g/a) |
| 3955(T/C) |
gacagtcctcttggaaaggtA |
CTCGACCGTTAGCAGCATGAtt |
(A1/A2) |
TCCGTCTGTTGAGTTAGGCC |
Taggttgatgcgtatccttc(a/g) |
| Primers and hybridization probes | |||||
| Common primer | TCATGCTGCTAACGGTCGAG | ||||
| Allele 1 specific primer and A1 | CCGAGATGTACCGCTATCGT | ||||
| Allele 2 specific primer and A2 | AGAGCGCATGAATCCGTAGT | ||||
| Hybridization control oligonucleotide | FI/TA – ttttttgaccgctgaagggctt | ||||
Padlock probes were designed to analyze sequence variations within the human ATP7B gene (GenBank accession no. NT_024524). Names of the probe pairs are given as the base pair numbers for the variable nucleotide positions, according to Petrukhin et al. (19), followed by the nucleotide variation in parentheses; allele 1 first, allele 2 second. For each padlock probe pair, the target-complementary sequences are in lower case and bold nucleotides in the target-complementary segments indicate mismatches introduced to prevent undesired self-complementary structures. The alternative nucleotide at the 3′-end of each padlock probe pair corresponds to alleles 1 and 2, respectively. Italic upper case indicates the segments where the common primer hybridizes and normal upper case indicates the locus tag sequences. Tag codes were selected from the GeneFlex™ Tag Array sequence collection (Affymetrix, Santa Clara, CA). The tag sequences specific for alleles 1 and 2 (A1 and A2) at any locus are located between the common and locus tag sequences. The A1 and A2 PCR primers, as well as a hybridization control oligonucleotide, were labeled with FITC or TAMRA. Spotting oligonucleotides for each locus and for the hybridization control are complementary to individual locus tag sequences and to the hybridization control oligonucleotide, respectively, with an additional 3′-(TC)7T-NH2 tail.
Ligation, exonuclease treatment and amplification
Genomic DNA was reduced in size by digestion using DraI and SspI (New England Biolabs) for 2 h in the recommended buffers. Aliquots of 10 µl of ligation reactions, containing 50 ng digested DNA or 2 µl of cDNA in 20 mM Tris–HCl pH 9.0, 100 mM KCl, 10 mM MgCl2, 1 mM EDTA, 1 mM DTT, 0.1% Triton X-100, 400 mU Tth ligase or Ampligase (Epicentre) and 400 pM each padlock probe were placed in a thermal cycler at 95°C for 5 min, then cycled 20 times between 95°C for 2 min and 55°C for 20 min, followed by 95°C for 2 min. Homozygous genotypes that were not present among our samples were instead represented by synthetic 40mer oligonucleotide targets added at 45 zmol to separate ligation reactions. After ligation, 10 µl of exonuclease mix was added to the reactions for a final concentration of 67 mM Tris–HCl pH 9.0, 50 mM KCl, 6.7 mM MgCl2, 0.05 µg/µl BSA, 0.35 U/µl exonuclease I (New England Biolabs) and 0.35 U/µl exonuclease III (Amersham Biosciences). Samples were incubated at 37°C for 2 h, followed by 95°C for 10 min. For PCR amplification, 6 µl of exonuclease reaction was combined with 24 µl of PCR mix, bringing the concentration to 30 mM Tris–HCl pH 8.8, 18 mM KCl, 3.0 mM MgCl2, 0.08% Triton X-100, 200 µM dNTP, 400 nM each of the three PCR primers and 0.1 U/µl HotStart Pfu polymerase (Stratagene). Reactions were placed in a thermal cycler at 95°C for 2 min, and then cycled 26 times between 98°C for 30 s and 55°C for 2 s. The competitor experiment (Fig. 3) was performed by mixing 45 zmol of circularized probes specific for the 1216 locus with the indicated molar excess of circularized probes for the remaining 12 loci. The probes were circularized using an excess of synthetic target oligonucleotides and then diluted to the desired concentrations. The probes were amplified and hybridized as described.
Figure 3.
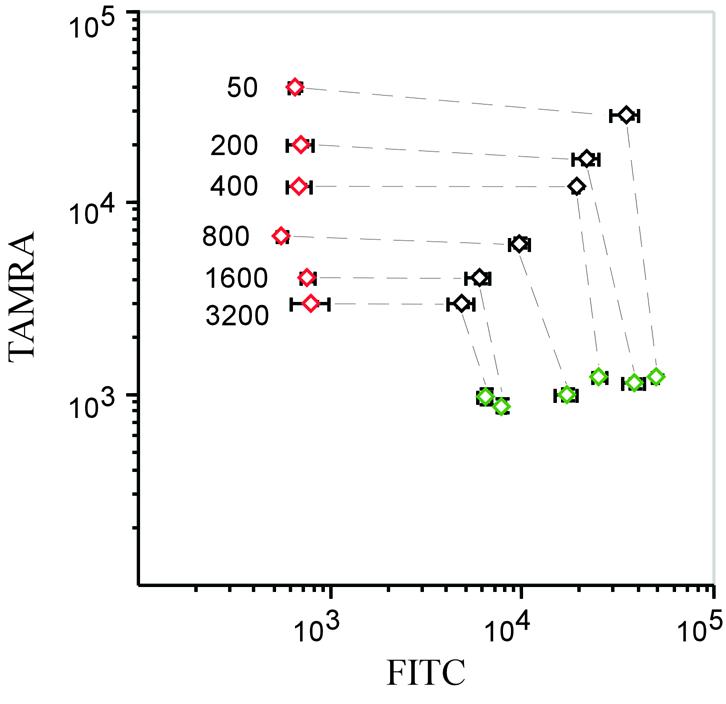
Effect on signal strength when more probes are analyzed. Constant amounts of circularized probes representing the three possible genotypes at the 1216 locus were mixed with increasing amounts of circularized competitor probes from the 12 other loci. Green and red diamonds represent the homozygous G and T alleles, respectively, while black diamonds represent heterozygotes. The ratios of competitor probes to those of the 1216 locus are indicated in the figure, and the corresponding genotypes are connected by lines. Error bars denote variation within each genotype. Scanner settings were optimized for the 1216 homozygous signals in the 50-fold reaction and the x-axis and y-axis scaled with FITC and TAMRA fluorescence intensities, respectively.
Oligonucleotide arrays
Amino-modified tag oligonucleotides were dissolved at 10 µM concentration in 150 mM sodium carbonate buffer pH 9 with 0.08% SDS and printed in triplicate on 3D-Link slides (Motorola) as 12 subarrays using a four pin GMS 417 Microarrayer (Genetic Microsystems Inc.). Slides were incubated in a 75% humidity chamber at room temperature for 2 h and then transferred to a phosphate-buffered saline bath containing 6.9 mM NaBH4 and 23% ethanol for 5 min, washed in dH2O, dried in a stream of pressurized air and finally stored at +4°C. Reusable reaction chambers demarcating individual subarrays were prepared as described by Pastinen et al. (12).
For hybridization, 30 µl amplification reactions were combined with 20 µl of 5× SSC, 0.1% Triton X-100, 22.5 mM EDTA, 0.2 nM PosHyb-FITC and TAMRA oligonucleotides as positive hybridization controls. They were heated at 100°C for 8 min, immediately placed on ice and 40 µl were transferred to the subarrays. The hybridization cassette was placed at 55°C for 4 h and the mask removed in a washing solution of 0.02× SSC, 0.1% Triton X-100. The slides were then transferred to a 0.02× SSC wash for 10 min and finally dried with pressurized air. Microarrays were scanned in a GSI 5000 Scanner and images were analyzed using QuantArray 2.0 software (GSI Lumonics). Local background was subtracted separately from the FITC and TAMRA signals and an average was calculated from spots within each triplicate.
RESULTS
The difficulty of genotyping large numbers of markers using PCR can be avoided if the genotyping assays are reformatted, via a padlock probe ligation reaction, to the amplification of large numbers of diagnostic tag sequences using a single set of primers.
Previous observations have established that padlock probes linked to a target strand are unsuitable as templates for DNA synthesis by rolling circle replication reactions (13). To investigate whether linkage of padlock probes to a target strand also compromises PCR, we used the single-stranded M13 cloning vector as a target for padlock probe ligation and then monitored the subsequent PCR amplification in real time. The target was circular or had been linearized ∼3.6 kb upstream and downstream of the probe ligation site (13). Amplification of probes having recognized linear ligation targets reached threshold fluorescence two cycle numbers earlier than the corresponding PCR of probes bound to circular targets, indicating that in this experiment, amplification of unlinked padlock probes was approximately 4-fold more efficient than amplification of linked probes (data not shown). This probably reflects that circularized padlock probes can slip off nearby ends of target DNA sequences under denaturing conditions (7). Therefore, in the following experiments all genomic DNA samples were fragmented using restriction enzymes that do not cleave the target sequences for the probes.
Pairs of padlock probes were designed to recognize the two alleles at individual loci. The segment between the target-complementary ends of the probes included sequence elements required for amplification, along with a unique locus-specific tag sequence. Pools of all probes were added to individual samples along with a thermostable DNA ligase. After ligation the reactions were treated with a mix of exonucleases to remove probes that had not been circularized or that might have undergone intermolecular ligation (Fig. 1A). Next, reacted probes were amplified by PCR with one common primer and two allele-specific fluorescence-labeled ones that allowed amplification products representing different alleles to be distinguished. Finally, the products were sorted by hybridization on a tag sequence microarray (Fig. 1B). Genotypes at individual loci were revealed by analyzing the allele-specific fluorescence signals at the corresponding microarray positions.
Figure 1.

Parallel padlock probe analysis of single nucleotide variation. (A) Padlock probes included two target-complementary sequences at both the 5′ and 3′ ends (grey) and the allele-specific nucleotide was positioned at the 3′-end. The target-specific ends were connected by the following sequence elements, 20 nt each, in 5′→3′ order: one sequence common for all probes used for amplification (black), either of two allele-specific tags (green or red) and, finally, a tag sequence unique for each locus (blue). Only probe arms correctly hybridized to a target sequence (black line) are efficiently joined by a ligase. After ligation, unreacted or dimerized probes are degraded by exonuclease treatment, preserving circularized ones. (B) Circularized probes at any locus serve as templates in a PCR with one primer specific for the common sequence (black) and two allele-specific primers, labeled with either FITC for allele 1 (green primer, marked F) or TAMRA for allele 2 (red primer, marked T). PCR products are then denatured and applied to a DNA microarray with complementary tag sequences, guiding the labeled strands to their corresponding positions. An overlaid scanning image is shown from an analysis of one individual (red color, TAMRA; green color, FITC; a combination of TAMRA and FITC appears as yellow). The spot position key identifies the investigated loci. Numbers represent locus nomenclature according to Petrukhin et al. (19).
Aliquots of 75 genomic DNA samples were individually combined with a set of 26 padlock probes designed to analyze 13 single nucleotide variants within the human ATP7B gene; seven mutations implicated in causing the recessive Wilson’s disease as well as six neutral polymorphisms (11). Only 1 fmol of each probe and 15 ng of genomic DNA were used in the fraction of each ligation reaction that was amplified and finally analyzed on the array (Fig. 2A). The data points formed clusters that were used to assign the genotypes. In a pilot experiment, 11 out of the 13 loci gave reliable results. On increasing the concentration of three of the probes 10-fold, the two remaining loci also yielded accurate results, illustrating that the call rate depends on probe quality. There was no need to redesign or resynthesize any of the probes. A subset of 29 samples were also genotyped by Sanger sequencing (11) or minisequencing, and results for all these 377 genotypes were in complete agreement with those obtained using the parallel padlock probe approach. The distinction between alleles at individual loci was not detectably influenced by the presence of probes for the other 12 loci, attesting to a lack of interference between probes (data not shown).
Figure 2.

(Opposite) Results from parallel gene analysis. (A) Seventy-five individuals were genotyped at 13 loci. Signal intensities from FITC- and TAMRA-labeled PCR products were plotted on logarithmic x- and y-axes, respectively. Locus identities are shown in the lower left corners. Individuals with confirmed genotypes that were homozygous for allele 1 at any locus are identified by green squares, homozygotes for allele 2 by red squares and heterozygotes by yellow squares. Blue diamonds represent individuals that have not been investigated by other genotyping techniques. Oligonucleotide targets representing all homozygous genotypes were also assayed, and included in the graphs if any of the homozygous genotypes were not represented in our set of samples (triangles). (B) Parallel genotyping of cDNA samples (open squares and triangles) from two liver necropsies and the corresponding genomic samples (filled squares and triangles). The results from two loci are plotted with the results from 75 genomic samples (A) shown as shaded diamonds to indicate the genotype clusters.
The same probe set was also used to analyze cDNA preparations from two liver necropsies. Thereby, expression data of transcribed ATP7B alleles could be related to genotyping data recorded using a DNA preparation from the same samples. Four loci were omitted from the liver analysis since the target-complementary arms of these probes extended over exon–intron junctions. A control reaction in which reverse transcriptase was omitted gave no signals, demonstrating that no contaminating genomic DNA was detected (data not shown). We observed no evidence of bias of allelic expression or splicing in the liver samples, which would have resulted in altered allele ratios at the two heterozygous loci (Fig. 2B). The results from the remaining homozygous loci were in agreement with their corresponding genomic DNA samples (data not shown).
To investigate how the signal strength from individual features of the arrays depended on the level of multiplexing, a dilution series was prepared where a constant amount of circularized tracer probes for one locus was mixed with increasing amounts of circularized competitor probes specific for the remaining 12 loci. Signals from the three possible genotypes of the tracer probes remained clearly distinct in the presence of a 3000-fold molar excess of competitor probes (Fig. 3), indicating that a far greater multiplicity of probes could be used in the present experimental set-up.
DISCUSSION
We present a strategy for parallel analyses of large sets of DNA or RNA sequence variants using padlock probes to detect and distinguish gene sequences in total genomic DNA or cDNA. The approach represents a scalable alternative to the highly multiplexed method we recently described (6), differing in a few aspects as described below.
In the method used by Hardenbol et al. (6), circularization of probes was affected in combined polymerization and ligation reactions, i.e. separate gap fill reactions were performed for each nucleotide triphosphate (14). These procedures require only one probe per polymorphism, eliminating probe pair imbalances and reducing probe cost, but four reactions must be performed per individual typed. In the present strategy a single ligation reaction was used to determine all genotypes in a sample which ensured that identical reaction conditions were used, but twice as many probes had to be prepared.
The combination of allele-specific incorporation of nucleotides followed by ligation in the previous study provides two levels of allele distinction. First, the correct nucleotide must be incorporated, and any misincorporated nucleotide would still hamper the ligation reaction. However, ligation reactions by themselves are known to distinguish single nucleotide variants by factors of thousands (15,16). Accordingly, the allele-specific padlock probe ligation reactions used in the present study also yield robust discrimination of alleles.
We confirm in this study that topologically linked padlock probes are poor substrates for polymerization reactions, but PCR is less affected than the previously studied isothermal rolling circle replication reaction (13). Here we have resolved the topological inhibition by fragmenting the genomic DNA before padlock probe hybridization and ligation, so that circularized probes can slip off the target strands during the denaturation step that precedes the polymerization reaction (7). An alternative approach was taken in Hardenbol et al. (6), where intact genomic DNA was used but probes were released from their target sequences by cleaving the probes at uracil residues after circularization, molecularly inverting the probes before amplification (6). In the present study the ligation reactions were repeated cyclically to increase circularization yield (7), something that may be less efficient using the uracil approach.
In the present study, amplification products from each individual were hybridized to individual tag sequence subarrays on a microscope slide prepared in-house. This array-of-arrays design permits analysis of many samples on each slide (12,17). Intermediary scalable levels of multiplexing can therefore be performed at moderate expense compared to the highly multiplexed format of the previous study, in which Affymetrix GeneFlex tag arrays with either 2000 or 16 000 features were used.
Parallel analyses with large sets of padlock probes and array-based readout can also allow investigation of transcribed sequences, for example to investigate splice variants or allelic imbalances due to effects such as promoter mutations, genomic imprinting, loss of heterozygosity or nonsense-mediated decay. Yeakley et al. used mRNA-templated ligation of pairs of oligonucleotide probes to investigate sets of splice variants (5). Padlock probes would be expected to provide additional advantages by virtue of their intramolecular dual recognition, allowing any intermolecular ligation products and also remaining unreacted probes to be degraded by exonuclease treatment (6,7,18). The exonuclease conditions used in this paper remove 99.9% of linear probe molecules with only minor loss of circularized probes (6).
Probe quality has a major influence on assay performance. Since the probes used herein and in Hardenbol et al. (6) are quite long, attention must be paid to probe quality. In this study the probes were synthesized and gel purified according to standard procedures by a commercial vendor. We estimate that ligation efficiency of the different full-length probes in this study ranged from 10 to 50%. The performance of the assay can be expected to be further improved with higher probe quality. It will be of importance to develop improved means to design and also to produce large sets of probes at high purity. This work is currently in progress in our group (J.Stenberg et al., unpublished results).
In conclusion, pools of padlock probes, each molecularly tagged to reflect its locus and allele specificity, are promising tools to interrogate minute DNA or RNA samples in extensive association studies or to investigate the genetic basis of responses to drug treatment and for highly precise transcript analyses. A general set of amplification primers and arrays can be used for different sets of padlock probes. The reaction kinetics of these unimolecular dual recognition probes allows lower probe concentrations to be used, reducing risks of amplification artifacts and providing for low cost, high throughput analyses of large sets of gene sequences.
Acknowledgments
ACKNOWLEDGEMENTS
Drs F. Barany and F. Pontén kindly provided Tth ligase and liver samples, respectively. The work was supported by the Borgstrom, Beijer and Wallenberg Foundations and by Polysaccharide Research AB (Uppsala), the Research Council of Sweden for Natural Science and Research Council of Sweden for Medicine, the Swedish Cancer Fund and by a long term EMBO fellowship to A.I.
REFERENCES
- 1.Syvanen A.C. (2001) Accessing genetic variation: genotyping single nucleotide polymorphisms. Nature Rev. Genet., 2, 930–942. [DOI] [PubMed] [Google Scholar]
- 2.Wang D.G., Fan,J.B., Siao,C.J., Berno,A., Young,P., Sapolsky,R., Ghandour,G., Perkins,N., Winchester,E., Spencer,J. et al. (1998) Large-scale identification, mapping and genotyping of single-nucleotide polymorphisms in the human genome. Science, 280, 1077–1082. [DOI] [PubMed] [Google Scholar]
- 3.Patil N., Berno,A.J., Hinds,D.A., Barrett,W.A., Doshi,J.M., Hacker,C.R., Kautzer,C.R., Lee,D.H., Marjoribanks,C., McDonough,D.P. et al. (2001) Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science, 294, 1719–1723. [DOI] [PubMed] [Google Scholar]
- 4.Oliphant A., Barker,D.L., Stuelpnagel,J.R. and Chee,M.S. (2002) BeadArray technology: enabling an accurate, cost-effective approach to high-throughput genotyping. Biotechniques, 32, 56–61. [PubMed] [Google Scholar]
- 5.Yeakley J.M., Fan,J.B., Doucet,D., Luo,L., Wickham,E., Ye,Z., Chee,M.S. and Fu,X.D. (2002) Profiling alternative splicing on fiber-optic arrays. Nat. Biotechnol., 20, 353–358. [DOI] [PubMed] [Google Scholar]
- 6.Hardenbol P., Baner,J., Jain,M., Nilsson,M., Namsaraev,E.A., Karlin-Neumann,G.A., Fakhrai-Rad,H., Ronaghi,M., Willis,T.D., Landegren,U. et al. (2003) Multiplexed genotyping with sequence-tagged molecular inversion probes. Nat. Biotechnol., 21, 673–678. [DOI] [PubMed] [Google Scholar]
- 7.Nilsson M., Malmgren,H., Samiotaki,M., Kwiatkowski,M., Chowdhary,B.P. and Landegren,U. (1994) Padlock probes: circularizing oligonucleotides for localized DNA detection. Science, 265, 2085–2088. [DOI] [PubMed] [Google Scholar]
- 8.Lizardi P.M., Huang,X., Zhu,Z., Bray-Ward,P., Thomas,D.C. and Ward,D.C. (1998) Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nature Genet., 19, 225–232. [DOI] [PubMed] [Google Scholar]
- 9.Antson D.O., Isaksson,A., Landegren,U. and Nilsson,M. (2000) PCR-generated padlock probes detect single nucleotide variation in genomic DNA. Nucleic Acids Res., 28, e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faruqi A.F., Hosono,S., Driscoll,M.D., Dean,F.B., Alsmadi,O., Bandaru,R., Kumar,G., Grimwade,B., Zong,Q., Sun,Z. et al. (2001) High-throughput genotyping of single nucleotide polymorphisms with rolling circle amplification. BMC Genomics, 2, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waldenström E., Lagerkvist,A., Dahlman,T., Westermark,K. and Landegren,U. (1996) Efficient detection of mutations in Wilsons disease by manifold sequencing. Genomics, 37, 303–309. [DOI] [PubMed] [Google Scholar]
- 12.Pastinen T., Raitio,M., Lindroos,K., Tainola,P., Peltonen,L. and Syvanen,A.C. (2000) A system for specific, high-throughput genotyping by allele-specific primer extension on microarrays. Genome Res., 10, 1031–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banér J., Nilsson,M., Mendel-Hartvig,M. and Landegren,U. (1998) Signal amplification of padlock probes by rolling circle replication. Nucleic Acids Res., 26, 5073–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abravaya K., Carrino,J.J., Muldoon,S. and Lee,H.H. (1995) Detection of point mutations with a modified ligase chain reaction (Gap-LCR). Nucleic Acids Res., 23, 675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo J., Bergstrom,D.E. and Barany,F. (1996) Improving the fidelity of Thermus thermophilus DNA ligase. Nucleic Acids Res., 24, 3071–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nilsson M., Banér,J., Mendel-Hartvig,M., Dahl,F., Antson,D.O., Gullberg,M. and Landegren,U. (2002) Making ends meet in genetic analysis using padlock probes. Hum. Mutat., 19, 410–415. [DOI] [PubMed] [Google Scholar]
- 17.Lindroos K., Sigurdsson,S., Johansson,K., Ronnblom,L. and Syvanen,A.C. (2002) Multiplex SNP genotyping in pooled DNA samples by a four-colour microarray system. Nucleic Acids Res., 30, e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang D.Y., Brandwein,M., Hsuih,T.C. and Li,H. (1998) Amplification of target-specific, ligation-dependent circular probe. Gene, 211, 277–285. [DOI] [PubMed] [Google Scholar]
- 19.Petrukhin K., Lutsenko,S., Chernov,I., Ross,B.M., Kaplan,J.H. and Gilliam,T.C. (1994) Characterization of the Wilson disease gene encoding a P-type copper transporting ATPase: genomic organization, alternative splicing and structure/function predictions. Hum. Mol. Genet., 3, 1647–1656. [DOI] [PubMed] [Google Scholar]