

Abstract
We report a 17-month-old African female with cutaneous and ophthalmologic features of oculocutaneous albinism type 2 as well as microcephaly, absent speech, tremulous movements. P gene mutations within the Angelman/Prader-Willi syndrome critical region at 15q11–q13 cause oculocutaneous albinism type 2. Co-morbid oculocutaneous albinism and Angelman syndrome were suspected and confirmed by cytogenetics. Phenotypic features of Angelman syndrome or Prader-Willi syndrome in a patient with albinism should prompt further investigation.
Keywords: Angelman syndrome, oculocutaneous albinism, P gene, OCA2, hypopigmentation, Prader-Willi syndrome
INTRODUCTION
Oculocutaneous Albinism
Oculocutaneous albinism is a genetically heterogeneous autosomal recessive disorder, categorized into four subtypes. Regardless of subtype, oculocutaneous albinism is not associated with developmental delay or seizures. Oculocutaneous albinism type 2, the focus of this paper, is caused by point mutations or deletions of the P gene, located within the Angelman syndrome and Prader-Willi syndrome critical region of 15q11-q13[1]. Oculocutaneous albinism type 2 is the most common form of albinism worldwide and is characterized by reduced pigmentation of the skin, hair, iris and retina. Individuals with oculocutaneous albinism type 2 are born with minimal to near-normal cutaneous pigmentation. Lack of normal melanin increases squamous cell and basal cell skin carcinoma risk. Hair color ranges from light yellow to brown and iris color from blue to brown[2]. Skin, hair and eye color can darken with age as yellow/orange pheomelanin accumulates over time. Lack of pigmentation in the eye results in foveal hypoplasia, horizontal nystagmus, visual impairment, photophobia and misrouting of the optic nerves which manifests as esotropic strabismus. Nystagmus may be congenital or develops within the first 3–4 months and persists throughout life. Visual acuity varies ranging from 20/30–20/400 and usually stabilizes in childhood without further deterioration [2].
Oculocutaneous albinism type 2 is particularly common in Africans with a frequency of 1/1000–1/7900[3, 4] and African-Americans of 1/10,000[4, 5]. In contrast, the frequency of oculocutaneous albinism type 2 in Caucasian-Americans is estimated to be 1/36,000[4]. A homozygous deletion of exon 7 of the P gene is the predominant cause of oculocutaneous albinism type 2 in Africans and African-Americans[6]; this 2.7-kb deletion causes a frameshift mutation, resulting in a truncated null allele.
Angelman Syndrome/Prader-Willi Syndrome
Angelman syndrome is characterized by severe speech impairment, developmental delay, gait ataxia and/or tremulous movements and behavioral features including frequent laughing, a happy demeanor and an excitable personality. Microcephaly by age two and seizure activity are also frequent. With a typically normal pregnancy, birth, physical appearance and biochemical testing, Angelman syndrome is not suspected until 6–12+ months of age when the child develops seizures and global delay[7]. The brain is also usually structurally normal though there may be mild cortical atrophy or dysmyelination[7].
The seizure types in Angelman syndrome can be varied to include both major and minor motor types, however most often they are myoclonic seizures[8]. The electroencephalogram (EEG) demonstrates characteristic changes consisting of high amplitude 2- to 3- Hz delta activity with intermittent spike and slow wave discharges, runs of rhythmic theta activity over a wide area and runs of rhythmic sharp theta activity of 5–6/s over the posterior third of the head, forming complexes with small spikes[9]. Seizures can be difficult to treat in infancy but their severity decreases significantly in later childhood[8]. Anticonvulsant medications such as valproic acid, topiramate, clonazepam, lamotrigine and ethosuximide are used more commonly[10]. However, benzodiazepines in combination with valproic acid or topiramate have been particulary effective[8]. Vigabatrin and tigabine, which increase brain GABA levels should be avoided due to the postulated abnormal GABA influence in Angelman syndrome seizures(7).
Prader-Willi syndrome is a distinct neurobehavioral disorder from Angelman syndrome. In the neonatal period, Prader-Willi syndrome is characterized by failure to thrive with significant feeding problems and hypotonia. This is followed by hyperphagia and obesity in childhood, mild to moderate mental retardation, persistent hypotonia, motor delay, short stature and hypogonadism. In both isolated Angelman syndrome or Prader-Willi syndrome, hypopigmentation is reported; however, typically much less severe than that in oculocutaneous albinism[11, 12].
The Angelman syndrome/Prader-Willi syndrome critical region is located at 15q11-q13. This region is subject to genomic imprinting, the process by which certain genes are activated or inactivated depending on their parent of origin. Different diseases can be caused by abnormal imprinting at the same locus as seen in Angelman syndrome and Prader-Willi syndrome. Failure of expression of the Angelman gene, UBE3A, on the maternally inherited allele results in Angelman syndrome. This expression failure is caused by maternal deletion of this locus (65–75%), point mutation in UBE3A (10%), uniparental disomy of two paternal chromosomes 15s present (5%), or an imprinting defect (5%)[13]. In 5–10% with the clinical diagnosis of Angelman syndrome, no genetic etiology is currently known. In Prader-Willi syndrome, the diagnosis is established by the absence of an active paternal allele via deletion (65–75%), uniparental maternal disomy (20–30%) or a non deletion/non uniparental disomy defect (~5%)[13].
We report a patient with characteristic cutaneous and ophthalmologic features of oculocutaneous albinism type 2, and features unattributable to albinism including microcephaly, absent speech, global developmental delay, tremulous jerky movements, and seizures. Subsequent evaluation revealed co-morbid conditions of oculocutaneous albinism type 2 and Angelman syndrome.
CASE REPORT
The proband, a 17-month old African female, was born at 39 weeks gestation to unrelated, healthy Yoruba Nigerian parents. Prior two pregnancies resulted in a 3 year old healthy male and a 10 week spontaneous abortion. Mother had standard prenatal care and there were no maternal illnesses or exposures. There was no family history of albinism, mental retardation, developmental delay or seizures.
Cutaneous diffuse depigmentation, nystagmus, retinal and iris depigmentation were noted directly after birth. The patient was referred for a developmental assessment at 15 months due to global delay (sat 12 months, not pulling to stand, absent verbalization/speech, no pincer grasp). Hearing evaluation and brain by MRI were normal.
At 17 months, she was generally fussy, sleepy and had lost the ability to sit independently after a 1–2 minute febrile seizure episode. Skin was pale and hair was coarse; nystagmus and gray irides with translucent red areas were present (Fig. 1). Head circumference was 44.5 cm (<5%, 50th% for a 9 month old), length 87cm (>95%) and weight 10kg (25%). She smiled spontaneously, but was otherwise non-interactive. Extremity movements were volitional, but jerky and tremulous.
FIGURE 1.
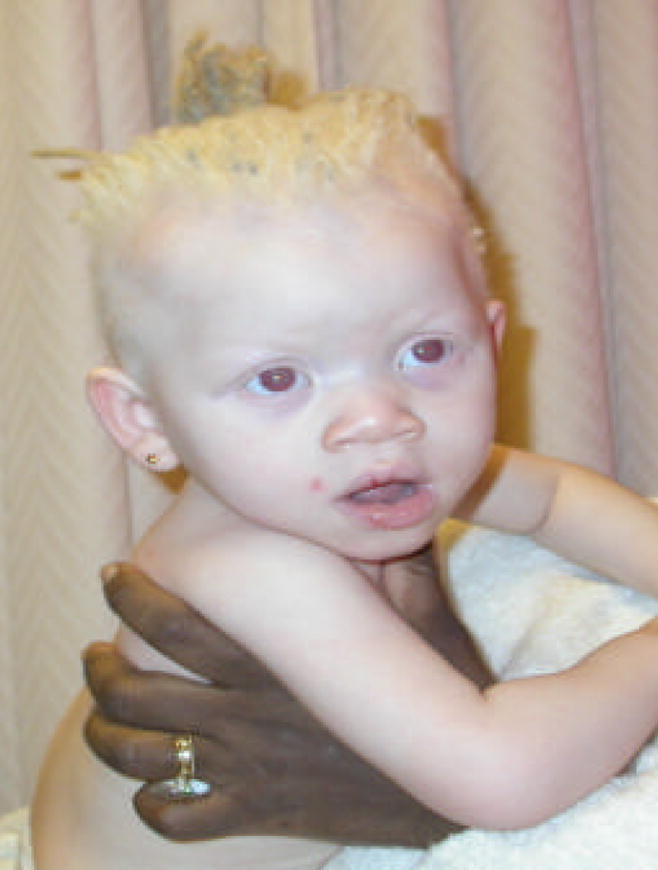
The patient at the age of 17 months. Note depigmentation of the skin compared to her mother who is holding her and course depigmented hair consistent with both albinism and her African descent.
At 19 months, over the course of a day, she developed multiple unprovoked and non focal generalized tonic-clonic seizures. These were described as synchronous jerking of all extremities for 15–20 seconds, followed by sleepiness and a difficulty to arouse. An EEG was markedly abnormal with diffuse slow activity and sharp activity posteriorly suggesting epileptiform activity. She was discharged on topiramate which has significantly reduced, but not eliminated the seizures.
Her clinical presentation was consistent with albinism and African descent, but also suspicious for co-morbid Angelman syndrome, with microcephaly, absent speech, jerky tremulous movements, seizures and global delay.
MATERIALS AND METHODS
Cytogenetics
Cytogenetic analysis was performed on thymidine synchronized peripheral blood lymphocytes. Chromosome preparations and Giemsa-banding followed standard procedures. Fluorescence in-situ hybridization was performed using SNRPN probe for the Angelman syndrome/Prader-Willi syndrome region on chromosome 15 that includes control probes for the centromere, CEP15, and LSI PML on 15q22 and for subtelomeres. (Vysis, Inc., Des Plaines, IL, USA).
Molecular Analysis, P gene
Genomic DNA was prepared from peripheral blood leukocytes [proband, mother][14] or mouthwash specimen [father] (Puregene DNA purification kit, Gentra Systems, Minneapolis, MN USA). Polymerase chain reaction (PCR) based screening for the P exon 7 2.7-kb deletion was performed as described by Durham-Pierre et al. (1994)[6](Fig. 2A).
FIGURE 2.

A. PCR detection of deletion. Primers MHB 71, 72 and 107 used in the PCR assays to amplify genomic DNA. Unaffected individuals have no deletion. Thus, a 240 bp fragment between MHB 71 and MHB 72 is amplified. The fragment for MHB 107 to MHB 71 is too large to amplify using our PCR conditions. Deletion of the 2.7-kb region of the P gene eliminates the primer sequence of MHB 72, resulting in amplification of a 420 bp fragment between MHB 71 and MHB 107.
B. Molecular analysis for the 2.7-kb deletion of the P gene showed that the affected child (P) carries only the deleted allele (420bp fragment), the mother (M) carries only the normal allele (240bp fragment) and the father (F) carries one deleted allele and one normal allele. Lane 1 is the size marker, lane C is a negative control.
RESULTS
Chromosome analysis revealed a microscopically visible deletion on one chromosome 15 with breakpoints at q11.2 and q13. Fluorescence in-situ hybridization (FISH) with the SNRPN probe confirmed the deletion, indicating absence of material within the Angelman syndrome/Prader-Willi syndrome critical region. The patient’s karyotype was 46,XX,del(15)(q11.2q13).ish del(15)(q11.2q11.2) (SNRPN-). FISH with subtelomere probes showed normal hybridization (results not shown).
Molecular analysis (Fig. 2B) revealed the proband is hemizygous for the exon 7 deletion inherited from her father who is a carrier of this allele. The mother has normal P gene alleles.
The paternal exon 7 P gene deletion in combination with a large de novo deletion of the Angelman syndrome critical region including the P gene from the maternal allele is responsible for this compound phenotype.
DISCUSSION
Approximately 1% of individuals with oculocutaneous albinism type 2 also present with clinical features of Prader-Willi syndrome or Angelman syndrome[15]. This co-morbid situation arises when a deletion of the Angelman syndrome/Prader-Willi syndrome critical region of chromosome 15q11-q13 includes the P gene on one allele and a P gene mutation or deletion occurs on the other allele. Our patient was found to have a paternally inherited deletion of exon 7 of the P gene and a large de novo deletion on the maternally inherited chromosome 15q including the Angelman syndrome critical region and the P gene on that allele, thus causing this compound phenotype of oculocutaneous albinism and Angelman syndrome.
Unlike the relatively rare occurrence of oculocutaneous albinism type 2 in patients with either Angelman syndrome or Prader-Willi syndrome, up to 50% of Angelman syndrome/Prader-Willi syndrome patients exhibit mild to moderate hypopigmentation[12]. Decreased pigmentation is most common in those patients with Angelman syndrome or Prader-Willi syndrome caused by a deletion[12]. Hemizygosity for the P gene within the Angelman syndrome/Prader-Willi syndrome critical region correlates significantly with the presence of hypopigmentation[12]. However, heterozygous carriers of P gene mutations alone do not have hypopigmentation. Hypotheses to explain this inconsistency include reduced expression of the intact P gene via an interaction with the larger 15q deletion resulting in reduced pigmentation or another yet undiscovered gene exists in that region that, in haploinsufficiency, is not adequate to create normal pigment.[12].
Although hypopigmentation, the primary feature of oculocutaneous albinism type 2, occurs in up to 50% of Angelman syndrome or Prader-Willi syndrome cases, severe speech impairment, seizure activity, gait/movement disorder and characteristic behavioral manifestations of Angelman syndrome or psychomotor retardation, infantile hypotonicity, hyperphagia, obesity and hypogonadism of Prader-Willi syndrome should not be present in an individual with isolated oculocutaneous albinism type 2. In young infants with albinism who may not yet manifest the entire constellation of Angelman syndrome or Prader-Willi syndrome features, neurodevelopmental delay may be the first clue to this compound diagnosis. Thus, our case demonstrates the necessity to further investigate for concurrent Angelman syndrome/Prader-Willi syndrome in a patient with developmental delay and albinism.
Acknowledgments
The authors wish to acknowledge Dr. Douglas Riegert-Johnson for his recruitment of the patient and the technical expertise of Maria Blazina, Shirley Johns, Susan Morsey and Elizabeth Squibb from the Cytogenetics Laboratory at Kennedy Krieger Institute, Baltimore, MD.
Footnotes
There are no financial relationships or conflicts of interests relevant to this article to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gardner JM, Nakatsu Y, Gondo Y, Lee S, Lyon MF, King RA, Brilliant MH. The mouse pink-eyed dilution gene: association with human Prader-Willi and Angelman syndromes. Science. 1992;257:1121–1124. doi: 10.1126/science.257.5073.1121. [DOI] [PubMed] [Google Scholar]
- 2.“Oculocutaneous Albinism Type 2 [www.genetests.org]
- 3.Okoro AN. Albinism in Nigeria. A clinical and social study. Br J Dermatol. 1975;92:485–492. [PubMed] [Google Scholar]
- 4.Durham-Pierre D, King RA, Naber JM, Laken S, Brilliant MH. Estimation of carrier frequency of a 2.7 kb deletion allele of the P gene associated with OCA2 in African-Americans. Hum Mutat. 1996;7:370–373. doi: 10.1002/(SICI)1098-1004(1996)7:4<370::AID-HUMU15>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 5.Lee ST, Nicholls RD, Bundey S, Laxova R, Musarella M, Spritz RA. Mutations of the P gene in oculocutaneous albinism, ocular albinism, and Prader-Willi syndrome plus albinism. N Engl J Med. 1994;330:529–534. doi: 10.1056/NEJM199402243300803. [DOI] [PubMed] [Google Scholar]
- 6.Durham-Pierre D, Gardner JM, Nakatsu Y, King RA, Francke U, Ching A, Aquaron R, del Marmol V, Brilliant MH. African origin of an intragenic deletion of the human P gene in tyrosinase positive oculocutaneous albinism. Nat Genet. 1994;7:176–179. doi: 10.1038/ng0694-176. [DOI] [PubMed] [Google Scholar]
- 7.“Angelman Syndrome” [www.genetests.org]
- 8.Guerrini R, Carrozzo R, Rinaldi R, Bonanni P. Angelman syndrome: etiology, clinical features, diagnosis, and management of symptoms. Paediatr Drugs. 2003;5:647–661. doi: 10.2165/00148581-200305100-00001. [DOI] [PubMed] [Google Scholar]
- 9.Rubin DI, Patterson MC, Westmoreland BF, Klass DW. Angelman’s syndrome: clinical and electroencephalographic findings. Electroencephalogr Clin Neurophysiol. 1997;102:299–302. doi: 10.1016/s0013-4694(96)96105-2. [DOI] [PubMed] [Google Scholar]
- 10.Nolt DH, Mott JM, Lopez WL. Assessment of anticonvulsant effectiveness and safety in patients with Angelman’s syndrome using an Internet questionnaire. Am J Health Syst Pharm. 2003;60:2583–2587. doi: 10.1093/ajhp/60.24.2583. [DOI] [PubMed] [Google Scholar]
- 11.King RA, Wiesner GL, Townsend D, White JG. Hypopigmentation in Angelman syndrome. Am J Med Genet. 1993;46:40–44. doi: 10.1002/ajmg.1320460109. [DOI] [PubMed] [Google Scholar]
- 12.Spritz RA, Bailin T, Nicholls RD, Lee ST, Park SK, Mascari MJ, Butler MG. Hypopigmentation in the Prader-Willi syndrome correlates with P gene deletion but not with haplotype of the hemizygous P allele. Am J Med Genet. 1997;71:57–62. doi: 10.1002/(sici)1096-8628(19970711)71:1<57::aid-ajmg11>3.0.co;2-u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet. 2001;2:153–175. doi: 10.1146/annurev.genom.2.1.153. [DOI] [PubMed] [Google Scholar]
- 14.Bellus GA, Hefferon TW, Ortiz de Luna RI, Hecht JT, Horton WA, Machado M, Kaitila I, McIntosh I, Francomano CA. Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am J Hum Genet. 1995;56:368–373. [PMC free article] [PubMed] [Google Scholar]
- 15.Spritz RA. Molecular genetics of oculocutaneous albinism. Hum Mol Genet. 1994;(3 Spec No):1469–1475. doi: 10.1093/hmg/3.suppl_1.1469. [DOI] [PubMed] [Google Scholar]