

Abstract
Organotypic cultures of rat cortex were used to test the hypotheses that nerve growth factor (NGF) is neuroprotective for immature cortical neurons and that ethanol abolishes this neuroprotection in a developmental stage-dependent manner. Samples were obtained on gestational day (G) 16 or postnatal day (P) 3 and cultured with ethanol (0 or 400 mg/dl) and NGF (0 or 30 ng/ml) for 72 hours. Dying neurons were identified as exhibiting terminal nick-end labeling, immunoreactivity for activated caspase 3, or condensed nuclear chromatin. Two cortical compartments were examined in fetal tissue; a superficial, cell-sparse marginal zone (MZ) and a cell-dense cortical plate (CP). At P3, the CP was subdivided into a cell-dense upper cortical plate (UCP) and a less densely packed lower cortical plate (LCP). Neuronal death in the MZ was affected by neither NGF nor ethanol at both ages. In the fetal CP, NGF did not affect the incidence of cell death, but ethanol increased it. Treatment with NGF caused an upregulation of the expression of Neg, a gene known to be affected by NGF and ethanol. NGF did not ameliorate the ethanol-induced death. In pups, ethanol increased the amount of death in the LCP. NGF did protect against this death. Neither ethanol nor NGF altered the incidence of cell death in the UCP. The laminar-dependent neuroprotection did not correlate with expression of NGF receptors or Neg. Thus, NGF can be protective against the neurotoxic effect of ethanol in the neonatal brain. This effect is site-selective and time-dependent and it targets post-migratory, differentiating neurons.
Keywords: fetal alcohol syndrome, ethanol, neurotrophin, apoptosis, p75, trk
Naturally occurring neuronal death (NOND) in the developing mammalian CNS is a well-orchestrated phenomenon. Neurons are overproduced and then a portion of them dies (Oppenheim, 1991; Burek and Oppenheim, 1996; Mooney and Henderson, 2006). Cell death is common among proliferating neuronal progenitors in the ventricular and subventricular zones (Blaschke et al., 1996; Thomiadou et al., 1997; Rehen and Chun, 2006) and among post-mitotic, post-migratory neurons in the cortical plate (e.g., Al-Ghoul and Miller, 1989; Ferrer et al., 1990; Finlay and Slattery, 1983; Miller, 1995; Spreafico et al., 1995; Mooney and Henderson, 2006). In the rat cortex, NOND peaks during the first postnatal week. The frequency of this death depends upon the time of origin of the neuron and upon its laminar location (e.g., Al-Ghoul and Miller, 1989; Wood et al., 1992; Miller, 1995).
NOND in the mammalian CNS is regulated by epigenetic factors, including neurotrophins. Among the neurotrophins, nerve growth factor (NGF) uniquely prevents the death of primary cultured, fetal cortical neurons (Seabold et al., 1998; Miller et al., 2003). NGF and its receptors, p75 and trkA, are expressed by developing (Cirulli et al., 1997; Conner and Varon, 1997; Dohrman et al., 1997; Seabold et al., 1998; Kwon et al., 2005) and mature cortical neurons (Conner et al., 1992; Pitts and Miller, 1995; 2000; Jacobs and Miller, 1999; Miller, 2000; Miller and Pitts, 2000; Bruns and Miller, 2007). The binding of NGF to trkA results in phosphorylation of the receptor, followed by activation of signaling pathways that enhance cell survival and neuronal differentiation (Bibel and Barde, 2000). In contrast, signaling via p75 can activate cell survival or cell death pathways.
NGF can be neuroprotective, i.e., it can reduce the amount of cell death in vitro that is normally detected within the first 48 hours of plating (Seabold et al., 1998; Miller et al., 2003). Ethanol blocks this effect. In other words, NGF fails to prevent the ethanol-induced death of primary cortical neurons (Seabold et al., 1998; Miller et al., 2003; Barclay et al., 2005). Conversely, NGF is protective against ethanol-induced death in cultured septal, hippocampal (Webb et al., 1997), and cerebellar neurons (Luo et al., 1997). These results reflect (a) site-specificity, (b) differential windows of vulnerability to ethanol, and/or (c) differences in responsiveness to NGF.
The present study tests the hypothesis that the neuroprotective effect of NGF and the neurotoxic effect of ethanol coincide with critical periods of cortical development. These periods concur with those of neuronogenesis and NOND which in rat neocortex occur during the third week of gestation (Brückner et al., 1976; Miller, 1988; Uylings et al., 1990) and the first postnatal week (Ferrer et al., 1990; Miller, 1995), respectively.
EXPERIMENTAL PROCEDURES
Animals
Pregnant Sprague-Dawley rats were obtained from Taconic (Germantown, NY). Animals were housed in a temperature- and humidity-controlled facility at the Syracuse Veterans Affairs Medical Center (VAMC) that was accredited by Assessment and Accreditation of Laboratory Animal Care. All procedures were approved by the Committee for Humane Use of Animals at Upstate Medical University and the Institutional Animal Care and Use Committee at the Syracuse VAMC.
Tissue preparation
Brains were collected from fetuses on gestational day (G) 16 or from pups on postnatal day (P) 3. G1 was defined as the first day a sperm-positive plug was found and G22 was uniformly designated as P0. To collect fetal tissue on G16, pregnant dams were anesthetized (100 mg/kg ketamine and 10.0 mg/kg xylazine) and four fetuses were removed from each dam. A mid-sagittal cut was made in the fetal cranium to expose the cerebral vesicles. Each vesicle was removed by making a circumferential cut at its base. To collect tissue from pups on P3, each animal was decapitated while anesthetized (100 mg/kg ketamine and 10 mg/kg xylazine). Brains were rapidly removed and cut into 400 μm coronal slices using a McIlwain Tissue Chopper (Mickle Lab Engineering, Gomshall UK). At each age, tissue was randomly assigned to one of four treatment groups: untreated controls, or those exposed to NGF (30 ng/ml), ethanol (400 mg/dl), or both NGF and ethanol. For each experiment, a total of five animals, each taken from a different litter, was assigned to each treatment group.
Tissue samples, vesicles from 16-day-old fetuses and slices from three-day-old pups, were collected in ice cold 100 mM phosphate buffer (PB) in saline (PBS) prior to transfer onto a sterile culture filter (PICMORG50, Millipore, Bedford MA). Tissue was incubated overnight (37°C, 6% CO2) in a medium (Minimum Essential Medium (MEM; GibcoBRL, Grand Island NY), 200 mM L-glutamine, and 24% glucose) supplemented with 25% fetal bovine serum (FBS). After 24 hours, the four treatment groups were made by replacing the FBS-laced medium with a serum-free medium containing human recombinant NGF (0 or 30 ng/ml; Sigma, St Louis MO) and ethanol (0 or 400 mg/dl). This concentration of ethanol was used because it mimics the effects caused by in vivo exposures that result in peak blood ethanol concentrations of 120−150 mg/dl. These include changes in cell proliferation (cf. Miller and Nowakowski, 1991; Jacobs and Miller, 2001) and migration (cf. Miller, 1993; Siegenthaler and Miller, 2004). Cultures were maintained in treatment for 72 hours and fresh medium was replaced every 24 hours.
Cultures were kept in humidified chambers containing 200 ml water and 60 ml CO2. For ethanol-treated cultures, the chambers also contained 1.5 ml ethanol. This stabilized the ethanol concentration in the medium (Luo and Miller, 1997). Samples of medium were extracted at the beginning and end of the experiment. Ethanol content was analyzed using the Analox GM7 (Analox Instruments, Lunenburg MA) and was found to remain constant over a 24 hr period.
Labeling
Tissue was fixed in 4.0% paraformaldehyde in 100 mM PB at room temperature for four hr, then cryoprotected in 30% sucrose in PB at 4°C. Tissue was cut into a complete series of 10 μm sections. Sections were processed for terminal uridylated nick-end labeling (TUNEL; Intergen, Purchase NY), protein immunohistochemistry, and in situ hybridization for Neg.
TUNEL was used to identify cells exhibiting fragmented DNA, a characteristic feature of apoptotic cells (Gavrieli et al., 1992; Jacobs and Miller, 2001). Sections were incubated in terminal deoxynucleotidyl transferase for one hour at 37°C, rinsed in PBS and then washed in horseradish peroxidase-conjugated anti-digoxigenin for 30 min. Labeled cells were identified by a wash in a solution of 0.010% hydrogen peroxide and 0.20% 3,3'-diaminobenzidine (DAB). Sections were counterstained with methyl green and coverslipped.
Sections from neonatal animals were stained immunhistochemically for the expression of p75, trkA, phosphorylated trk (p-trk), or activated caspase 3. All steps were performed at room temperature. Non-specific labeling was quenched by a wash in 5.0% bovine serum albumin (BSA) in PB containing 0.10% Tween 20 (TPBS). Sections were incubated for one hour in a solution of primary antibody (anti-p75, 05−446, Upstate Biotech, 1:100 in TPBS), anti-trkA (a generous gift from Louis F. Reichardt, 1:200), anti-p-trk, (sc-8058; Santa Cruz, 1:100), or anti-activated caspase 3 (AF835; R&D Systems, 1:1000). Subsequently, unbound antibody was removed by a wash in 5.0% non-fat milk in TPBS. Sections were processed with a Vectastain Elite kit (Vector, Burlingame CA). Non-specific labeling was examined by omitting either the primary or the secondary antibody.
In situ hybridization was used to detect the expression of the mRNA for Neg. Neg is a gene identified in primary cultures of cortical neurons that is (a) up-regulated by treatment with NGF and (b) down-regulated by concurrent treatment with ethanol (Miller et al., 2003). Sections were incubated with a digoxigenin-labeled anti-sense probe to Neg for 18 hr at 55°C. Non-annealed probe was removed by a series of stringent washes, then tissue was treated with RNase to degrade any remaining probe.
Localization of the digoxigenin-labeled probed was performed using immunohistochemistry similar to that described above. After blocking, an alkaline phosphatase-conjugated anti-digoxigenin antibody was applied to the tissue for 18 hr at 4°C. Tissue was rinsed, then incubated in the chromagen nitroblue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate (Roche) for 2−4 hr at room temperature. Slides were dehydrated, cleared, and coverslipped. Two controls were performed; sections were incubated with the sense probe to Neg, or to a sense or anti-sense probe for actin mRNA.
Analysis
In the fetal tissue, analyses focused on two cortical segments; the marginal zone (MZ; the relatively acellular layer at the external aspect of the cortex) and the cell body dense cortical plate (CP). In the sections derived from postnatal animals, the CP was subdivided into the upper cortical plate (UCP; a cell body dense region subjacent to the MZ) and the lower cortical plate (LCP; the less densely packed region below the UCP).
The frequency of dead/dying cells was calculated as the total number of TUNEL-positive cells plus the total number of cells exhibited condensed chromatin divided by the total number of cells counted. Cell counts were generated separately for (1) TUNEL-positive cells (cells exhibiting any DAB staining), (2) cells with condensed nuclei but were DAB-negative, i.e., pyknotic cells, and (3) viable cells (cells that were TUNEL-negative and were not pyknotic). Cell density was determined using an unbiased stereological method (Gunderson and Jensen, 1987; Mooney and Miller, 2003). The number of cells in a counting frame (50 μm × 50 μm) was counted. To ensure that the counts accurately represented a section, a minimum of 10 counting frames was used, each was randomly placed over the image within a compartment of the developing cortex. Using this procedure, a minimum of 100 cells was counted in each organotypic preparation.
The data were compiled and means (± standard errors of the means) were calculated. A comparison among multiple groups was performed using one-way analysis of variance (ANOVA). Where the ANOVA showed a significant effect, post-hoc testing was done using the Student-Newman-Keuls test.
RESULTS
Fetal preparations
Two distinct cortical regions (the relatively cell body-sparse MZ and the cell body-dense CP) were discerned in cultured cortical vesicles from 16-day-old fetuses (Fig. 1). Treatment with NGF or ethanol had no apparent effect on this organization. Cell packing density in the MZ was significantly (F 3, 19 = 6.137; p<0.05) affected by treatment (Fig. 2). Density was similar in control and NGF-treated cultures, but, as shown previously (Mooney and Miller, 2003), exposure to ethanol significantly (p<0.05) increased the cell packing density in the MZ. Concurrent administration of NGF and ethanol resulted in a density similar to that seen after treatment with ethanol alone. Thus, NGF did not offset the ethanol-induced change. In contrast, the cell packing density in the CP was unaffected by treatment with NGF and/or ethanol.
Figure 1. Appearance of fetal cerebral cortex in slice cultures.

Slices were obtained from fetal rats on gestational day (G) 16 were labeled for fragmented DNA (using the TUNEL method). The marginal zone (MZ) and cortical plate (CP) were apparent in samples from all four treatment groups: those treated with nerve growth factor (NGF; 0 or 30 ng/ml) and ethanol (Et; 0 or 400 mg/dl) (upper panel). In the slices taken from animals on postnatal day (P) 3 (middle and lower panels), the CP can be subdivided into upper (UCP) and lower (LCP) compartments on the basis of cell packing density. Tissue was labeled for fragmented DNA (using the TUNEL method, middle panel) or activated caspase-3 (lower panel). Solid arrows indicate TUNEL- or active caspase-3-positive cells. Open arrows designate cells with condensed nuclear chromatin. IZ, intermediate zone. Scale bars are 25 μm.
Figure 2. Quantification of cortical plate parameters at G16.
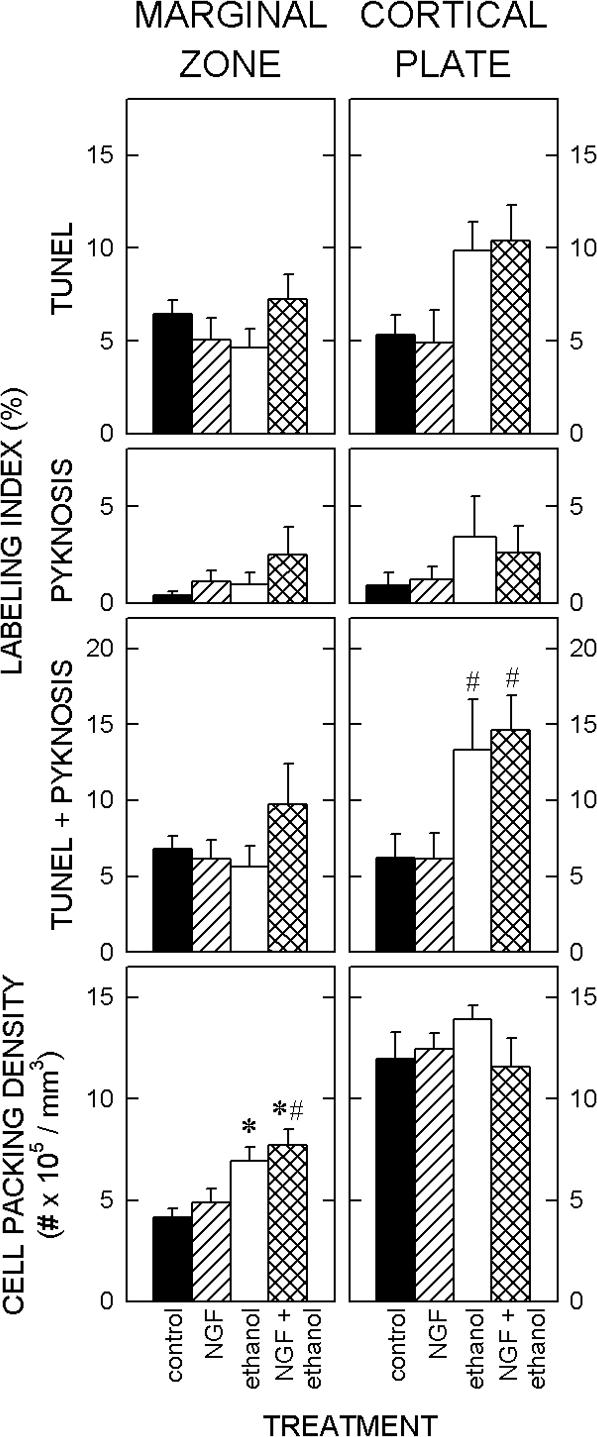
The top three panels show the effects of nerve growth factor (NGF) and ethanol on the density of cells that exhibit terminal uridylated nick-end labeling (TUNEL) and nuclear condensations. The cell packing densities in the marginal zone (left) and cortical plate (right) of the fetal slices are shown (bottom).
Marginal Zone. The labeling index shows the proportion of dead/dying cells, i.e., TUNEL-positive cells and pyknotic cells. The labeling index in the MZ was unaffected by exposure to ethanol or by administration of NGF. Bottom. The cell packing density in the MZ of the fetal cultures was significantly higher in cultures treated with ethanol compared to untreated cultures. NGF alone did not significantly affect cell density, and it also failed to ameliorate the ethanol-induced increase in cell packing density.
Cortical Plate. Top. Ethanol induced an increase in the frequency of TUNEL-positive cells, however, the total amount of cell death induced by exposure to ethanol failed to reach statistical significance. Bottom. Cell packing density in the CP was unaffected by exposure to ethanol or by administration of NGF.
Each value represents the mean of five cultures. T-bars are standard errors of the means. Each * denotes a significant (p<0.05) difference relative to the untreated control. A # signifies a significant difference relative to the NGF-treated cultures.
The frequency of dead/dying cells, i.e., TUNEL-positive or pyknotic cells, was determined separately for the MZ and the CP. Treatment with NGF and/or ethanol did not significantly alter this frequency in the MZ (Fig. 2). On the other hand, the frequency of dead/dying cells in the CP doubled following treatment with ethanol, in the presence or absence of NGF.
The pattern of expression of activated caspase 3 immunoreactivity paralleled TUNEL labeling (data not shown). That is, immunopositive cells were apparent in both cortical layers in all treatment groups. The frequency of labeling was higher in the CP of slices treated with ethanol regardless of the presence or absence of NGF.
p75 immunolabeling was apparent throughout the depth of the cortical wall, i.e., in the MZ and the CP. Treatment did not affect the pattern of staining. TrkA was expressed in control tissue, as well as that treated with ethanol and/or NGF (Fig. 3). Trk receptors were evenly distributed through both regions. Treatment did not appear to alter the pattern of expression of either receptor. Phosphorylated trk was also expressed throughout the developing cortical plate, in a similar pattern as the non-phosphorylated isoform. Treatment failed to alter the pattern of expression of p-trk in the cortical wall.
Figure 3. Receptor expression at G16 in control-treated cultures.
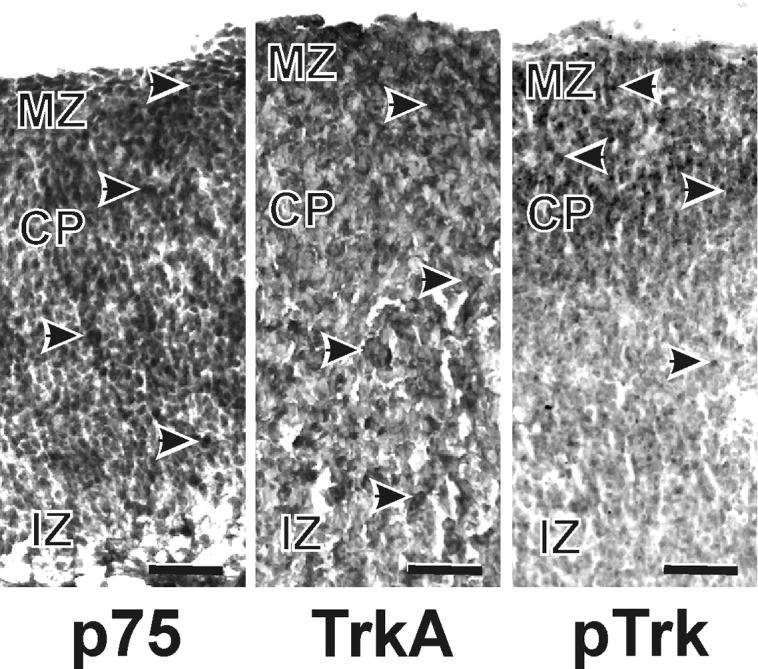
Cells immunopositive for the neurotrophin receptors, trkA, p75, and phosphorylated (p) trk, were identifiable in all three compartments of the cerebral wall; the marginal zone (MZ), cortical plate (CP) and the intermediate zone (IZ). Scale bars are 25 μm.
A low amount of Neg was expressed in the MZ and CP of fetal cortices (Fig. 4). Neg expression increased following three days of treatment with NGF, i.e., relative to the amount of Neg expressed in controls. In contrast, Neg expression was low in ethanol-treated slices (like that detected in controls) and ethanol eliminated the NGF-induced increase in Neg expression. The implication was that exogenous NGF activated normal neural signal transduction pathways in the organotypic model.
Figure 4. Neg expression at G16.

In situ hybridization for Neg revealed no labeling in control-, ethanol-, or ethanol + NGF-treated tissue. In contrast, treatment with NGF alone resulted in up-regulation of neg expression. Scale bars are 25 μm.
Neonatal slices
Three distinct cortical regions were discriminable after culturing the slices from 3-day-old pups for three days: the relatively cell body-sparse MZ, cell body-dense UCP, and less dense LCP (Figs. 1). Treatment with neither NGF nor ethanol affected this organization. The cell packing density was not significantly altered by treatment with NGF or ethanol in any of the three compartments of the cortical wall (Fig. 5).
Figure 5. Quantification of cortical plate parameters at P3.
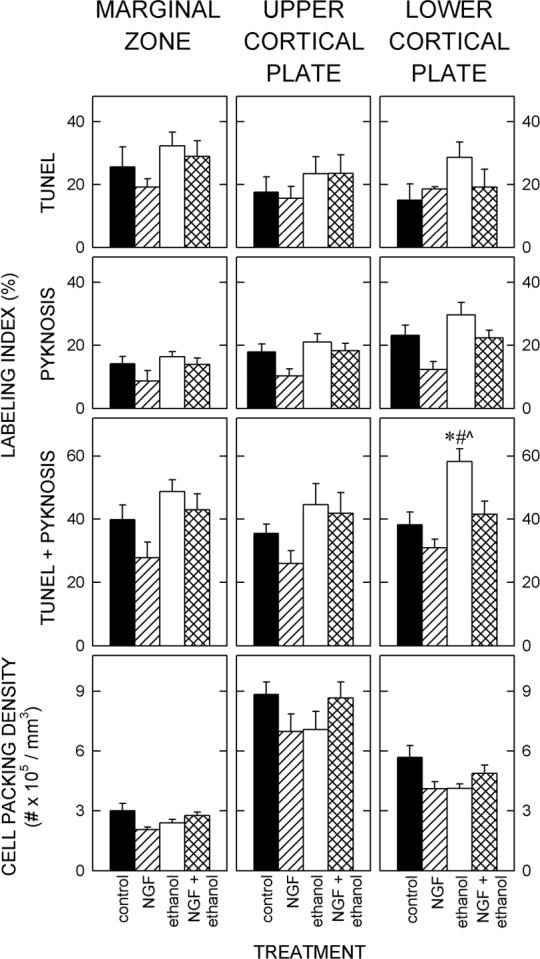
Marginal zone. Top. The frequency of dead/dying cells was not significantly affected by treatment with ethanol or NGF. Bottom. The cell packing density in the MZ was not different among control cultures or those treated with NGF, ethanol, or ethanol and NGF.
Upper cortical plate. Top. The expression of indicators of cell death (TUNEL and pyknotic cells) in the UCP was not significantly affected by exposure to ethanol or treatment with NGF. Bottom. The density of cells in the lower cortical plate was unaffected by treatment.
Lower cortical plate. Top. The frequencies of TUNEL-positive cells and pyknotic cells were increased in the LCP following exposure to ethanol. Concurrent exposure to NGF ameliorated this effect. Bottom. Treatment did not alter the density of cells in the LCP.
Each value represents the mean of five cultures and T-bars are standard errors of the means. An *, #, and ^ denote significant (p<0.05) differences relative to the untreated controls, to the NGF-treated cultures, and to the cultures treated with ethanol and NGF, respectively.
In each of the three segments of developing cortex in controls, about ⅓ of the cells were dead/dying (Fig. 5). Neither NGF nor ethanol affected this frequency in the MZ or the UCP. In the LCP, however, an ANOVA showed a significant effect of treatment (F3, 19 = 10.352; p<0.001). Exposure to ethanol significantly (p<0.05) increased the frequency of dead/dying cells in the LCP. Following concurrent administration of NGF, the frequency of cell death in the LCP was similar to that in the control. Thus, NGF blocked the toxic effects of ethanol.
Immunolabeling with an antibody to active caspase 3 showed a pattern of expression similar to that seen with TUNEL, i.e., cells in all three layers were immunopositive (Fig. 1). Their frequency in all three cortical segments also increased following exposure to ethanol.
NGF receptors, p75 and trkA, were present throughout the cortical plate (Fig. 6). p75 immunoexpression was apparent in the UCP and LCP, but was only rarely seen in the MZ, whereas trkA was expressed in all three cortical segments. There was no apparent effect of ethanol or NGF on expression of either receptor. Expression of p-trk was similar to that of trkA, i.e., it was apparent in all three compartments of the cortical anlage. Treatment did not appear to have an effect on its expression.
Figure 6. Expression of neurotrophin receptors and neg at P3.

Immunohistochemical labeling of the neurotrophin receptors showed that p75-positive cells were only localized in the cortical plate, not in the marginal zone. In contrast, trkA- and phosphorylated trk-positive cells were apparent throughout the cortical wall. In situ hybridization for Neg labeled cells in all three segments of the cortical wall. Treatment with NGF did not alter Neg expression. Nomenclature as for Figure 5. Scale bars are 50 μm.
In situ hybridization showed Neg was present throughout the cortical wall in all four treatment groups (Fig. 6). Treatment did not alter its expression pattern.
DISCUSSION
Methodological considerations
Three methods were used in the present study to identify cells as dead/dying; TUNEL, pyknosis, and immunolabeling for activated caspase 3. Each method has advantages and limitations.
(1) TUNEL identifies cells with poly-adenylated tails found on fragmented DNA. DNA fragmentation is a feature of apoptotic cells that is associated with activation of endogenous nucleases (Wyllie, 1980). False-positive labeling may result from failure to optimize conditions (such as incorrect sample fixation or inappropriate concentrations of reagents) or may occur in tissue undergoing active RNA synthesis (Saraste, 1999).
(2) Pyknotic cells are cells with condensed or fragmented chromatin. This is a hallmark of apoptosis (Kerr et al., 1972). Unfortunately, such cells can be confused with glia that have small heterochromatic nuclei, e.g., oligodendrocytes and young astrocytes.
(3) Activated caspase 3 is an effector of cell death (e.g., Mooney and Henderson, 2006). Caspase 3 can also be activated during the cell cycle and cell differentiation (Oomann et al., 2004; 2005; 2006; Launay et al., 2005). As a death effector, activation of caspase 3 is up-stream of activation of nucleases. Thus, expression of activated caspase 3 can occur prior to, in concert with, or without TUNEL labeling.
Neither the extent of overlap between TUNEL, pyknosis, and immunolabeling for activated caspase 3 nor the time course of cell death are known in this model. In other models, depending on the structure and the insult, morphological changes indicative of cell death may be found between 6 and 24 hours after insult (e.g., McCarthy et al., 1997; Saraste and Pulkki, 2000).
Windows of vulnerability
Developing cortical neurons are susceptible to ethanol-induced death at both ages examined in the present study. Based on the incidence of markers for cell death, fetal tissue appears more vulnerable to ethanol than tissue taken from three-day-old pups. In fetal cultures, ethanol doubles the incidence of death throughout the cortical wall. Neither the NOND nor the ethanol-induced cell death in fetal cultures is mitigated by concurrent administration of NGF. This lack of protection from ethanol-induced death is similar to the non-response previously reported in primary cultures of cortical neurons taken from fetal tissue (Seabold et al., 1998; Miller et al., 2003; Barclay et al., 2005).
In contrast to the relatively uniform effect of ethanol on fetal cortical plate, tissue from neonates exhibits a region-specific response. That is, only neurons in the LCP are vulnerable to ethanol-induced death. This ethanol-induced death is completely blocked by concurrent administration of NGF. The neuroprotective effect of NGF is specific; it does not rescue cells in any of the three compartments from NOND, but it does specifically ameliorate the ethanol-induced death in the LCP. The implication of this finding is that the mechanism of the ethanol-induced death differs from that of NOND.
The neurotoxic effects of ethanol apparently target post-migratory, early differentiating neurons. The UCP, which contains neurons that are refractive to ethanol exposure, is comprised primarily of migrating neurons and neurons that have only recently begun their differentiation. In contrast, the LCP contains neurons that were born earlier and are further through their sequence of maturation. At least a subset of these LCP neurons is vulnerable to ethanol. Thus, there is a temporal component to ethanol-vulnerability which may correlate to a stage of differentiation.
Role of neurotrophins
The specificity of NGF-induced neuroprotection in neonates may also be related to the maturational state of the cells. The spatial specificity of the neuroprotection does not correlate with the expression of receptors. That is, both p75 and trkA are present throughout the LCP and the UCP. Although there is evidence of receptor expression in the developing cortical plate, activation of downstream pathways is unknown. It is possible that a downstream effector(s) has a temporal sequence of expression, or there is a co-factor(s) that is expressed with zonal specificity.
The ability of NGF to ameliorate cell death does not correlate with trk receptor activation, as phosphorylated trk is present throughout the laminar extent of the developing cortex and NGF only rescues cells in the LCP from ethanol-induced cell death. It is possible that signaling through p75 is important for cortical survival. The role of p75 during development is time- and area-dependent. For example, p75 can mediate cell death in specific populations, including oligodendrocytes and sympathetic neurons (e.g., Casaccia-Bonnefil et al., 1996; Bamji et al., 1998). In other models, e.g., cultured hippocampal neurons or PC12 cells, p75 mediates survival (Roux et al., 2001; Bui et al., 2002; Culmsee et al., 2002). In a similar vein, primary cortical neurons exposed to ethanol show a downregulation of p75 expression that is coincident with an increased incidence of cell death (Seabold et al., 1998). In the current model, p75 apparently mediates survival as the addition of an anti-p75 antibody to the culture medium, which presumably inhibits receptor activation, results in disintegration of the slice preparation (data not shown). Likely, this is the result of widespread cell death.
The expression of Neg, a gene proposed to promote survival in primary cortical neurons (Miller et al., 2003), is up-regulated in fetal cultures by the addition of NGF to the medium. This occurs in the absence of any survival-promoting effect of NGF in these organotypic cultures. In contrast, in cultures from neonates, Neg is constitutively expressed and its expression does not appear to be altered by the addition of NGF. Thus, Neg expression is NGF-responsive in fetal tissue, but it does not correlate with NGF-mediated protection or survival in organotypic cultures of fetal or neonatal forebrain.
In the present study, exposure to ethanol doubled the incidence of cell death in the fetal cortical plate. Despite evidence that the concurrently administered NGF is biologically available, it is unable to protect against the ethanol-induced death. In contrast, in the neonatal tissue, only the LCP is affected by ethanol. This death is ameliorated by concurrent administration of NGF. Thus, ethanol and NGF specifically target post-migratory, differentiating neurons in the neonatal tissue. That NGF can protect against ethanol-induced cell death in the neonatal LCP but not NOND in fetal or neonatal tissue implies that the mechanism(s) by which ethanol induces cell death is different from the mechanism(s) of NOND in this model.
ACKNOWELEDGEMENTS
We thank Jennifer Jaynes and Terri Novak for the immunohistochemical labeling of tissue. This work was supported by grants from the National Institute of Alcohol Abuse and Alcoholism (AA015413 to SMM and AA06916 and AA07568 to MWM) and from the Department of Veterans Affairs (MWM) .
Research support provided by grants from the National Institutes of Health (AA015413 to SMM; AA06916 and AA07568 to MWM) and the Department of Veterans Affairs (MWM).
ABBREVIATIONS
- ANOVA
one way analysis of variance
- CNS
central nervous system
- CP
cortical plate
- DAB
3,3'-diaminobenzidine
- FBS
fetal bovine serum
- G
gestational day
- LCP
lower cortical plate
- mRNA
messenger ribonucleic acid
- MZ
marginal zone
- NGF
nerve growth factor
- NOND
naturally occurring neuronal death
- P
postnatal day
- PB
100mM phosphate buffer
- PBS
phosphate buffered saline
- p-trk
phosphorylated tyrosine receptor kinase
- TPBS
phosphate buffered saline containing tween
- trk
tyrosine receptor kinase
- TUNEL
terminal uridylated nick-end labeling
- UCP
upper cortical plate
- VAMC
Veteran's Affairs Medical Center
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Al-Ghoul WM, Miller MW. Transient expression of Alz-50 immunoreactivity in developing rat neocortex: a marker for naturally occurring neuronal death? Brain Res. 1989;481:361–367. doi: 10.1016/0006-8993(89)90815-9. [DOI] [PubMed] [Google Scholar]
- Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol. 1998;140:911–923. doi: 10.1083/jcb.140.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay DC, Hallbergson AF, Montague JR, Mudd LM. Reversal of ethanol toxicity in embryonic neurons with growth factors and estrogen. Brain Res Bull. 2005;67:459–465. doi: 10.1016/j.brainresbull.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Bibel M, Barde Y-A. Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 2000;14:2919–2937. doi: 10.1101/gad.841400. [DOI] [PubMed] [Google Scholar]
- Blaschke AJ, Weiner JA, Chun J. Programmed cell death is a universal feature of embryonic and postnatal neuroproliferative regions throughout the central nervous system. J Comp Neurol. 1998;396:39–50. doi: 10.1002/(sici)1096-9861(19980622)396:1<39::aid-cne4>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Brückner G, Mares V, Biesold D. Neurogenesis in the visual system of the rat. An autoradiographic investigation. J Comp Neurol. 1976;166:245–255. doi: 10.1002/cne.901660208. [DOI] [PubMed] [Google Scholar]
- Bruns MB, Miller MW. Functional nerve growth factor and trkA autocrine/paracrine circuits in adult rat cortex are revealed by episodic ethanol exposure and withdrawal. J Neurochem. 2007;100:1155–1168. doi: 10.1111/j.1471-4159.2006.04301.x. [DOI] [PubMed] [Google Scholar]
- Bui NT, Konig HG, Culmsee C, Bauerbach E, Poppe M, Krieglstein J, Prehn JH. p75 neurotrophin receptor is required for constitutive and NGF-induced survival signalling in PC12 cells and rat hippocampal neurones. J Neurochem. 2002;81:595–605. doi: 10.1046/j.1471-4159.2002.00841.x. [DOI] [PubMed] [Google Scholar]
- Burek MJ, Oppenheim RW. Programmed cell death in the developing nervous system. Brain Pathol. 1996;6:427–446. doi: 10.1111/j.1750-3639.1996.tb00874.x. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV. Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature. 1996;383:716–719. doi: 10.1038/383716a0. [DOI] [PubMed] [Google Scholar]
- Cirulli F, Shooter EM, Levine S. Developmental expression of the NGF receptor p140trk in the septohippocampal system of the rat: a quantitative analysis. Intl J Dev Neurosci. 1997;15:901–909. doi: 10.1016/s0736-5748(97)00020-8. [DOI] [PubMed] [Google Scholar]
- Conner JM, Muir D, Varon S, Hagg T, Manthorpe M. The localization of nerve growth factor-like immunoreactivity in the adult rat basal forebrain and hippocampal formation. J Comp Neurol. 1992;319:454–462. doi: 10.1002/cne.903190310. [DOI] [PubMed] [Google Scholar]
- Conner JM, Varon S. Developmental profile of NGF immunoreactivity in the rat brain: a possible role of NGF in the establishment of cholinergic terminal fields in the hippocampus and cortex. Dev Brain Res. 1997;101:67–79. doi: 10.1016/s0165-3806(97)00051-5. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Gerling N, Lehmann M, Nikolova-Karakashian M, Prehn JH, Mattson MP, Krieglstein J. Nerve growth factor survival signaling in cultured hippocampal neurons is mediated through TrkA and requires the common neurotrophin receptor P75. Neuroscience. 2002;115:1089–1108. doi: 10.1016/s0306-4522(02)00539-0. [DOI] [PubMed] [Google Scholar]
- Dohrman DP, West JR, Pantazis NJ. Ethanol reduces expression of the nerve growth factor receptor, but not nerve growth factor protein levels in the neonatal rat cerebellum. Alcohol Clin Exp Res. 1997;21:882–893. [PubMed] [Google Scholar]
- Ferrer I, Bernet E, Soriano E, del Rio T, Fonesca M. Naturally occurring cell death in the cerebral cortex of the rat and removal of dead cells by transitory phagocytes. Neuroscience. 1990;39:451–458. doi: 10.1016/0306-4522(90)90281-8. [DOI] [PubMed] [Google Scholar]
- Finlay BL, Slattery M. Local differences in the amount of early cell death in neocortex predict adult local specializations. Science. 1983;219:1349–1351. doi: 10.1126/science.6828866. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen HJ, Jensen EB. The efficiency of systematic sampling in stereology and its prediction. J Microsc. 1987;147:229–263. doi: 10.1111/j.1365-2818.1987.tb02837.x. [DOI] [PubMed] [Google Scholar]
- Jacobs JS, Miller MW. Expression of nerve growth factor, p75, and the high affinity neurotrophin receptors in the adult rat trigeminal system: evidence for multiple trophic support systems. J Neurocytol. 1999;28:571–595. doi: 10.1023/a:1007019422675. [DOI] [PubMed] [Google Scholar]
- Jacobs JS, Miller MW. Proliferation and death of cultured fetal neocortical neurons: effects of ethanol on the dynamics of cell growth. J Neurocytol. 2001;30:391–401. doi: 10.1023/a:1015013609424. [DOI] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Brit J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H-J, Lee K-Y, Park I-K, Park M-S, Lee M-Y, Kim M-K. Expression of tyrosine kinase A in the cerebral cortex of postnatal developing rat. J Vet Sci. 2005;6:185–189. [PubMed] [Google Scholar]
- Launay S, Hermine O, Fontenay M, Kroemer G, Solary E, Garrido C. Vital functions for lethal caspases. Oncogene. 2005;24:5137–5148. doi: 10.1038/sj.onc.1208524. [DOI] [PubMed] [Google Scholar]
- Luo J, West JR, Pantazis NJ. Nerve growth factor and basic fibroblast growth factor protect rat cerebellar granule cells in culture against ethanol-induced cell death. Alcohol Clin Exp Res. 1997;21:1108–1120. [PubMed] [Google Scholar]
- Luo J, Miller MW. Differential sensitivity of human neuroblastoma cell lines to ethanol: correlations with their proliferative responses to mitogenic growth factors and expression of growth factor receptors. Alcohol Clin Exp Res. 1997;21:1186–1194. [PubMed] [Google Scholar]
- McCarthy NJ, Whyte MKB, Gilbert CS, Evan GI. Inhibition of Ced-3/ICE-related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J Cell Biol. 1997;136:215–227. doi: 10.1083/jcb.136.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MW. Effect of prenatal exposure to ethanol on the development of cerebral cortex: I. Neuronal generation. Alcohol Clin Exp Res. 1988;12:440–449. doi: 10.1111/j.1530-0277.1988.tb00223.x. [DOI] [PubMed] [Google Scholar]
- Miller MW. Relationship of time of origin and death of neurons in rat somatosensory cortex: Barrel versus septal cortex and projection versus local circuit neurons. J Comp Neurol. 1995;355:6–14. doi: 10.1002/cne.903550104. [DOI] [PubMed] [Google Scholar]
- Miller MW. Expression of nerve growth factor and its receptors in the somatosensory-motor cortex of Macaca nemestrina. J Neurocytol. 2000;29:453–469. doi: 10.1023/a:1007207527842. [DOI] [PubMed] [Google Scholar]
- Miller MW, Jacobs JS, Yokoyama R. Neg, a nerve growth factor-stimulated gene expressed by fetal neocortical neurons that is downregulated by ethanol. J Comp Neurol. 2003;460:212–222. doi: 10.1002/cne.10651. [DOI] [PubMed] [Google Scholar]
- Miller MW, Pitts AF. Neurotrophin receptors in the somatosensory cortex of the mature rat: co-localization of p75, trk, isoforms and c-neu. Brain Res. 2000;852:355–366. doi: 10.1016/s0006-8993(99)02176-9. [DOI] [PubMed] [Google Scholar]
- Mooney SM, Henderson GI. Intracellular pathways of neuronal death. In: Miller MW, editor. Brain Development: Normal Processes and the Effects of Alcohol and Nicotine. Oxford UP; New York: 2006. pp. 91–103. [Google Scholar]
- Mooney SM, Miller MW. Ethanol-induced neuronal death in organotypic cultures of rat cerebral cortex. Dev Brain Res. 2003;147:135–141. doi: 10.1016/j.devbrainres.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Oomman S, Finckbone V, Dertien J, Attridge J, Henne W, Medina M, Mansouri B, Singh H, Strahlendorf H, Strahlendorf J. Active caspase-3 expression during postnatal development of rat cerebellum is not systematically or consistently associated with apoptosis. J Comp Neurol. 2004;476:154–173. doi: 10.1002/cne.20223. [DOI] [PubMed] [Google Scholar]
- Oomman S, Strahlendorf H, Finckbone V, Strahlendorf J. Non-lethal active caspase-3 expression in Bergmann glia of postnatal rat cerebellum. Dev Brain Res. 2005;160:130–145. doi: 10.1016/j.devbrainres.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Oomman S, Strahlendorf H, Dertien J, Strahlendorf J. Bergmann glia utilize active caspase-3 for differentiation. Brain Res. 2006;1078:19–34. doi: 10.1016/j.brainres.2006.01.041. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Ann Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Pitts AF, Miller MW. Expression of nerve growth factor, p75, and trk in the somatosensory and motor cortices of mature rats: evidence for local trophic support circuits. Somatosens Mot Res. 1995;12:329–342. doi: 10.3109/08990229509093666. [DOI] [PubMed] [Google Scholar]
- Pitts AF, Miller MW. Expression of nerve growth factor, brain-derived neurotrophic factor, and neurotrophin-3 in the somatosensory cortex of the mature rat: coexpression with high-affinity neurotrophin receptors. J Comp Neurol. 2000;418:241–254. doi: 10.1002/(sici)1096-9861(20000313)418:3<241::aid-cne1>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Roux PP, Bhakar AL, Kennedy TE, Barker PA. The p75 neurotrophin receptor activates Akt (protein kinase B) through a phosphatidylinositol 3-kinase-dependent pathway. J Biol Chem. 2001;276:23097–23104. doi: 10.1074/jbc.M011520200. [DOI] [PubMed] [Google Scholar]
- Rehen SK, Chun JJM. Cell death. In: Miller MW, editor. Brain Development: Normal Processes and the Effects of Alcohol and Nicotine. Oxford University Press; New York: 2006. pp. 73–90. [Google Scholar]
- Saraste A. Morphologic criteria and detection of apoptosis. Herz. 1999;24:189–195. doi: 10.1007/BF03044961. [DOI] [PubMed] [Google Scholar]
- Saraste A, Pulkki K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc Res. 2000;45:528–537. doi: 10.1016/s0008-6363(99)00384-3. [DOI] [PubMed] [Google Scholar]
- Seabold GK, Luo J, Miller MW. Effect of ethanol on neurotrophin-mediated cell survival and receptor expression in cultures of cortical neurons. Dev Brain Res. 1998;108:139–145. doi: 10.1016/s0165-3806(98)00043-1. [DOI] [PubMed] [Google Scholar]
- Spreafico R, Frassoni C, Arcelli P, Selvaggio M, De Biasi S. In situ labeling of apoptotic cell death in the cerebral cortex and thalamus of rats during development. J Comp Neurol. 1995;363:281–295. doi: 10.1002/cne.903630209. [DOI] [PubMed] [Google Scholar]
- Thomiadou D, Mione MC, Cavanagh JF, Parnavelas JG. Apoptosis and its relation to the cell cycle in the developing cerebral cortex. J Neurosci. 1997;17:1075–1085. doi: 10.1523/JNEUROSCI.17-03-01075.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uylings HB, van Eden CG. Qualitative and quantitative comparison of the prefrontal cortex in rat and in primates, including humans. Prog Brain Res. 1990;85:31–62. doi: 10.1016/s0079-6123(08)62675-8. [DOI] [PubMed] [Google Scholar]
- Webb B, Heaton MB, Walker DW. Ethanol effects on cultured embryonic hippocampal neuronal calcium homeostasis are altered by nerve growth factor. Alcohol Clin Exp Res. 1997;21:1643–1652. [PubMed] [Google Scholar]
- Wood JG, Martin S, Price DJ. Evidence that the earliest generated cells of the murine cerebral cortex form a transient population in the subplate and marginal zone. Dev Brain Res. 1992;66:137–140. doi: 10.1016/0165-3806(92)90150-u. [DOI] [PubMed] [Google Scholar]
- Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284:555–556. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]