

Abstract
Hepatocyte growth factor (HGF) is a potent inducer of motility in epithelial cells. Since we have previously found that activation of the epidermal growth factor receptor (EGFR) is an absolute prerequisite for induction of motility of corneal epithelial cells after wounding, we investigated whether induction of motility in response to HGF is also dependent on activation of the EGFR. We now report that HGF induces transactivation of the EGFR in an immortalized line of corneal epithelial cells, in human skin keratinocytes, and in Madin-Darby canine kidney cells. EGFR activation is unconditionally required for induction of motility in corneal epithelial cells, and for induction of a fully motile phenotype in Madin-Darby canine kidney cells. Activation of the EGFR occurs through amphiregulin and heparin-binding epidermal growth factor-like growth factor. Early after HGF stimulation, blocking EGFR activation does not inhibit extracellular-signal regulated kinase 1/2 (ERK1/2) activation by HGF, but the converse is seen after approximately one hour, indicating the existence of EGFR-dependent and -independent routes of ERK1/2 activation. In summary, HGF induces transactivation of the EGFR in epithelial cells, and this is a prerequisite for induction of full motility.
Keywords: Hepatocyte growth factor, epidermal growth factor, cell motility, cornea, transactivation, wound healing
Introduction
Hepatocyte Growth Factor (HGF)/scatter factor is a strong inducer of motility in epithelial cells [for reviews see: 1-4]. It is predominantly produced by mesenchymal cells, and interacts with a receptor, MET, that is present in most epithelia. HGF has numerous effects: during wound healing it stimulates re-epithelization, stimulates angiogenesis, promotes deposition of extracellular matrix, and modulates immune function. HGF/MET provides a strong mitogenic signal, and MET was originally isolated as an oncogene. HGF/MET signaling is also a major contributor to malignant progression of common human tumors, increasing their tendency to invade and metastasize over time. For these reasons there is considerable interest in developing drugs to target HGF/MET signaling to combat cancer [for reviews see: 5-8].
We and others have previously reported that the epidermal growth factor receptor (EGFR) is activated upon wounding sheets of corneal epithelial cells, as it is in vivo, and that its activation is necessary for healing [9-11]. The classic mode of EGFR activation occurs by binding to any of a small number of ligands [12]. Activation of some receptors may lead to transactivation of the EGFR, and in this way other receptors may commandeer the EGFR and its down-stream signaling pathways [13, 14]. Since HGF strongly stimulates migration of corneal epithelial cells [15-17] it seemed reasonable to hypothesize that EGFR signaling might be part of the mechanism whereby HGF induces motility. We therefore studied the role of EGFR in HGF signaling in corneal epithelial cells.
Materials and Methods
Materials
HCLE cells were kindly provided by Dr. Irene Gipson, human epidermal keratinocytes were from Cascade Biologicals, and MDCK type II cells were a gift from Dr. Ora Weisz. HGF was from Chemicon, tyrphostin AG 1478 was from Alexis Biochemicals, and GM6001 was from EMD Biosciences. Anti-amphiregulin and anti-HB-EGF were from R&D Systems. Antibodies against phospho-ERK1/2, MET, and the EGFR phosphorylated on tyrosine 1173 were from Santa Cruz Biotechnology. Antibodies to Ecadherin were from BD Transduction Laboratories. Antibodies against MET phosphorylated on tyrosine 1234/1235 were from Cell Signaling. Alexa Fluor® 546-conjugated phalloidin was from Invitrogen™.
Tissue culture, growth factor stimulation, and measurement of amphiregulin release
HCLE cells were grown in keratinocyte serum-free medium (Gibco-Invitrogen) supplemented with 25 μg/ml bovine pituitary extract, 0.2 ng/ml epidermal growth factor (EGF), and 0.3 mM CaCl2. Stimulations were performed after incubating sub-confluent cells overnight in the same medium without added EGF. Stimulations with HGF (10 ng/ml) or EGF (100 ng/ml) were for 10 minutes. Human epidermal keratinocytes were treated similarly except that pituitary extract was omitted for the overnight starvation. MDCK cells were grown to confluence in Dulbecco's Modified Eagle's Medium with 10% fetal calf serum, and starved overnight in the same medium with 2% fetal calf serum. Chemical inhibitors were added 15 minutes prior to stimulation, and neutralizing antibodies were added 4-6 hours before stimulation. Amphiregulin release was measured using the DuoSet ELISA (R & D systems). The cells were starved and incubated as described above. Supernatants were clarified by centrifugation at 5000xg for 1 minute and assayed according to the manufacturer's instructions.
Wounding assay
HCLE cells were grown in the presence of agarose strips [10]. When near confluence, cells were stratified by transfer to a high Ca2+ medium (Dulbecco's Modified Eagles Medium: F12 1:1, with 10% new-born calf serum and 10 ng/ml EGF). Three days later, cells were transferred to the same medium with 2% new-born calf serum and no EGF. The following day, the strips were removed, and wounds allowed to heal for 14 hours. MDCK cells were grown in DMEM with 10% fetal calf serum, and transferred to 2% fetal calf serum the day before the experiment. Experiments with mitomycin C demonstrated that HGF-induced healing of wounds in MDCK cells was due to cell migration.
Immunofluorescence, microscopy, and immunoblotting
Immunofluorescence labeling was performed using formaldehyde-fixed cells, and microscopy was performed as described previously [10]. Images were captured on an Olympus 1 × 70 inverted microscope equipped with a Bio-Rad Radiance 2000® confocal system and Lasersharp® software using a 40x objective. Phase contrast microscopy was performed using a Nikon TS100 microscope with a 20x objective and the “Spot” imaging system (Diagnostics Instruments, Inc.). Western blotting was performed according to standard procedures, and blots were developed using the SuperSignal® Dura detection kit (Pierce). Ponceau S staining of blots and phalloidin staining of fixed cells were performed as described [18]. The autoradiograms were scanned with the BioRad Fluor-S™ MultiImager and densitometry analysis performed with Quantity One software (BioRad). Statistical analysis was performed using the unpaired students's t test.
Results and discussion
HGF induces transactivation of the EGFR in corneal epithelial cells
HCLE cells are human corneal limbal epithelial cells that have been immortalized by a three-step process involving abrogation of p16INK4A/Rb and p53 functions, and overexpression of the catalytic subunit of the telomerase holoenzyme [19]. They express the keratins K3 and K12 which are specific markers for the corneal epithelium. When grown in low Ca2+ concentrations, they grow as monolayers, but stratify upon transfer to high Ca2+. They therefore serve as a model for native corneal epithelial cells which are stratified in their resting state, but spread out to form monolayers when they cover wounds.
Unstratified HCLE cells were stimulated with HGF and extracts were immunoblotted with an antibody that recognizes the EGFR phosphorylated on tyr-1173. As expected, HGF induced strong activation of its receptor MET (Fig. 1A). Importantly, HGF also induced phosphorylation of the EGFR, reaching a maximum after 10 minutes (Fig. 1A and C). Stimulations were routinely performed in the presence of pituitary extract (which is used in the growth medium of the cells) since these conditions were found to yield the strongest activations, but increases were also apparent after cells had been incubated overnight without this supplement. The EGF receptor was also phosphorylated in response to HGF in their stratified state, but to a lesser extent. This may simply be due to the fact that added HGF only reaches the uppermost cell layer in the multilayered epithelium. Tyr-1173 is autophosphorylated by the receptor, and the EGFR kinase inhibitor tyrphostin AG 1478 blocked tyr-1173 phosphorylation completely. Interestingly, tyrphostin AG 1478 significantly decreased phosphorylation of MET by HGF (Fig 1A). The EGFR is known to transactivate MET [20-22], and the result suggests that part of the stimulation of MET in response to HGF is routed through the EGFR.
Fig. 1.

Activation of the EGFR by HGF in corneal epithelial cells. (A) HCLE cells were treated with 10 ng/ml HGF for 10 minutes, the extracts were immunoblotted with an antibody that recognizes the EGFR phosphorylated on tyr-1173, and the blots were stripped and subsequently blotted with an antibody that recognizes the total amount of EGFR. The same blots were immunoblotted with an antibody that recognizes activated ERK1/2. The extracts were also immunoblotted with antibodies against MET and MET phosphorylated on tyr-1234/1235. Where indicated, 10 μM tyrphostin AG 1478 was added for 15 minutes before treatment. The bar graph shows the results of densitometry of the autoradiograms. The values in this and the following figures are means of triplicates or more, and the error bars are standard deviations. Analysis by the students's t test for unpaired samples showed significant increase of the signal from the EGFR in response to HGF (*, P<0.005). The signal from MET in response to HGF was significantly reduced in the presence of tyrphostin AG 1478 (**, P<0.005) (B) Cells were treated with 100 ng/ml EGF, and processed similarly. (C) Cells were treated for the indicated times and extracts immunoblotted as in (A). pERK1/2 was significantly reduced in the presence of tyrphostin AG 1478 (*, P<0.0005).
The activation of extracellular-signal regulated kinase 1/2 (ERK1/2) was monitored by immunoblotting with an antibody that recognizes the activated, phosphorylated forms of the kinases. As expected, HGF clearly activates ERK1/2. Early after stimulation by HGF, ERK1/2 activation was not reduced in the presence of tyrphostin AG 1478 (Fig. 1C), whereas EGF-induced activation was completely blocked by tyrphostin AG 1478 (Fig. 1B), suggesting that activation of ERK1/2 by HGF occurs independently of EGFR signaling. At one hour and later, tyrphostin AG 1478 blocks activation of ERK1/2, indicating that it is at later times regulated by the EGFR. In most cells, ERK is absolutely required for motility, and we have found that addition of the ERK inhibitor UO126 completely blocks healing of wounds in sheets of HCLE cells. The requirement for EGFR activity in these cells could therefore reflect a requirement for ERK activation.
Activation of the EGFR by HGF occurs through the triple membrane-passing signaling (TMPS) mode
Transactivation of the EGFR can occur intracellularly through reactive oxygen species, which are thought to inactivate tyrosine phosphatases that deactivate the receptor, and through p60src-mediated mechanisms [23-27]. Heterodimerization of the EGFR with unrelated receptors may also provide an intracellular mode of transactivation [28-30]. Alternatively, activation of certain receptors, particularly G-protein coupled receptors, can result in proteolytic cleavage of precursors of the EGF family of ligands located in the cell membrane, which can then bind and activate the EGFR; this is known as the triple membrane-passing signaling mechanism [13, 14]. When the broad-range metalloprotease inhibitor GM6001 was added to the cells, activation of the EGFR by HGF was inhibited (Fig. 2A), indicating that a proteolytic event is required for activation. GM6001 does not block activation of the receptor by EGF, indicating that the compound does not act by inducing general toxicity in these cells [10]. Addition of the LA1 antibody, which binds to the extracellular domain of human EGFR [31], also abrogated HGF stimulation of the receptor, supporting that HGF induces activation of the EGFR through a mechanism that requires ligand binding.
Fig. 2.
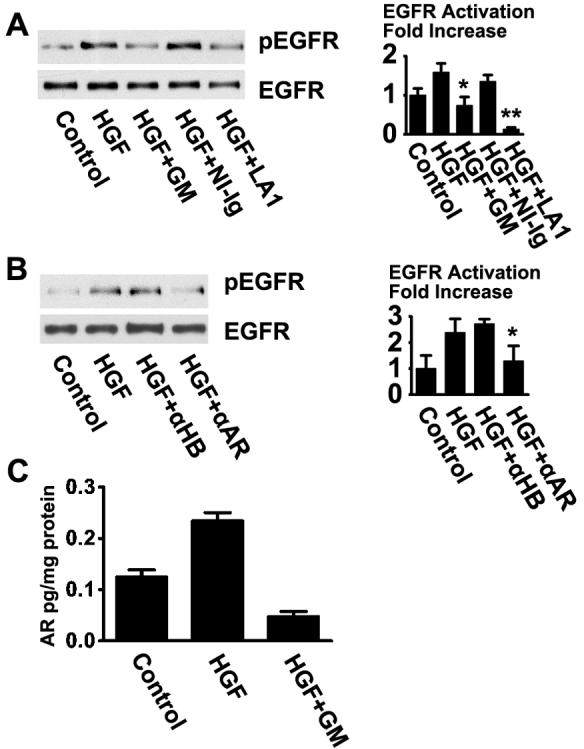
Activation of the EGFR by HGF occurs through the TMPS mode of activation in HCLE cells. (A) HCLE cells were pre-incubated with 50 μM GM6001 (GM), 20 μg/ml non-immune immunoglobulin (NI-Ig), or 20 μg/ml of the EGFR-neutralizing LA1 antibody (LA1), and immunoblotted with an anti-pEGFR(tyr-1173) antibody. The signals were significantly reduced in the presence of GM6001 and LA1 antibody (*, P< 0.01; **, P<0.0005). (B) Cells were pretreated with 20 μg/ml of neutralizing antibodies against HB-EGF (αHB) or amphiregulin (αAR) and immunoblotted as in A. The signals in response to the anti-amphiregulin antibody was significantly reduced (*, P<0.05). (C) Amphiregulin was measured in supernatants from HCLE cells. Amphiregulin was measured in supernatants from HCLE cells following one hour treatments and results were normalized to total protein content of whole cell extracts. Equal loading of the lanes in the blots in this and the following figures were monitored by protein staining with Ponceau S Red.
We further tested neutralizing antibodies against ligands of the EGFR. As is seen in figure 2B, antibodies against amphiregulin clearly inhibited activation of the receptor, whereas an antibody against heparin-binding EGF-like growth factor (HB-EGF) had less effect. Antibodies against tumor necrosis factor-α and EGF were without effect (data not shown). The results suggest that HGF-mediated EGFR activation is predominantly mediated through amphiregulin signaling under these conditions.
To directly measure release of ligands into the tissue culture supernatant, unstratified HCLE cells were cultured and starved as in other experiments, then were incubated for one hour with no treatment, with 10ng/ml HGF, or with HGF and 50 μM GM6001. Cell-free supernatants were collected, assayed, and protein contents of whole cell extracts were determined for normalization. As is seen in figure 2C, HGF induced a two-fold increase in release of soluble amphireguin. Importantly, the amount of amhiregulin detected in the supernatant was reduced in the presence of the protease inhibitor GM6001, supporting the notion that it is released as a result of proteolytic activity. HB-EGF was not detected in the tissue culture supernatants, even after addition of phorbol myristic acetate, which is a strong inducer of cleavage of the precursors of ligands of the EGFR. This negative result could either be due to inadequate sensitivity of the assay system, or it could be due to the fact that HB-EGF binds tenaciously to the surfaces of cells.
We have previously reported that wounding induces transactivation of the EGFR through PLD [18]. However, in contrast to wounding, we find no activation of PLD after stimulation with HGF in HCLE cells. Src and protein kinase C have also been implicated as upstream activators of EGF receptor transactivation [13], but the their inhibitors, pp2 and bisindolymaleimide, do not block the EGFR transactivation by HGF (data not shown). Thus, the signaling intermediates between the MET and the proteolysis of the ligands for the EGFR remain to be determined.
Requirement of EGFR activation for HGF-induced motility in HCLE cells
A prominent feature of HGF is its ability to induce scattering of colonies of epithelial cells. HCLE cells undergo marked scattering upon treatment with HGF (Fig. 3A). Scattering induced by HGF is inhibited by tyrphostin AG 1478 and the LA1 anti-EGFR antibody, but not by control immunoglobulin (data not shown), strongly supporting a role for the TMPS mechanism in HGF-induced scattering. Inhibition of scattering was also seen by addition of anti-amphiregulin antibody. Addition of EGFR ligands such as EGF, HB-EGF, or amphiregulin induced scattering of colonies to an extent similar to that seen with HGF (Fig. 3A, and data not shown).
Fig. 3.
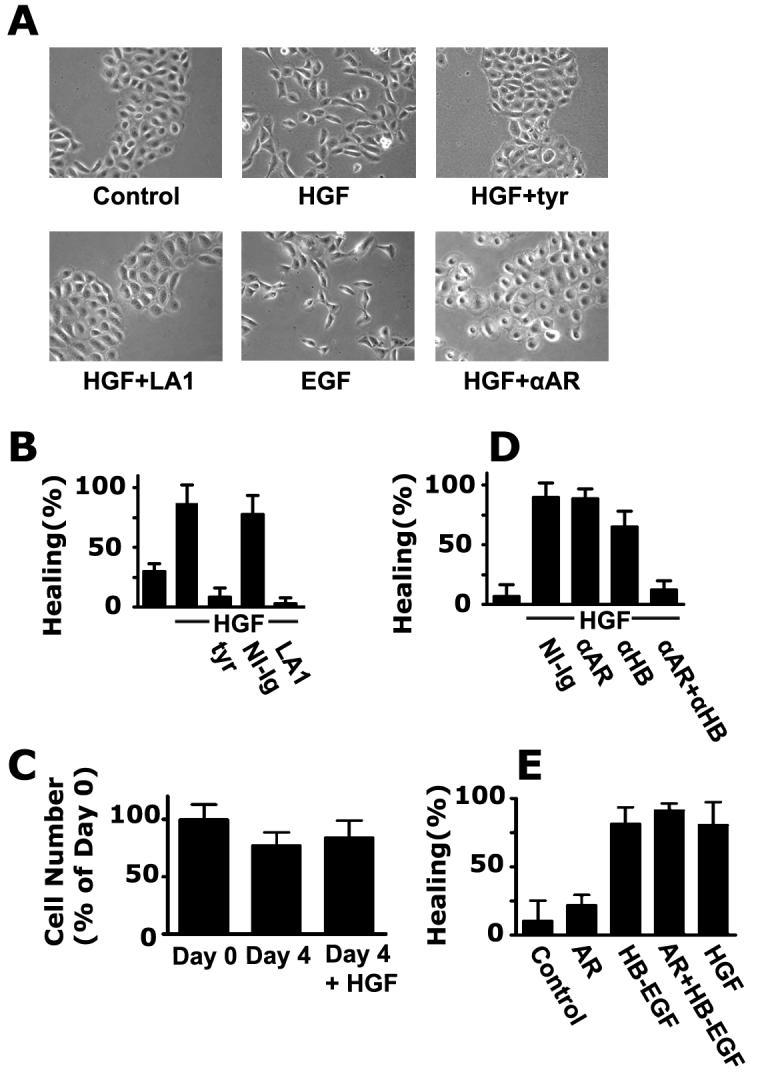
Activation of the EGFR is absolutely required for induction of motility by HGF in HCLE cells. (A) HCLE cells were grown for four days to form colonies, and were incubated with 10 ng/ml HGF for five hours or were left untreated. Where indicated, stimulation was performed in the presence of 10 μM tyrphostin AG 1478 (tyr), 20 μg/ml LA1 antibody, 100 ng/ml EGF, or 20 μg/ml anti-amphiregulin antibody (αAR), and photographed in phase-contrast. (B). Wound healing was monitored as described [10] in the presence of the indicated reagents. The means of quadruplicate measurements are shown and the error bars are standard deviations in this and the following panels. (C) HCLE cells were seeded and treated as in the wound-healing assays. They were counted at day 0, or incubated for four days with or without 10 ng/ml HGF and counted again. (D) Wound healing was performed similarly to (B) in the presence of 20 μg/ml of the indicated antibodies. (E) Wound-healing was performed in the presence of 50 ng/ml amphiregulin or HB-EGF, or 10 ng/ml HGF, as indicated.
Epithelia generally move as integral sheets, and a healing of wounds in confluent cell layers is presumably a more relevant model for healing. As a more quantitative measure of motility, we also evaluated the role of TMPS in wound healing. Unstratified HCLE cells heal very slowly, but upon stratification, HGF-accelerated wound closure is readily detected (Fig. 3B). It should be noted that the HCLE cells do not divide when stratified (Fig. 3C), so healing reflects cell movement rather than cell division. When tyrphostin AG 1478 or LA1 antibody was added, healing was totally abrogated, demonstrating an absolute requirement of EGFR activation.
The effects of ligand-neutralizing antibodies on wound healing were also examined. As is seen in figure 3D, addition of anti-amphiregulin antibody had little detectable effect, whereas some effect was seen with the anti-HB-EGF antibody. The combination of both antibodies strongly blocked HGF-induced wound closure, demonstrating that these two ligands are the major determinants of cell motility. In figure 3E, we illustrate the effect of the ligands on wound closure. Amphiregulin or EGF (not shown) were not as potent in accelerating healing as was HGF, however, HB-EGF induced a similar degree of healing (Fig 3E). This may be related to the fact that the two former ligands only activate the EGFR, whereas HB-EGF also activates the EGFR-related receptor ErbB4 [12].
Clearly, the activity of HGF is dependent on activation of the EGFR in HCLE cells though amphiregulin or HB-EGF signaling. The two ligands obviously both can participate in mediating HGF activity, but their relative roles seem to vary according to the parameter that is assayed. For instance, when unstratified cells are stimulated with HGF for 10 minutes, amphiregulin is the dominant ligand that induces EGFR phosphorylation (Fig 2B). However, in the wounding assays, which are long-term assays with stratified cells, the antibody-blocking showed that both are important.
HGF-induced transactivation of the EGFR in human epidermal keratinocytes and Madin-Darby canine kidney (MDCK) cells
To determine whether HGF-induced EGFR activation is particular to HCLE cells we also tested other epithelial cells. The EGFR was found to be activated by HGF in human epidermal keratinocytes. Activation was inhibited by the protease inhibitor GM6001 and by the LA1 antibody, strongly suggesting that it occurs through a TMPS mechanism as in HCLE cells (Fig. 4).
Fig 4.
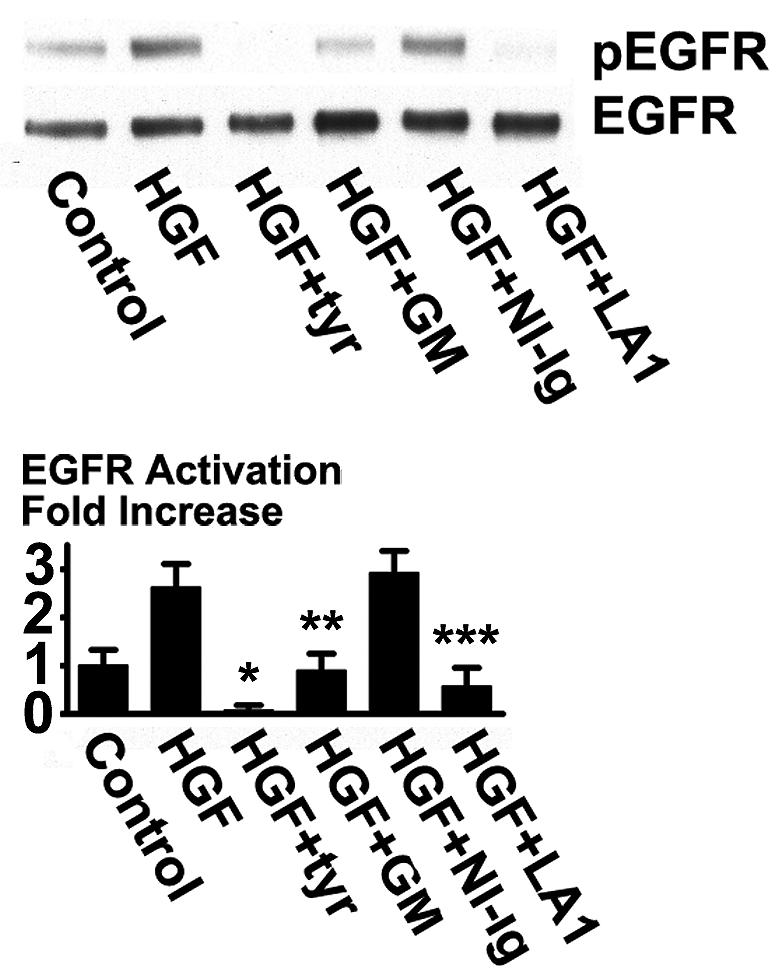
Transactivation of the EGFR by HGF in human skin keratinocytes. Human skin keratinocytes were stimulated with 10 ng/ml HGF for 10 minutes in the presence of 50 μM GM6001 (GM), 20 μg/ml non-immune IgG (NI-Ig), or 20 μg/ml LA1 antibody and immunoblotted with an anti-pEGFR(tyr-1173) antibody. The signals were significantly reduced in the presence of tyrphostin AG1478, GM6001, and LA1 antibody (*, P< 0.001; **, P< 0.01; ***P< 0.005).
MDCK cells are commonly used as a generic model of epithelial cells. HGF increased phosphorylation of tyr-1173 in the EGFR, and the increase was blocked by tyrphostin AG 1478 and GM6001, again pointing to a TMPS mode of activation (Fig. 5A). Since the LA1 antibody is human-specific, its effect on MDCK cells could not be evaluated. HGF induces scattering strongly in MDCK cells, but blocking EGFR activation with tyrphostin AG 1478 in the presence of HGF had interesting effects: the cells flattened out, numerous lamellipodia were seen at the peripheries of the colonies, but the cells did not dissociate (Fig. 5B). The cytoskeleton in MDCK cells undergoes marked reorganization in response to HGF: the prominent actin cable that surrounds the colonies is dissolved, cortical f-actin disappears, and stress fibers tend to become less prominent [32, 33]. In the presence of tyrphostin AG1478, stress fibers were very prominent, and the actin cable at the circumference was intact, but almost complete dissolution of cortical actin was observed (Fig. 5C). Tyrphostin AG 1478 did not interfere with the internalization of E-cadherin that occurs in response to HGF (data not shown). Tyrphostin AG 1478 partially reduced healing in a wounding assay (Fig. 5D). Stimulation of MDCK cells with EGF or HB-EGF only induced scattering very weakly, as has been previously reported [34, 35], and had little effect on the healing rate in sheets of MDCK cells (data not shown).
Fig 5.
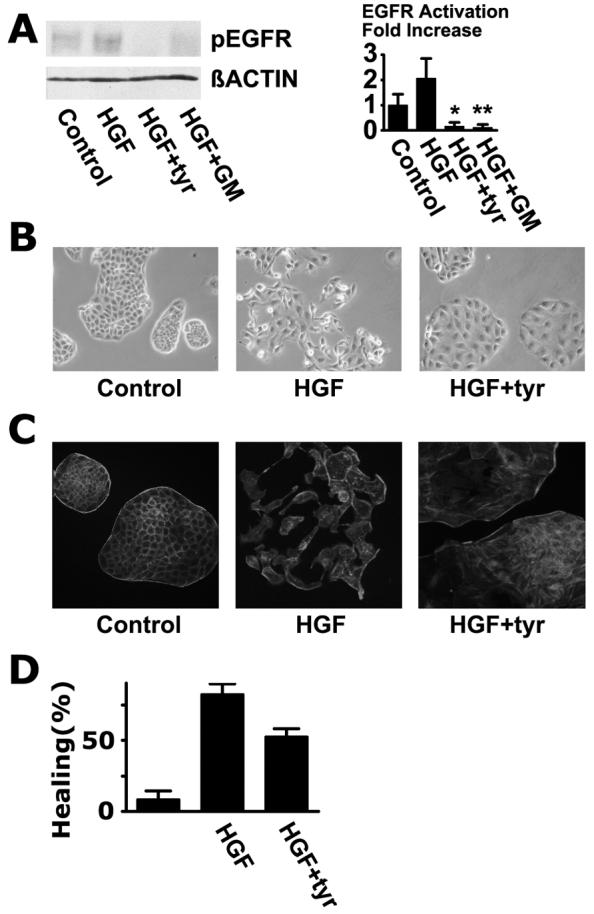
Transactivation of the EGFR is required for induction of a fully motile phenotype in MDCK cells. (A) Activation of the EGFR in MDCK cells under conditions similar to those shown in Fig. 1B except that 20 ng/ml HGF was used. The signals were significantly reduced in the presence of tyrphostin AG1478 and GM6001 (*, P< 0.0005; **, P< 0.001) (B) Phase contrast photo-micrographs of control MDCK colonies, colonies stimulated with 20 ng/ml HGF for 7 hours, and colonies stimulated with HGF in the presence of 10 μM tyrphostin AG 1478 (tyr). (C) Cells were stained with Alexa Fluor® 546-labeled phalloidin. (D) Wounding assay was performed as in Fig. 3B. The means of quadruplicate measurements are shown and the error bars are standard deviations.
Taken together, these data suggest that transactivation of the epidermal growth factor by HGF through the TMPS mode is a general phenomenon in epithelial cells. In HCLE cells, EGFR activation is absolutely required for induction of motility in response to HGF, and induction of the fully motile phenotype in MDCK cells also requires EGFR signaling. Cell-specific differences were also seen when ligands for the EGFR were added. EGF, amphiregulin and HB-EGF induced full scattering in HCLE cells, and HBEGF induced healing of wounds in sheets of the cells as potently as did HGF. In contrast, the ligands had little effects in MDCK cells suggesting that HGF induces an additional array of signals in these cells.
Interactions between the HGF receptor and the EGFR have been reported previously. Scheving et al. described that addition of an inhibitor of the EGFR kinase blocks HGF-induced DNA synthesis in hepatocytes [36]. It is not clear whether this reflects a requirement of basal EGFR activity, since stimulation of the EGFR by HGF was not reported. Conversely, stimulation of the EGFR was found to result in tyrosine phosphorylation of MET in several cancer cell lines either through a TMPS type of mechanism, or through production of reactive oxygen species [20-22].
HGF is important in inducing motility in the corneal epithelium so wounds can be covered rapidly [37, 38]. Our results show that HGF promotes motility in part by activating the EGFR, and in this way HGF appropriates the well-described motility-inducing signaling pathways of the EGFR. Increased activity of the EGFR is a major contributor to malignant transformation and tumor progression, and many mechanisms for its activation have been identified [39-41]. We suggest that the increased HGF/MET signaling that occurs regularly during progression of common human tumors [1-3] contributes to EGFR activation. As a consequence, pharmacological interference with EGFR signaling may be useful in managing tumors driven by aberrant HGF signaling.
Acknowledgements
We thank Dr. Irene Gipson for providing the HCLE cell line. This work was supported by the National Institutes of Health Grants EY013463 and EY08098, and grants from Research to Prevent Blindness and The Eye and Ear Foundation (Pittsburgh, PA).
Abbreviations
- HGF
hepatocyte growth factor
- EGFR
epidermal growth factor receptor
- EGF
epidermal growth factor
- TMPS
triple membrane-passing signal
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–25. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 2.Boccaccio C, Comoglio PM. Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer. 2006;6:637–45. doi: 10.1038/nrc1912. [DOI] [PubMed] [Google Scholar]
- 3.Matsumoto K, Nakamura T. Hepatocyte growth factor and the Met system as a mediator of tumor-stromal interactions. Int J Cancer. 2006;119:477–83. doi: 10.1002/ijc.21808. [DOI] [PubMed] [Google Scholar]
- 4.Conway K, Price P, Harding KG, Jiang WG. The molecular and clinical impact of hepatocyte growth factor, its receptor, activators, and inhibitors in wound healing. Wound Repair Regen. 2006;14:2–10. doi: 10.1111/j.1743-6109.2005.00081.x. [DOI] [PubMed] [Google Scholar]
- 5.Christensen JG, Burrows J, Salgia R. c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2005;225:1–26. doi: 10.1016/j.canlet.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 6.Jiang WG, Martin TA, Parr C, Davies G, Matsumoto K, Nakamura T. Hepatocyte growth factor, its receptor, and their potential value in cancer therapies. Crit Rev Oncol Hematol. 2005;53:35–69. doi: 10.1016/j.critrevonc.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 7.Corso S, Comoglio PM, Giordano S. Cancer therapy: can the challenge be MET? Trends Mol Med. 2005;11:284–92. doi: 10.1016/j.molmed.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Mazzone M, Comoglio PM. The Met pathway: master switch and drug target in cancer progression. Faseb J. 2006;20:1611–21. doi: 10.1096/fj.06-5947rev. [DOI] [PubMed] [Google Scholar]
- 9.Zieske JD, Takahashi H, Hutcheon AE, Dalbone AC. Activation of epidermal growth factor receptor during corneal epithelial migration. Invest Ophthalmol Vis Sci. 2000;41:1346–55. [PubMed] [Google Scholar]
- 10.Block ER, Matela AR, SundarRaj N, Iszkula ER, Klarlund JK. Wounding induces motility in sheets of corneal epithelial cells through loss of spatial constraints: role of heparin-binding epidermal growth factor-like growth factor signaling. J Biol Chem. 2004;279:24307–12. doi: 10.1074/jbc.M401058200. [DOI] [PubMed] [Google Scholar]
- 11.Xu KP, Ding Y, Ling J, Dong Z, Yu FS. Wound-induced HB-EGF ectodomain shedding and EGFR activation in corneal epithelial cells. Invest Ophthalmol Vis Sci. 2004;45:813–20. doi: 10.1167/iovs.03-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris RC, Chung E, Coffey RJ. EGF receptor ligands. Exp Cell Res. 2003;284:2–13. doi: 10.1016/s0014-4827(02)00105-2. [DOI] [PubMed] [Google Scholar]
- 13.Fischer OM, Hart S, Gschwind A, Ullrich A. EGFR signal transactivation in cancer cells. Biochem Soc Trans. 2003;31:1203–8. doi: 10.1042/bst0311203. [DOI] [PubMed] [Google Scholar]
- 14.Ohtsu H, Dempsey PJ, Eguchi S. ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am J Physiol Cell Physiol. 2006;291:C1–10. doi: 10.1152/ajpcell.00620.2005. [DOI] [PubMed] [Google Scholar]
- 15.Wilson SE, He YG, Weng J, Zieske JD, Jester JV, Schultz GS. Effect of epidermal growth factor, hepatocyte growth factor, and keratinocyte growth factor, on proliferation, motility and differentiation of human corneal epithelial cells. Exp Eye Res. 1994;59:665–78. doi: 10.1006/exer.1994.1152. [DOI] [PubMed] [Google Scholar]
- 16.Daniels JT, Limb GA, Saarialho-Kere U, Murphy G, Khaw PT. Human corneal epithelial cells require MMP-1 for HGF-mediated migration on collagen I. Invest Ophthalmol Vis Sci. 2003;44:1048–55. doi: 10.1167/iovs.02-0442. [DOI] [PubMed] [Google Scholar]
- 17.McBain VA, Forrester JV, McCaig CD. HGF, MAPK, and a small physiological electric field interact during corneal epithelial cell migration. Invest Ophthalmol Vis Sci. 2003;44:540–7. doi: 10.1167/iovs.02-0570. [DOI] [PubMed] [Google Scholar]
- 18.Mazie AR, Spix JK, Block ER, Achebe HB, Klarlund JK. Epithelial cell motility is triggered by activation of the EGF receptor through phosphatidic acid signaling. J Cell Sci. 2006;119:1645–54. doi: 10.1242/jcs.02858. [DOI] [PubMed] [Google Scholar]
- 19.Gipson IK, Spurr-Michaud S, Argueso P, Tisdale A, Ng TF, Russo CL. Mucin gene expression in immortalized human corneallimbal and conjunctival epithelial cell lines. Invest Ophthalmol Vis Sci. 2003;44:2496–506. doi: 10.1167/iovs.02-0851. [DOI] [PubMed] [Google Scholar]
- 20.Jo M, Stolz DB, Esplen JE, Dorko K, Michalopoulos GK, Strom SC. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J Biol Chem. 2000;275:8806–11. doi: 10.1074/jbc.275.12.8806. [DOI] [PubMed] [Google Scholar]
- 21.Pai R, Nakamura T, Moon WS, Tarnawski AS. Prostaglandins promote colon cancer cell invasion; signaling by cross-talk between two distinct growth factor receptors. Faseb J. 2003;17:1640–7. doi: 10.1096/fj.02-1011com. [DOI] [PubMed] [Google Scholar]
- 22.Fischer OM, Giordano S, Comoglio PM, Ullrich A. Reactive oxygen species mediate Met receptor transactivation by G protein-coupled receptors and the epidermal growth factor receptor in human carcinoma cells. J Biol Chem. 2004;279:28970–8. doi: 10.1074/jbc.M402508200. [DOI] [PubMed] [Google Scholar]
- 23.Aslan M, Ozben T. Oxidants in receptor tyrosine kinase signal transduction pathways. Antioxid Redox Signal. 2003;5:781–8. doi: 10.1089/152308603770380089. [DOI] [PubMed] [Google Scholar]
- 24.Frank GD, Eguchi S. Activation of tyrosine kinases by reactive oxygen species in vascular smooth muscle cells: significance and involvement of EGF receptor transactivation by angiotensin II. Antioxid Redox Signal. 2003;5:771–80. doi: 10.1089/152308603770380070. [DOI] [PubMed] [Google Scholar]
- 25.Luttrell DK, Luttrell LM. Not so strange bedfellows: G-protein-coupled receptors and Src family kinases. Oncogene. 2004;23:7969–78. doi: 10.1038/sj.onc.1208162. [DOI] [PubMed] [Google Scholar]
- 26.Lezama R, Diaz-Tellez A, Ramos-Mandujano G, Oropeza L, Pasantes-Morales H. Epidermal growth factor receptor is a common element in the signaling pathways activated by cell volume changes in isosmotic, hyposmotic or hyperosmotic conditions. Neurochem Res. 2005;30:1589–97. doi: 10.1007/s11064-005-8837-5. [DOI] [PubMed] [Google Scholar]
- 27.Mossman BT, Lounsbury KM, Reddy SP. Oxidants and signaling by mitogen-activated protein kinases in lung epithelium. Am J Respir Cell Mol Biol. 2006;34:666–9. doi: 10.1165/rcmb.2006-0047SF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saito Y, Haendeler J, Hojo Y, Yamamoto K, Berk BC. Receptor heterodimerization: essential mechanism for platelet-derived growth factor-induced epidermal growth factor receptor transactivation. Mol Cell Biol. 2001;21:6387–94. doi: 10.1128/MCB.21.19.6387-6394.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005;65:11118–28. doi: 10.1158/0008-5472.CAN-04-3841. [DOI] [PubMed] [Google Scholar]
- 30.Morgillo F, Woo JK, Kim ES, Hong WK, Lee HY. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res. 2006;66:10100–11. doi: 10.1158/0008-5472.CAN-06-1684. [DOI] [PubMed] [Google Scholar]
- 31.Johnson GR, Kannan B, Shoyab M, Stromberg K. Amphiregulin induces tyrosine phosphorylation of the epidermal growth factor receptor and p185erbB2. Evidence that amphiregulin acts exclusively through the epidermal growth factor receptor at the surface of human epithelial cells. J Biol Chem. 1993;268:2924–31. [PubMed] [Google Scholar]
- 32.Ridley AJ, Comoglio PM, Hall A. Regulation of scatter factor/hepatocyte growth factor responses by Ras, Rac, and Rho in MDCK cells. Mol Cell Biol. 1995;15:1110–22. doi: 10.1128/mcb.15.2.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imamura H, Takaishi K, Nakano K, Kodama A, Oishi H, Shiozaki H, Monden M, Sasaki T, Takai Y. Rho and Rab small G proteins coordinately reorganize stress fibers and focal adhesions in MDCK cells. Mol Biol Cell. 1998;9:2561–75. doi: 10.1091/mbc.9.9.2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang CC, Chen HC. Sustained activation of extracellular signal-regulated kinase stimulated by hepatocyte growth factor leads to integrin alpha 2 expression that is involved in cell scattering. J Biol Chem. 2001;276:21146–52. doi: 10.1074/jbc.M010669200. [DOI] [PubMed] [Google Scholar]
- 35.Singh AB, Tsukada T, Zent R, Harris RC. Membrane-associated HB-EGF modulates HGF-induced cellular responses in MDCK cells. J Cell Sci. 2004;117:1365–79. doi: 10.1242/jcs.01037. [DOI] [PubMed] [Google Scholar]
- 36.Scheving LA, Stevenson MC, Taylormoore JM, Traxler P, Russell WE. Integral role of the EGF receptor in HGF-mediated hepatocyte proliferation. Biochem Biophys Res Commun. 2002;290:197–203. doi: 10.1006/bbrc.2001.6157. [DOI] [PubMed] [Google Scholar]
- 37.Grierson I, Heathcote L, Hiscott P, Hogg P, Briggs M, Hagan S. Hepatocyte growth factor/scatter factor in the eye. Prog Retin Eye Res. 2000;19:779–802. doi: 10.1016/s1350-9462(00)00015-x. [DOI] [PubMed] [Google Scholar]
- 38.Klenkler B, Sheardown H. Growth factors in the anterior segment: role in tissue maintenance, wound healing and ocular pathology. Exp Eye Res. 2004;79:677–88. doi: 10.1016/j.exer.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 39.Sebastian S, Settleman J, Reshkin SJ, Azzariti A, Bellizzi A, Paradiso A. The complexity of targeting EGFR signalling in cancer: from expression to turnover. Biochim Biophys Acta. 2006;1766:120–39. doi: 10.1016/j.bbcan.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 40.Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–16. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 41.Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]