

Abstract
Tularemia is caused by the Gram-negative facultative intracellular bacterium Francisella tularensis, which has been classified as a Category A Select Agent – a likely bioweapon. The high virulence of F. tularensis and the threat of engineered antibiotic resistant variants warrant the development of new therapies to combat this disease. We have characterized 14 anti-Francisella hybridoma antibodies derived from mice infected with F. tularensis live vaccine strain (LVS) for potential use as immunotherapy of tularemia. All 14 antibodies cross-reacted with virulent F. tularensis type A clinical isolates, eight bound to a purified preparation of LVS LPS, and six bound to five protein antigens, identified by proteome microarray analysis. An IgG2a antibody, reactive with the LPS preparation, conferred full protection when administered either systemically or intranasally to BALB/c mice post challenge with a lethal dose of intranasal LVS; three other antibodies prolonged survival. These anti-Francisella hybridoma antibodies could be converted to chimeric versions with mouse V regions and human C regions to serve as components of a recombinant polyclonal antibody for clinical testing as immunotherapy of tularemia. The current study is the first to employ proteome microarrays to identify the target antigens of anti-Francisella monoclonal antibodies and the first to demonstrate the systemic and intranasal efficacy of monoclonal antibodies for post exposure treatment of respiratory tularemia.
Keywords: Francisella tularensis, antibodies, lipopolysaccharide, proteome microarray, mice
1. Introduction
Francisella tularensis, a Gram negative facultative intracellular bacterium that causes tularemia, has been classified as a category A select agent – a likely biowepon, due to its high infectivity (10 inhaled CFU can cause respiratory disease in humans) and high mortality rate (up to 30% of cases for untreated respiratory tularemia) [1,2]. F. tularensis comprises four subspecies: tularensis (type A), holarctica (type B), novicida, and mediasiatica. F. tularensis tularensis (type A) and F. tularensis holarctica (type B) cause most cases of human disease; type A, found predominantly in North America, being the more virulent of the two [1,2]. An attenuated F. tularensis type B strain, designated live vaccine strain (LVS), is partially protective against pathogenic F. tularensis in humans [3] but is highly virulent in mice [4].
The genomes of two type A strains, Schu S4 and FSC198, two type B strains, OSU 18 and LVS, and the novicida strain U112 have been sequenced (BioHealthBase BioDefense Public Health Database, www.biohealthbase.org). Schu S4 was found to contain 1804 predicted coding sequences [5], most of which have been expressed as recombinant proteins and used to generate a proteome microarray chip. A smaller chip, containing 244 proteins most often identified by human and mouse anti-tularemia immune sera, has also been generated [6] The current study provides a first demonstration of the use of these chips to identify the target antigens of F. tularensis-specific monoclonal antibodies.
Tularemia is usually treated by i.v. then oral administration of antibiotics but infection is still associated with considerable morbidity and up to 2–3% mortality in treated patients [7]. (http://www.uos.harvard.edu/ehs/fs_tularemia.shtml). And LVS, the partially protective vaccine, is not currently licensed [8]. These considerations, combined with the threat of engineered multiple antibiotic-resistant strains for bioterrorism, suggest the need for additional strategies to combat tularemia. One such strategy could be antibody immunotherapy.
Antibodies, in the form of serum from immunized goats or horses, were used in the management of tularemia in the pre-antibiotic era [9]. Although some reports claimed that antibodies were ineffective and even harmful, others described a significant reduction in both morbidity and mortality, with the greatest benefit obtained when the antibodies were given within two weeks of the onset of symptoms [9]. Mice treated with human or mouse immune serum to LVS showed protection from infection with LVS [4,10–12] and mice treated with mouse immune serum to F. tularensis LPS showed protection from infection with LVS and delay in time of death after infection with the type A F. tularensis strain Schu S4 [13]. LVS-specific mouse serum and LVS-specific human IgG were also shown to partially protect mice against virulent type B strains [3,12]. Similarly, pre-treatment with the mouse IgG2a hybridoma antibody FB11, specific for F. tularensis LPS, was shown to partially protect mice and guinea pigs against subcutaneous (s.c.) infection with a virulent F. tularensis type B strain [14]. And recently, LVS-specific mouse serum was shown to confer protection against intranasal LVS infection even when given 24–48 h post-exposure [15].
These reports are encouraging, even though no immune serum or IgG protection of mice against type A F. tularensis strains has been shown [3]; because only a fraction of antibodies in immune sera and IgG preparations is likely to be specific for F. tularensis and an even smaller fraction is expected to comprise antibodies to protective antigens or of an optimal isotype. Single hybridoma antibodies, even of an optimal isotype, are also not expected to protect against type A F. tularensis strains because all antibody molecules would compete against the same epitope.
To further explore the possibility of using antibodies as therapy for tularemia, we have generated anti-Francisella hybridomas from LVS-infected mice, to identify antibodies that could constitute components of a therapeutic recombinant chimeric polyclonal antibody with mouse V regions and human C regions for clinical use. Unlike immune serum or IgG preparations, all antibodies in a recombinant polyclonal antibody preparation would be of the optimal isotype and specific for protective F. tularensis antigens. In the current study, we analyzed 14 anti-LVS hybridoma antibodies for their target antigens and for their efficacy against intranasal LVS infection in mice, and identified four antibodies with therapeutic or prophylactic potential.
2. Materials and Methods
2.1. Generation of hybridomas
All animal studies have been reviewed and approved by the Boston University Medical Center Institutional Animal Care and Use Committee. BALB/cJ and C57BL/6J female mice were obtained from the Jackson Laboratory, and 8–10 week old mice were infected with LVS by the intranasal (i.n.), intradermal (i.d.), or i.p. routes. Splenocytes were prepared from euthanized mice by lysis of erythrocytes with 0.83% NH4Cl, and used in polyethylene glycol-mediated fusions with Sp2/0-Ag14 mouse myeloma cells [16] as previously described for spheroplast fusions with mouse myeloma cells [17]. Hybridomas were obtained in 96-well tissue culture plates in IMDM (GIBCO) supplemented with 20% FBS, 10% Hybridoma Enhancing Supplement (Sigma-Aldrich) and HAT (13.9 μg/ml hypoxanthine, 6 μg/ml aminopterin, and 7.2 μg/ml thymidine), and cell supernatants were screened for binding to whole LVS bacteria by ELISA as described below. Cells from positive wells were subcloned in 96-well plates, and single clones recovered and gradually adapted to growth in serum free medium (SFM, HYQ® SFM4Mab™ - Utility, HyClone) supplemented with 2% FBS.
2.2. Cell Lines, monoclonal antibodies, and bacterial strains
Hybridoma cell lines generated in our laboratory were cultured in IMDM/10% FBS or SFM/2% FBS. Mouse hybridoma cell line CO17-1A [18], producing an IgG2a antibody specific for a glycoprotein on human colorectal cancer cells [19] was obtained from Dr. Dorothee Herlyn of the Wistar Institute (Philadelphia, PA) and cultured in IMDM/10% FBS. All cell cultures were maintained at 37°C in a humidified environment of 5% CO2/95% air. Protein G-purified mouse IgG2a monoclonal antibody FB11, specific for the O-antigen polysaccharide chain of F. tularensis LPS [20], was purchased from GeneTex® Inc. F. tularensis holarctica strain LVS was obtained from Jeannine Petersen (Centers for Disease Control and Prevention, Fort Collins, CO). It was certified to be type B (F. tularensis subspecies holarctica) based on the amplification of PCR products [21] with primer set pdpDh but not with primer sets hyp4, hypR, hyp6 or pdpDn (Madico, G. et al., unpublished data). To prepare LVS stocks, bacteria were grown on chocolate agar plates (REMEL, Inc.) at 37°C in a humidified environment of 100% air for 2.5 days and pools of single colonies were scraped and resuspended in PBS. Eight heat-inactivated (2 h 80°C) F. tularensis clinical isolates (strains KU29, KU37, KU40, KU42, KU44, KU47, KU49 and KU54) [22] were the gift of Michael Parmely and Rebecca Horvat (University of Kansas Medical Center, Kansas City, KS). To identify their subspecies, a sample of DNA from each strain was prepared and analyzed by PCR (Madico G. et al., unpublished data). The following species- or subspecies-specific primers were used: hyp4 (F. tularensis subsp. tularensis), pdpDh (F. tularensis subsp. holarctica), hypR (F. tularensis subsp. mediasiatica and F. tularensis subsp. tularensis), pdpDn (F. tularensis subsp. novicida), and hyp6 (F. tularensis subsp. tularensis and Francisella philomiragia). Strains KU29, KU37, KU40, KU42, KU44, KU47, KU49 and KU54 were designated type A (F. tularensis subsp. tularensis) based on the amplification of PCR products with primers hyp4, hypR, and hyp6 but not with primer pdpDh or pdpDn. Preparations from these eight strains were pooled to test reactivity of antibodies by ELISA.
2.3. Preparation of antibodies
Hybridoma cell supernatants were used as sources of antibodies. Supernatants from hybridoma cells grown in SFM supplemented with 2% FBS were concentrated 10–20 fold on Centricon® Plus-70 centrifugal filter devices (Millipore). Purified FB11 antibody was dialyzed against PBS then SFM to remove the sodium azide used as preservative by the manufacturer, then supplemented with 20% FBS.
2.4. Purification of LPS
LVS bacterial lawns were grown at 37°C in a humidified environment of 5% CO2/95% air for 48 h on 200 cysteine heart agar (CHA) plates supplemented with 5% defibrinated rabbit blood and LPS was extracted from the bacteria with hot phenol as described [23–25].
2.5. ELISA for antigen-binding, isotyping, and quantification of hybridoma antibodies
For antigen-binding ELISA, EIA/RIA High Binding polystyrene 96-well plates (Corning) were coated with 50 μl/well of 0.04 OD600/ml of bacteria in 50 mM sodium carbonate buffer pH 9.6 by leaving the plates uncovered to dry overnight in a laminar flow hood; or with 2 μg/ml of purified LPS preparation in the same buffer by 3 h incubation at 37°C followed by overnight incubation at 4°C. Plates coated with buffer only were used as nonspecific control in the assay. Coated plates were washed twice with PBS supplemented with 0.05% Tween 20 (PBST) and blocked for 1 h at room temperature with either PBST - 2% non-fat dry milk (2% MPBST) or PBST - 10% BSA. Antibody-containing hybridoma supernatants or purified antibodies in the respective blocking agent, or only blocking agent, were added to the blocked plates and the plates were incubated, with shaking at 160 rpm, for 1 h at room temperature. After 3 washes with PBST, 50 μl/well of blocking agent containing one or more of three peroxidase-conjugated antibodies (anti-mouse IgG, anti-mouse IgM, anti-mouse IgA, diluted 1/4,000) (Sigma-Aldrich) was added and the plates were incubated, with shaking, for 1 h at room temperature. After 4 washes with PBST, the ELISA was developed by addition of 60 μl/well of TMB substrate (Kirkegaard & Perry Labs) and 15 min incubation at room temperature in the dark. The reaction was stopped with 60 μl/well of 0.2 M H2SO4, and absorbance at 450 nm measured in a microplate reader. For isotyping, plates were coated with LVS bacteria as described above, and assayed with the Mouse MonoAb ID Kit (HRP) from Zymed Laboratories according to the manufacturer’s instructions. For Ig quantification, plates were coated with biotin-conjugated (incidental for the current assay) anti-mouse IgG (Fc specific) or anti-mouse IgM (μ chain specific) (Sigma-Aldrich) diluted 1/2,000 in 50 mM sodium carbonate buffer pH 9.6 at 4°C overnight. After blocking as described above, 3-fold serial dilutions of hybridoma supernatants or purified antibodies were added, and the reactions visualized using peroxidase-conjugated anti-mouse IgG or anti-mouse IgM secondary antibodies, as appropriate, as described above for the antigen-binding ELISA. Serial dilutions of known concentrations of mouse IgG or IgM (Sigma-Aldrich) were included and their OD values were plotted against their concentrations. The linear portions of the curves were re-plotted by the least squares method in Microsoft Excel® and the least squares equations used to calculate the Ig concentrations in the hybridoma samples.
2.6. Bacterial cell lysate
Two OD600 of LVS in PBS were centrifuged at 1,800 × g for 10 min and the bacterial pellet was resuspended in 400 μl of QIAGEN P1 buffer without RNase A (50 mM Tris-HCl pH 8.0, 10 mM EDTA). After 5 min at room temperature, 400 μl of lysis buffer [1% NP-40, 10 mM Tris-HCl pH 7.4, 0.14 M NaCl, and one Protease Inhibitor cocktail tablet (Roche Diagnostics, Indianapolis, IN) per 4 ml of buffer], was added and the mixture kept at room temperature for 1 h till clear.
2.7. Immunoblot analysis
SDS-PAGE of LVS lysate (prepared from 0.55 OD600 of LVS per gel), that has or has not been treated with Proteinase K (PK), or of purified LPS preparation (15 μg per gel) was performed under reducing conditions (5 min boiling in SDS sample buffer supplemented with 5% 2-mercaptoethanol) using precast preparative 4–15% polyacrylamide gradient gels (2-D/Prep) and Broad Range Prestained SDS-PAGE molecular weight standards from Bio-Rad Laboratories. After electrotransfer, nitrocellulose membranes were blocked with 10% MPBST and used directly or “aged” by storage at 4°C in minimal volumes of PBST supplemented with 1% BSA then re-blocked. Blocked membranes were cut into strips and each strip incubated for 2–3 h at room temperature with a different hybridoma antibody diluted in 10% MPBST. After three washes with PBST, the strips were incubated for 1–2 h at room temperature with alkaline phosphatase-conjugated anti-mouse IgG + IgM secondary antibodies (Promega for anti-IgG and SouthernBiotech for anti-IgM) diluted 1:1,000 and 1:1,200 respectively in 10% MPBST. The strips were washed 2X with PBST, 1X with TBST, and 1X with TBS, then developed with Western® Blue Stabilized Substrate for alkaline phosphatase (Promega).
2.8. Proteome microarray analysis
Proteome microarray chips consisting of 244 F. tularensis strain Schu S4 recombinant proteins were fabricated by PCR amplification of each open reading frame, in vivo recombination cloning, and in vitro transcription/translation and microarray chip printing onto nitrocellulose coated glass slides, as described previously [6,26] The arrays were rehydrated for 30 min in Protein Array Blocking Buffer (Whatman) containing Escherichia coli lysate at a final concentration of 30% and probed with concentrated hybridoma supernatants overnight at 4°C with constant agitation. The slides were then washed five times in TBST, and incubated in biotin-conjugated goat anti-mouse Ig diluted 1:200 in blocking buffer; the secondary antibodies were obtained from Jackson Immuno Research and were either anti-IgG subclass specific (G1, G2a, G2b or G3) or anti-IgM or a mixture of anti-IgG + anti-IgM, depending on the experiment. The slides were washed five times in TBST and bound antibodies were detected by incubation with streptavidin-conjugated PBXL-3 (Martek). The slides were then washed three times in TBST and three times in TBS followed by a final water wash. The slides were air dried under brief centrifugation and examined in a Perkin Elmer ScanArray Express HT microarray scanner. Fluorescence signal intensities were quantified using QuantArray software. All signal intensities were corrected for background.
2.9. LD50 determination and antibody efficacy studies in the mouse model
BALB/cJ female mice (5–6 wk old) obtained from Jackson Laboratories were used for in vivo efficacy evaluation of hybridoma antibodies against LVS infection. For intranasal (i.n.) inoculation, freshly grown LVS bacteria were diluted in PBS to the desired CFU/ml concentration based on OD600, using the formula 1 OD600 = 2×109 CFU/ml, then the actual CFU/ml determined, retrospectively, by plating serial dilutions of the LVS suspension on chocolate agar plates. Forty μl of LVS suspension were instilled drop-wise with a micropipette in the right nostril of each mouse under anesthesia with 60 mg/kg ketamine (Fort Dodge Animal Health) and 6 mg/kg xylazine (Beb Venue Laboratories) administered i.p. Three-fold serial dilutions of LVS were used for LD50 determination. The LD50 was calculated as described [27]. For body weight determination, mice were weighed daily on a battery-operated scale. For i.n. administration of antibody, 40 μl of antibody or mock control in SFM/20% FBS were instilled in the left nostril while the mouse was still under anesthesia. For i.p. administration of antibody or mock control, 0.4 ml injections were given 1 h after the i.n. LVS inoculation. Survival was monitored daily for 28 days and biweekly thereafter.
Data were analyzed for mean time to death of the mice that died, median survival, and statistical significance with GraphPad Prism Version 4.0 (GraphPad Software, San Diego, CA). A survival curve representing percent survival as a function of time was created for every group using the method of Kaplan and Meier, in which for each dead mouse the day of death is entered in the X column and “1” is entered in the Y column; and for each surviving mouse on the end day of the experiment the end day is entered in the X column and “0” is entered in the Y column. For statistical analysis, the log rank test was used to compare two or more Kaplan-Meier survival curves and the log rank test for trend was used to compare the linear trend among three or more groups. P values of less than 0.05 were considered statistically significant.
3. Results
3.1. Generation and isotyping of anti-LVS hybridomas
Hybridomas were generated from BALB/c and C57BL/6 mice following infection with LVS, by fusion of splenocytes with Sp2/0-Ag14 mouse myeloma cells, and cell supernatants were tested for reactivity to whole LVS bacteria and for Ig isotype by ELISA. Supernatants with OD values of at least 0.25 (5–6 times background) were considered positive for LVS binding. Twenty eight anti-LVS hybridoma antibodies were obtained, 26 from BALB/c and two from C57BL/6 mice. The BALB/c anti-LVS antibodies comprised 16 IgM and 10 IgG antibodies of the IgG1, IgG2a, IgG2b and IgG3 isotypes, and the two C57BL/6 anti-LVS antibodies were typed as IgM; all antibodies were found to contain κL chain (Fig. 1). One C57BL/6-derived hybridoma antibody (Ab19), which did not bind LVS, was recovered and used as negative control (Fig. 1). Fourteen of the 28 hybridomas were subcloned and their antibodies (bolded in Fig. 1) were further analyzed in the current study.
Fig. 1.
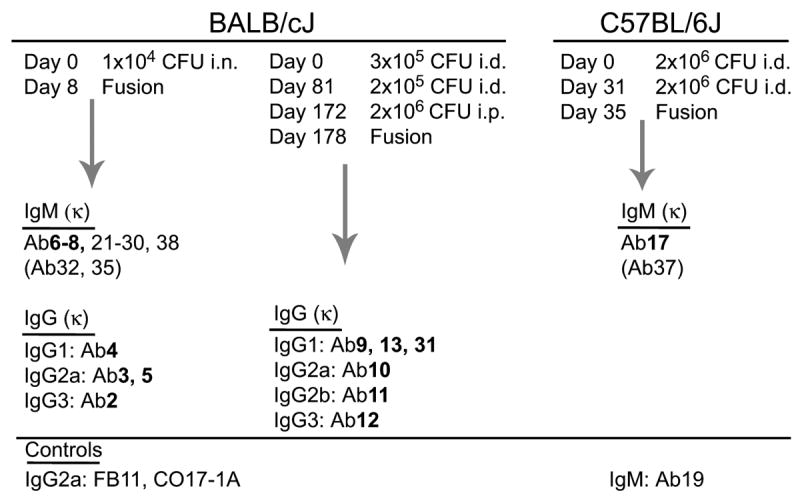
Derivation of anti-LVS hybridoma antibodies. Antibodies are grouped based on the mouse strain of origin (BALB/cJ or C57BL/6J), the immunization protocol and day of fusion, and the isotype (determined with an isotyping ELISA kit). Also listed are the non-LVS binding hybridoma antibody 19, used as negative control and IgM isotype control; the commercially available hybridoma antibody FB11, specific for the O-antigen polysaccharide chain of F. tularensis LPS, used as positive control; and the anti-human colorectal cancer hybridoma antibody CO17-1A, used as IgG isotype control. Antibodies further characterized in the current study are bolded.
3.2. Reactivity of anti-LVS hybridoma antibodies with Type A clinical isolates and with a purified preparation of LVS LPS
To determine if the 14 hybridoma antibodies cross-react with F. tularensis type A antigens, the antibodies were tested by ELISA, at 1 and 0.04 μg/ml concentrations, for binding to a mixture of eight heat-killed type A F. tularensis clinical isolates. Because these virulent type A strains were available only after 2 h heat-killing at 80°C, a preparation of LVS that had been heated for 2 h at 80°C was included in the experiment for comparison (Fig. 2 Left). In a separate experiment (using another batch of LVS) the same antibodies were also tested for binding to the TG1 strain of E. coli, another Gram-negative bacterium and for binding to a purified preparation of LVS LPS (Fig. 2 Right). The commercially available IgG2a hybridoma antibody FB11, specific for the O-antigen polysaccharide chain of F. tularensis LPS, served as positive control; our non-LVS-binding IgM hybridoma antibody Ab19 and the IgG2a hybridoma antibody CO17-1A, specific for a glycoprotein on human colorectal cancer cells, served as negative controls. The reactivity of all the 14 LVS-reactive hybridoma antibodies with the type A strains was similar to their reactivity with both live and heat-killed LVS, except that Ab11 showed very little if any reactivity on either heat-killed LVS or heat-killed type A strains (Fig. 2 Left), suggesting that its target epitope is heat-sensitive. None of the 14 hybridoma antibodies reacted with TG1 (Fig. 2 Right) or with uncoated wells (data not shown), but eight (Ab2–9) reacted with the purified preparation of LPS (Fig. 2 Right). To confirm the reactivity of Ab10–13, 17 and 31 with type A strains and their lack of reactivity with the purified LPS preparation, these antibodies were re-tested at 10 and 100 μg/ml concentrations. As shown in Fig. 3, all six antibodies, including Ab11, reacted with both heat-killed LVS and heat-killed type A strains; Ab10 showing especially high reactivity on heat-killed type A strains. None of the six antibodies reacted with the LPS preparation even at the 100 μg/ml concentration (Fig. 3), demonstrating that the relatively high concentrations do no cause non-specific binding. That the binding of some of the antibodies does not increase with increasing antibody concentration (Fig. 3) suggests a relatively low abundance of their target antigens on the bacteria. Other factors that may influence binding in these ELISAs include the reactivity of different Ig isotypes with the secondary (anti-IgM + anti-IgG) antibodies and the affinity of each hybridoma antibody for its target antigen.
Fig. 2.
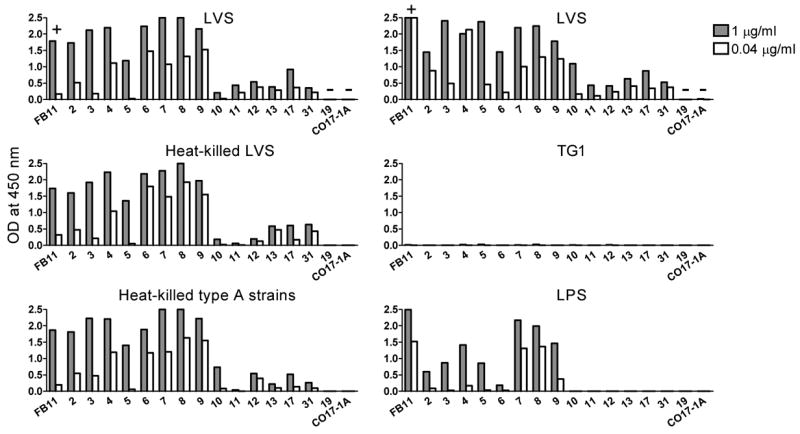
Reactivity of hybridoma antibodies with virulent F. tularensis type A clinical isolates and a purified LVS LPS preparation. Hybridoma supernatants containing 1 and 0.04 μg/ml of antibody were compared by ELISA for binding to LVS, heat-killed LVS, a mixture of eight heat-killed F. tularensis type A clinical isolates (Left), or for binding to LVS, equal OD of E. coli TG1 (or equal CFU which yielded the same results) and LPS. The names (numbers) of the antibodies are indicated. FB11, the positive control, is indicated by “+” and antibodies 19 and CO17-1A, the negative controls, are indicated by “−”. OD values that exceeded the capacity of the spectrophotometer (2.5) were recorded as 2.5. Background values (of wells that received no hybridoma supernatants, generally 0.04–0.05 OD) were subtracted from all other OD values.
Fig. 3.
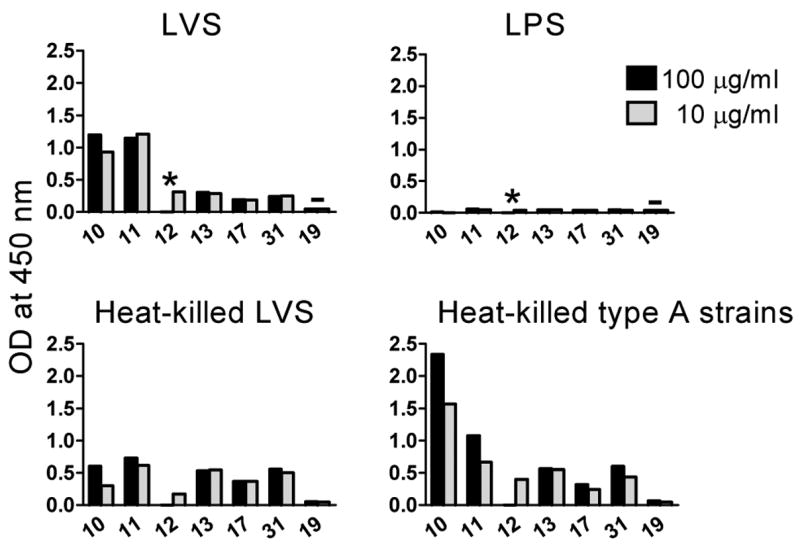
ELISA reactivity of high concentrations of non-LPS-binding hybridoma antibodies. Hybridoma supernatants containing 10 and 100 μg/ml of antibody were processed as described in the legend to Fig. 2.
3.3. Immunoblot reactivity of hybridoma antibodies
The eight hybridoma antibodies that reacted with LVS LPS in ELISA also bound to LVS lysate immunoblots, showing the characteristic LPS ladder pattern (Fig. 4A), which reflects the incremental sizes of LPS chains. Steps of the ladders identified by our hybridoma antibodies coincided with steps identified by the commercially available antibody FB11, specific for the O-antigen polysaccharide chain of F. tularensis LPS. However, all our hybridoma antibodies lacked the lower part of the FB11 ladder and two (Ab7 and 8) also showed strong reactivity (and always smearing) with higher molecular weight bands than those identified by FB11. Furthermore, FB11 bound to all steps of the ladder with similar intensity whereas our hybridoma antibodies showed a gradient of decreasing intensities from higher to lower molecular weight steps (Fig. 4A). On immunoblots of the purified LPS preparation, all antibodies, including FB11, identified higher molecular weight bands; but the lower molecular weight bands were absent from or barely visible in the ladders of Ab3–6 and 9 (Fig. 4B). Interestingly, the ladder identified by Ab2 on the LPS immunoblot matched more closely the ladder identified by FB11 (Fig. 4B). LPS immunoblot ladders comparable to ours were reported by others for anti-F. tularensis hybridoma antibodies [28–31]
Fig. 4.
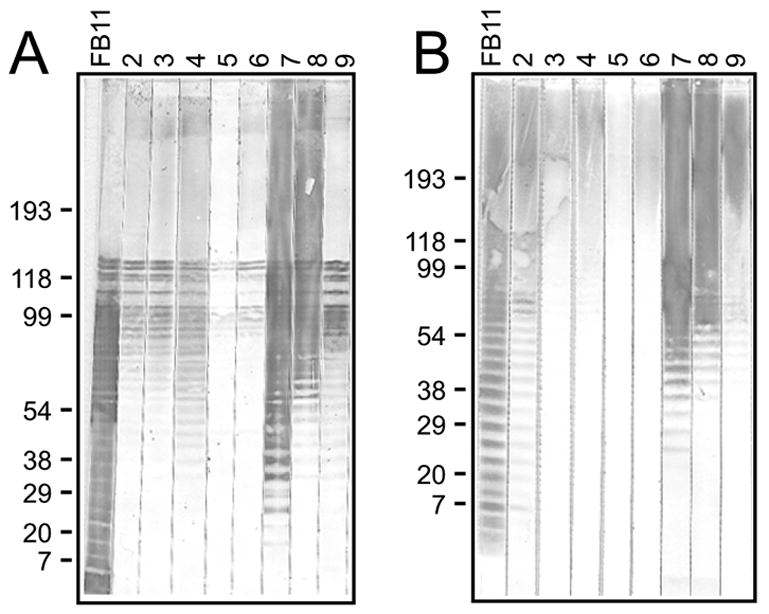
Immunoblot analysis of LPS-binding hybridoma antibodies. LVS lysate (A) or purified LPS preparation (B) was subjected to SDS-PAGE under reducing conditions and transferred to a nitrocellulose membrane. The membrane was cut into strips and each strip probed with the indicated hybridoma antibody at 2.5 μg/ml for Ab2–6, and 0.5 μg/ml for FB11 and Ab7–9. FB11, specific for the O-antigen polysaccharide chain of F. tularensis LPS was used as standard. The most clearly visible of several similar immunoblots are shown, and the estimated positions of molecular weight markers (in kDa) are indicated.
The remaining six hybridoma antibodies (10, 11, 12, 13, 17, and 31) showed a total of five different reactivity patterns on LVS lysate immunoblots (Fig. 5A); antibodies 13 and 31 identifying the same band. The reactivity of all six antibodies was abolished by pre-treatment of the LVS lysate with PK (Fig. 5B), suggesting that the target antigens of these antibodies are proteins. The reactivity of Ab7, which identified an LPS ladder by immunoblot (Fig. 4), was unaffected by the PK treatment of the LVS lysate (Fig. 5).
Fig. 5.
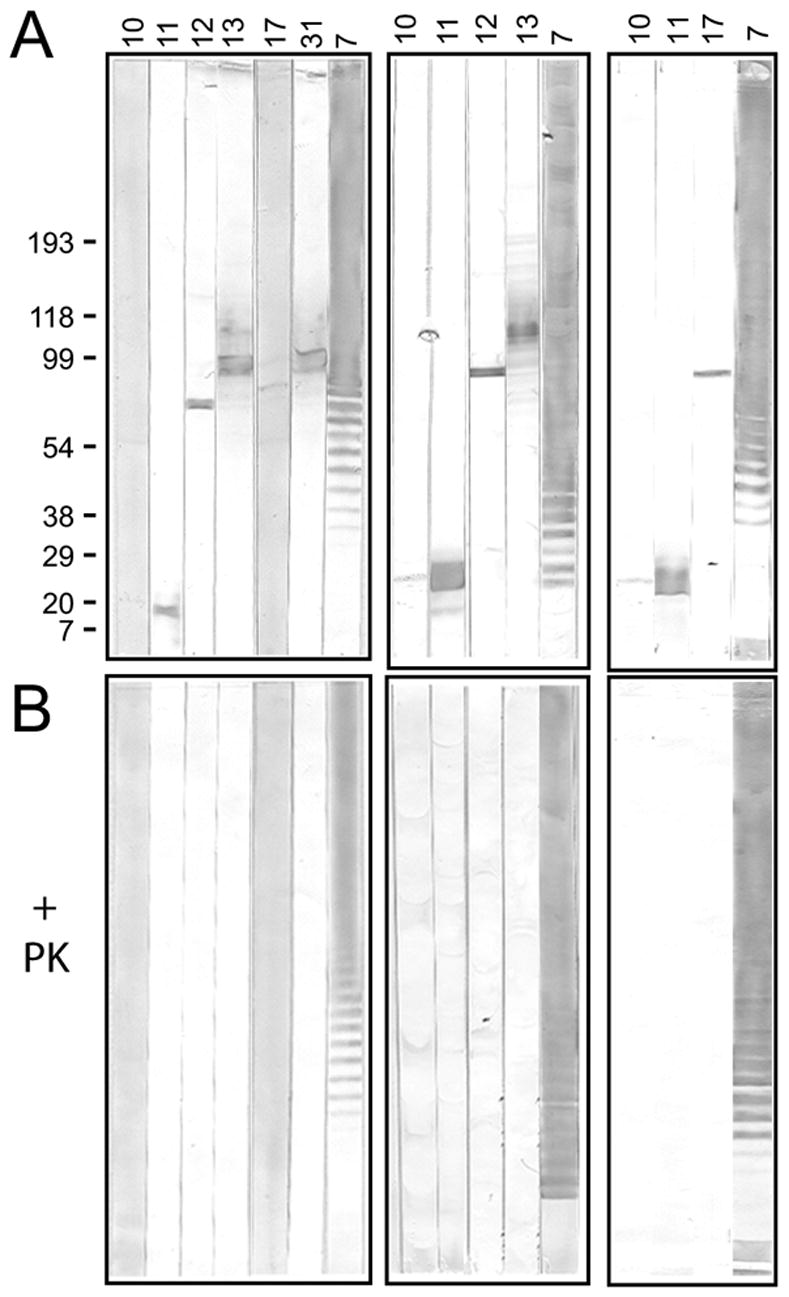
Proteinase K (PK) sensitivity of target antigens of anti-LVS hybridoma antibodies. One half of a sample of LVS lysate was treated with PK, then each the untreated and PK-treated samples were subjected to SDS-PAGE under reducing conditions and transferred to nitrocellulose membranes. The membranes were cut into strips and each strip probed with the indicated hybridoma antibody. Ab7, an LPS-binder, was used as negative control for PK-treatment. The untreated (A) and PK-treated (B) membrane strips for each antibody were developed simultaneously and for the same time. Immunoblots from three separate experiments are shown; the nitrocellulose membranes in the middle and right immunoblots were “aged” for two weeks prior to use to allow visualization of the bands identified by antibodies 10 and 17. The positions of molecular weight markers (in kDa) are indicated for the left immunoblot.
3.4. Proteome microarray identification of the target antigens of protein-specific hybridoma antibodies
To identify the putative protein antigens of the non-LPS-binding hybridoma antibodies, the antibodies were tested by proteome microarray analysis on a type A (strain Schu S4) chip, and the identified FTT (assigned Francisella tularensis tularensis) numbers and predicted functions (from BioHealthBase BioDefense Public Health Database http://www.biohealthbase.org and the PSORTdb Database http://db.psort.org) were noted. As shown in Fig. 6, identification of the target antigens of IgG antibodies 10, 11, 12, 13 and 31 was unequivocal, with both antibodies 13 and 31 recognizing FTT1269c, chaperone protein DnaK (heat shock protein family 70 protein) [32–39]. Antibody 10 recognized FTT0143, the 50S ribosomal protein L7/L12 [34,38,39]. Antibody 11 recognized FTT1778c, a hypothetical membrane protein. And antibody 12 recognized FTT1696, chaperone protein GroEL (heat shock protein family 60 protein) [32–40]. Identification of the target antigens of antibody 17, an IgM antibody, was more difficult. The antigen recognized by antibody 17 was determined to be FTT0233c, an inner membrane protein homologous to YidC of Escherichia coli, [41,42] by analysis of two separate chips (see Fig. 6).
Fig. 6.
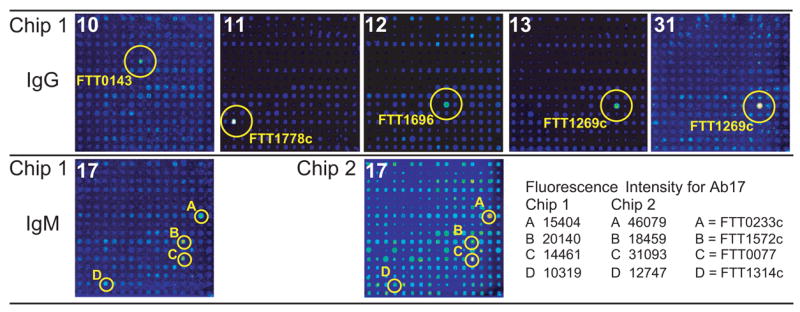
Identification of the target antigens of hybridoma antibodies by proteome microarray analysis. Separate pads of Chip 1 were probed with the indicated IgG or IgM hybridoma antibodies at antibody concentrations ranging from 9 to 141 μg/ml, followed by isotype-specific secondary antibodies. Antibody 17, at a concentration of 150 μg/ml, was re-tested on Chip 2. Microarray spots giving the highest fluorescence intensity signals on each pad are circled and the FTT numbers of the corresponding proteins are indicated. The fluorescence intensities of the circled spots are indicated for antibody 17.
Further validation of the antigens identified by proteome microarray analysis came from comparing their predicted molecular weights with the apparent molecular weights determined by immunoblot analysis, listed in Table 1 along with other properties of the proteins. The apparent molecular weights for all five target antigens were in the range of the predicted molecular weights.
Table 1.
Properties of protein-specific anti-F. tularensis hybridoma antibodies
| Ab No. | Apparent m.w.a (kDa) | Schu S4 FTTb | Predicted m.w.c (kDa) | Predicted function |
|---|---|---|---|---|
| 10 | <7 | 0143 | 12.8 | 50S ribosomal protein L7/L12 |
| 11 | <7 | 1778c | 13.7 | Hypothetical membrane protein |
| 12 | 59 | 1696 | 57.4 | Chaperone protein, GroEL |
| 13 | 83 | 1269c | 69.3 | Chaperone protein, DnaK |
| 17 | 61 | 0233c | 61.9 | Inner membrane protein, YidC |
| 31 | 83 | 1269c | 69.3 | Chaperone protein, DnaK |
Apparent molecular weights were determined from immunoblots (Fig. 5A) by comparison to molecular weight markers
FTT numbers and predicted functions were obtained from BioHealthBase BioDefense Public Health Database http://www.biohealthbase.org and the PSORTdb Database http://db.psort.org
Predicted molecular weights were calculated based on amino acid sequence by an online Protein Calculator tool http://www.scripps.edu/cgi-bin/cdputnam/protcalc3
3.5. Efficacy of hybridoma antibodies against intranasal LVS infection of mice
To determine if single or multiple hybridoma antibodies can confer protection against F. tularensis, we used i.n. LVS infection as an inhalational mouse model of tularemia. Intranasal infection with F. tularensis LVS and Schu S4 has been used by others as an inhalational model in lieu of aerosol exposure [4,15,43–46] and has been found to yield similar results with regard to LD50 and time to death [44]. In this model, the bacteria enter the lungs, as in aerosol exposure, but the intranasal route is advantageous from a safety and ease of use standpoint [44]. Infection spreads from the lungs to other organs including the liver and spleen [44]. To establish the i.n. mouse model in our laboratory, we inoculated groups of 10 five-week-old BALB/c mice, i.n., with three-fold serial dilutions of LVS and determined the number of survivors for each group (Table 2). The i.n. LD50 for LVS was determined to be 5 × 103 CFU, with LD100 between 1×104 and 2.9×104 CFU (Table 2). The higher LVS doses correlated with more severe disease, as assessed qualitatively by fur ruffling and inactivity (data not shown), and quantitatively, through daily weight measurements (Fig. 7). The lone survivor at 1×104 CFU did not recover up to day 28 when the experiment was terminated.
Table 2.
LD50 determination for intranasnal LVS inoculation
| LVS inoculum (CFU)a | No. of surviving mice/total | Mean time to death ± SD (days) | Median survival (days) |
|---|---|---|---|
| 3.6×102 | 10/10 | Undefinedb | Undefined |
| 1.0×103 | 10/10 | Undefined | Undefiend |
| 3.2×104 | 8/10 | Undefined | Undefiend |
| 1.0×104 | 1/10 | 9.2 ± 4.3 | 9 |
| 2.9×104 | 0/10 | 10.3 ± 1.3 | 10 |
| 8.7×104 | 0/10 | 8.5 ± 1.4 | 8 |
| 2.6×105 | 0/10 | 7.9 ± 0.7 | 8 |
Mice were inoculated i.n. with the indicated number of LVS CFU
Survival exceeds 50% at the longest time point
Fig. 7.
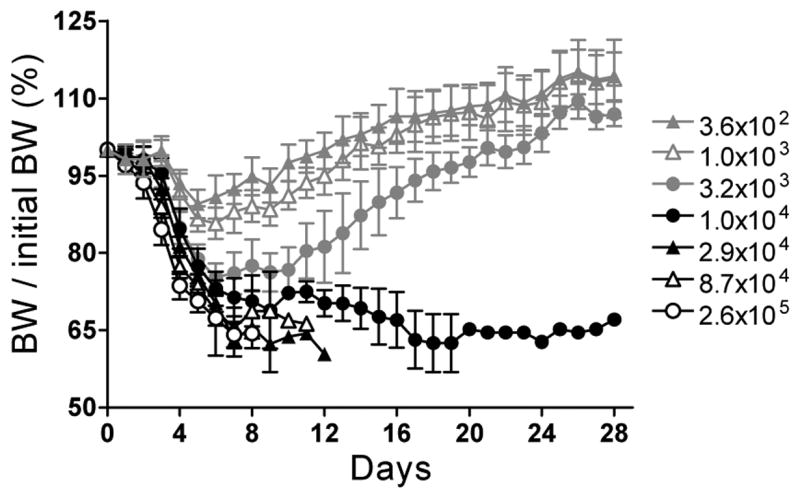
Changes in body weight (BW) of mice inoculated i.n with LVS. Groups of 10 five-week-old BALB/cJ mice were exposed to the indicated number of LVS CFU and weighed daily. The daily BW of each mouse was divided by its initial (pre-LVS exposure) BW and the mean BW ratio (in %) ± SD was plotted for each group as a function of time.
For initial efficacy testing of single hybridoma antibodies, we chose 2×104 CFU as a sensitive dose, which will likely result in 100% mortality for control mice but may allow survival of mice treated with potentially efficacious antibodies. However, as described in Materials and Methods, we used fresh LVS preparations diluted to the intended dose based on OD600 with subsequent determination of actual CFU, and therefore the LVS dose varied between 2×104 and 4×104 CFU in these experiments. In the first experiment we tested four of the LPS-binding antibodies of different isotypes. BALB/c mice were inoculated i.n. with 4×104 CFU of LVS, then 1h later injected i.p. with 100 μg of LPS-binding or isotype control antibody or PBS, and monitored daily for survival. The LPS-binding antibodies tested included one IgG1 (Ab9), one IgG2a (Ab3), one IgG3 (Ab2) and one IgM antibody (Ab7); the latter was tested in both the 100 μg and a 600 μg dose (6X). All mice in the control groups died by day 8 post infection (Table 3 Experiment 1). In contrast, the IgG2a antibody treatment prolonged survival by 2–7 days for three of the five mice and led to complete recovery with long term survival for the other two mice compared to PBS or CO17-1A isotype control. The IgG1 antibody prolonged survival up to day 18 post infection, with mean time to death of 13.8 days but did not lead to recovery for any of the mice (Table 3 Experiment 1). The IgG3 antibody prolonged survival by one and a half days but the IgM antibody had no effect on survival, even when used at the 600 μg dose (Table 3 Experiment 1). The relative efficacy of the LPS-binding IgG antibodies was IgG2a > IgG1 > IgG3 (p = 0.018 by the log rank test for trend).
Table 3.
Efficacy of hybridoma antibodies against intranasal LVS infection of BALB/c mice
| LVS inoculation/Ab treatmenta | No. of surviving mice/total | Mean time to death ± SD (days) | Median survival (days) | p value of log rank test |
|---|---|---|---|---|
| Experiment 1 | ||||
| 4×104 CFU i.n./100 μg i.p. | versus isotype control | |||
| 3 (G2a) | 2/5 | 13.7 ± 2.5 | 16 | 0.003 |
| 9 (G1) | 0/5 | 13.8 ± 4.7 | 16 | 0.033 |
| 2 (G3) | 0/5 | 9.4 ± 1.5 | 9 | 0.011 |
| 7 (M) | 0/5 | 8.2 ± 1.3 | 8 | 0.333 |
| 7 (M)/600 μg | 0/5 | 7.6 ± 0.9 | 7 | 0.918 |
| CO17-1A (G control) | 0/5 | 7.6 ± 0.5 | 8 | NAb |
| 19 (M control) | 0/5 | 7.6 ± 0.5 | 8 | NA |
| PBS | 0/5 | 8.0 ± 0.0 | 8 | NA |
| Experiment 2 | ||||
| 2×104 CFU i.n./200 μg i.p. | versus Sp2/0 | |||
| FB11 (G2a) | 5/5 | NA | undefinedc | 0.003 |
| 3 (G2a) | 5/5 | NA | undefined | 0.003 |
| 10 (G2a) | 0/5 | 7.4 ± 1.5 | 7 | 0.133d |
| 3+10 (G2a) | 5/5 | NA | undefined | 0.003 |
| 11 (G2b) | 0/5 | 10.6 ± 2.7 | 11 | 0.192 |
| 13+31 (G1) | 0/5 | 7.4 ± 1.5 | 7 | 0.044d |
| 12 (G3) | 1/5 | 12.0 ± 6.6 | 13 | 0.206 |
| 10+11+13+31 (G) | 0/5 | 10.2 ± 3.1 | 10 | 0.422 |
| 3+10+11+13+31 (G) | 5/5 | NA | undefined | 0.003 |
| Sp2/0 (fusion partner) | 0/5 | 9.2 ± 1.3 | 10 | NA |
| Experiment 3A | ||||
| 2×104 CFU i.n./50 μg i.n. | versus isotype control | |||
| 3 (G2a) | 5/5 | NA | undefined | 0.001 |
| CO17-1A (G2a) | 0/5 | 15.6 ± 5.9 | 17 | NA |
| Experiment 3B | ||||
| 2×105 CFU i.n./50 μg i.n. | versus isotype control | |||
| 3 (G2a) | 3/5 | 16.0 ± 0.0 | undefined | 0.003 |
| CO17-1A (G2a) | 0/5 | 7.8 ± 1.6 | 9 | NA |
Mice were inoculated i.n. with the indicated number of LVS CFU, then treated i.p. (1 h later) or i.n. (within minutes) with supernatants containing the indicated Ab and dose or with PBS or Sp2/0 supernatant
Not applicable
Survival exceeds 50% at the longest time point
The p values are different for Ab10 and Ab13+Ab31 because of differences in the day of death of individual mice in these two groups compared to the day of death in the Sp2/0 control group
In a second experiment, the IgG2a LPS-binding Ab3 was re-tested at 200 μg per mouse i.p. 1 h post infection with 2×104 CFU of LVS along with other hybridoma antibodies (Table 3 Experiment 2). The other hybridoma antibodies included the commercial IgG2a anti-LPS antibody FB11 and the protein-specific anti-LVS IgG antibodies 10, 11, 12, 13, and 31. Of the protein-specific antibodies, only the IgG3 Ab12, specific for the chaperone protein GroEL, conferred partial protection, with long term survival for one mouse and prolongation of survival by up to 12 days for the other mice, with a mean time to death of 12 days. However, this effect was not statistically significant compared to the group that received supernatant of the Sp2/0-Ag14 cell line used as fusion partner due to the large variation in day of death for the Ab12-treated mice that died (Table 3 Experiment 2). Both antibodies 3 and FB11 conferred full protection, with 100% of mice showing long-term survival. Thus, of our hybridoma antibodies, Ab3 was most efficacious, conferring full protection even when administered at 40 μg per mouse in a mixture with four other antibodies (10, 11, 13, and 31) that were otherwise ineffective either alone or as a group (Table 3 Experiment 2).
Full protection by Ab3 was also achieved at 50 μg per mouse administered i.n. within minutes of i.n. infection with 2×104 CFU of LVS (Table 3 Experiment 3A). Intranasal Ab3 conferred only partial protection against 2×105 CFU of LVS, with prolongation of survival for all mice, long-term survival for 60% of mice, and a mean time to death of 16 days compared to 7.8 days for the isotype control antibody CO17-1A (Table 3 Experiment 3B).
4. Discussion
We have described the derivation and analysis of 14 anti-LVS hybridoma antibodies, all of which cross-react with virulent F. tularensis type A strains. This extensive cross-reactivity is not surprising in view of the high genomic sequence and LPS similarity between F. tularensis type A and type B subspecies (BioHealthBase BioDefense Public Health Database, www.biohealthbase.org) [2,47]. Eight of the antibodies bound to a purified preparation of LVS LPS and showed LPS ladder patterns on immunoblots. These ladder patterns overlapped only partially with that of the commercial hybridoma antibody FB11, specific for the O-antigen polysaccharide chain of F. tularensis LPS, suggesting that our hybridoma antibodies may bind to different LPS-associated epitopes.
The LPS-binding IgG2a hybridoma Ab3 conferred full protection against i.n. LVS infection, when administered post challenge, both i.p. at 200 μg per mouse and i.n. at 50 μg per mouse, suggesting that either i.v. or i.n. administration of antibodies might be feasible in the clinical setting. The latter would be more easily administered in the field, with no need for medical personnel, and would require lesser doses because the therapeutic/prophylactic agent would be delivered directly to the lung, the site of bacterial entry, in case of a bioterrorist attack with aerosolized Francisella. Furthermore, the commercial IgG2a hybridoma antibody FB11, which likely binds to another epitope than Ab3 based on the immunoblot reactivity pattern, also conferred full protection at 200 μg per mouse i.p., suggesting that IgG2a is an efficacious isotype, at least among LPS-binding antibodies. The IgG1 Ab9 and, to a lesser extent, the IgG3 Ab2 LPS-binding antibodies tested i.p. at the 100 μg dose showed some efficacy, prolonging survival by 6 and 1.5 days respectively; the IgM LPS-binding Ab7 (at either 100 or 600 μg) had no efficacy against LVS infection. The lack of efficacy of the IgM antibody agrees with a previous report that IgG, but not IgM, anti-LVS serum antibodies were responsible for transfer of protection against LVS to naïve recipient mice [11]. Furthermore, the superior therapeutic efficacy of IgG2a antibodies has been previously shown. Mouse IgG2a, like IgG1 in humans, is the most effective isotype at binding to Fc receptors on effector cells [48–50], leading to elimination of microbes through opsonophagocytosis [51]. And recently, mouse anti-Francisella antibody responses in which the IgG2a subclass predominates have been correlated with protection against F. tularensis and with an IL-12/IFN-γ Th1 response [6]. Consistent with these results, immune serum-mediated protection of mice against lethal intranasal LVS infection has been shown to depend on the presence of FcγR on phagocytes [15]. Despite these arguments, the relative efficacy of our hybridoma antibodies may reflect both isotype and antigen-binding specificity and/or affinity, as none of the LPS-binding antibodies tested in the mouse model were identical in both VH and VL region nucleotide sequence (data not shown).
The post-exposure efficacy of LPS-binding antibodies against intranasal LVS infection of mice extends previous findings in which pre-treatment with anti-LPS immune serum [13] or the hybridoma antibody FB11 [14] protected mice against i.p. or s.c. infection with F. tularensis live vaccine strains. It is also consistent with the high density surface exposure of LPS, the main constituent of the outer membrane of Gram-negative bacteria [52].
The six non-LPS-binding hybridoma antibodies were identified as protein-binders based on their Proteinase K-sensitive reactivity on LVS lysate immunoblots, and their five target antigens were identified by proteome microarray analysis. This study provides a first example of the use proteome microarrays to identify the target antigens of anti-Francisella monoclonal antibodies. This approach could be used not only to select monoclonal antibodies of potential interest in therapeutic or diagnostic applications, but also to identify potential vaccine candidates or to determine the expression and function of microbial components. Four of the five target proteins of our hybridoma antibodies (the hypothetical membrane protein FTT1778c, the chaperone heat shock proteins GroEL and DnaK, and the inner membrane protein YidC) have been recently identified, using the same proteome microarray chip, to be among 48 immunodominant antigens in LVS immunized mice [6]. The chaperone, heat shock proteins GroEL and DnaK (the target antigens of Ab12 and of Ab13 and Ab31 respectively) were previously identified as potentially surface-exposed LVS proteins by mass spectrometry [37,39] and were found in culture filtrates of LVS and of a fresh clinical isolate [36]. GroEL and DnaK were identified as immunoreactive proteins both in sera of mice immunized with LVS [34] and in sera from tularemia patients [38,39]. GroEL was also reported to stimulate IFN-γ secreting CD4+ and CD8+ T cells in LVS-infected mice [36] and, along with DnaK and the heat shock protein Cpn-10, to be a target of human alpha-beta T cells [33]. However, one study [40] concluded that the protection of mice against F. tularensis type B strains by i.d. immunization with GroEL may be due to copurified LPS.
It is noteworthy that of our five IgG hybridoma antibodies (the IgM antibody was not tested), only Ab12, specific for the chaperone protein GroEL, partially protected BALB/c mice against i.n. LVS infection although the effect was not statistically significant. Because Ab12 is of the IgG3 isotype, like the least effective LPS-binding Ab2, we expect that recombinant DNA conversion of Ab12 into an IgG2a version [53,54] will increase its efficacy. Of the other anti-LVS IgG hybridoma antibodies, the DnaK-specific Ab13 and Ab31 (both IgG1) were ineffective against LVS even though the LPS-binding IgG1 Ab9 showed some efficacy. Similarly, the IgG2a Ab10, specific for the 50S ribosomal protein L7/L12 showed no protection, consistent with the expected cytoplasmic location of the target antigen, even though L7/L12 was previously described as potentially surface-exposed and as an immunoreactive protein in sera from mice immunized with LVS or F. tularensis novicida [34], and in sera from tularemia patients [38,39]. The induction of antibodies with no protective function during infection underscores the advantage of the hybridoma and recombinant DNA technologies over the use of serum antibodies for immunotherapy, as the former allow for selection of antibodies of only the desired specificities, in addition to their conversion into antibodies with C regions of desired isotypes and species [53,54].
Combining IgG2a-converted efficacious monoclonal antibodies from the ones described in the current study and from monoclonal antibodies yet to be discovered, may allow the assembly of a sufficiently potent recombinant polyclonal antibody to protect mice, and eventually humans, even against type A F. tularnensis infection. A recombinant polyclonal antibody would target multiple bacterial epitopes, which would enhance effector functions and decrease the likelihood of emergence of antigen-escape variants, as we previously suggested [54,55]. Such recombinant polyclonal antibodies are already being produced for clinical therapeutic use [56], and the first such product has recently entered Phase I clinical trials (for treatment of hemolytic disease of the newborn and/or idiopathic thrombocytopenic purpura) (http://www.symphogen.com).
For clinical therapeutic applications for tularemia, we envision the use of recombinant chimeric polyclonal antibodies with mouse V regions and human C regions of the optimal isotype, rather than all-human antibodies, because of the relatively low immunogenicity of chimeric antibodies [57] and the ease of generating hybridoma antibodies from mice optimally immunized with whole microbes or microbial components. Whether chimeric or all-human, the results of the current study support an important role for recombinant polyclonal antibodies in the armamentarium against intracellular pathogens in general and F. tularensis in particular.
Acknowledgments
We thank Nancy Nowak for excellent technical assistance with LPS purification; Damiana Chiavolini for instruction on intranasal inoculation of mice; Louis Vaickus for helping develop the immunoglobulin quantification assay; Jeannine Petersen for LVS; Michael Parmely and Rebecca Horvat for the F. tularensis clinical isolates; Dorothee Herlyn for the CO17-1A cell line; Mercio Perrin and members of the Boston University Tularemia Group including John Murphy, Damiana Chiavolini, and Frank C. Gibson III for discussion; Robina Folland and Richard P. Trevino, MPH, for administrative support; and Konstantin Kandror for Russian-English translation of two references. J. Sharon has a significant financial interest in Symphogen A/S, a company dedicated to the production of recombinant polyclonal antibodies for clinical use. Philip Felgner is a founder of ImmPORT Therapeutics and chairman of its scientific board of directors. This work was supported by Grants U19 AI 56543 and U01 AI 56464 from the National Institute of Allergy and Infectious Diseases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sjostedt A. Virulence determinants and protective antigens of Francisella tularensis. Curr Opin Microbiol. 2003;6:66–71. doi: 10.1016/s1369-5274(03)00002-x. [DOI] [PubMed] [Google Scholar]
- 2.McLendon MK, Apicella MA, Allen LA. Francisella tularensis: taxonomy, genetics, and Immunopathogenesis of a potential agent of biowarfare. Annu Rev Microbiol. 2006;60:167–185. doi: 10.1146/annurev.micro.60.080805.142126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffin KF, Oyston PC, Titball RW. Francisella tularensis vaccines. FEMS Immunol Med Microbiol. 2007;49:315–323. doi: 10.1111/j.1574-695X.2007.00219.x. [DOI] [PubMed] [Google Scholar]
- 4.Fortier AH, Slayter MV, Ziemba R, Meltzer MS, Nacy CA. Live vaccine strain of Francisella tularensis: infection and immunity in mice. Infect Immun. 1991;59:2922–2928. doi: 10.1128/iai.59.9.2922-2928.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Larsson P, Oyston PC, Chain P, Chu MC, Duffield M, Fuxelius HH, Garcia E, Halltorp G, Johansson D, Isherwood KE, Karp PD, Larsson E, Liu Y, Michell S, Prior J, Prior R, Malfatti S, Sjostedt A, Svensson K, Thompson N, Vergez L, Wagg JK, Wren BW, Lindler LE, Andersson SG, Forsman M, Titball RW. The complete genome sequence of Francisella tularensis, the causative agent of tularemia. Nat Genet. 2005;37:153–159. doi: 10.1038/ng1499. [DOI] [PubMed] [Google Scholar]
- 6.Eyles JE, Unal B, Hartley MG, Newstead SL, Flick-Smith H, Prior JL, Oyston PC, Randall A, Mu Y, Hirst S, Molina DM, Davies DH, Milne T, Griffin KF, Baldi P, Titball RW, Felgner PL. Immunodominant Francisella tularensis antigens identified using proteome microarray. Proteomics. 2007;7:2172–2183. doi: 10.1002/pmic.200600985. [DOI] [PubMed] [Google Scholar]
- 7.Dennis DT, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Friedlander AM, Hauer J, Layton M, Lillibridge SR, McDade JE, Osterholm MT, O’Toole T, Parker G, Perl TM, Russell PK, Tonat K. Tularemia as a biological weapon: medical and public health management. Jama. 2001;285:2763–2773. doi: 10.1001/jama.285.21.2763. [DOI] [PubMed] [Google Scholar]
- 8.Oyston PC, Sjostedt A, Titball RW. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat Rev Microbiol. 2004;2:967–978. doi: 10.1038/nrmicro1045. [DOI] [PubMed] [Google Scholar]
- 9.Foshay L. Tularemia: A summary of certain aspects of disease including methods for early diagnosis and the results of serum treatment in 600 patients. Medicine. 1940;19:1–83. [Google Scholar]
- 10.Drabick JJ, Narayanan RB, Williams JC, Leduc JW, Nacy CA. Passive protection of mice against lethal Francisella tularensis (live tularemia vaccine strain) infection by the sera of human recipients of the live tularemia vaccine. Am J Med Sci. 1994;308:83–87. doi: 10.1097/00000441-199408000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Rhinehart-Jones TR, Fortier AH, Elkins KL. Transfer of immunity against lethal murine Francisella infection by specific antibody depends on host gamma interferon and T cells. Infect Immun. 1994;62:3129–3137. doi: 10.1128/iai.62.8.3129-3137.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stenmark S, Lindgren H, Tarnvik A, Sjostedt A. Specific antibodies contribute to the host protection against strains of Francisella tularensis subspecies holarctica. Microb Pathog. 2003;35:73–80. doi: 10.1016/s0882-4010(03)00095-0. [DOI] [PubMed] [Google Scholar]
- 13.Fulop M, Mastroeni P, Green M, Titball RW. Role of antibody to lipopolysaccharide in protection against low- and high-virulence strains of Francisella tularensis. Vaccine. 2001;19:4465–4472. doi: 10.1016/s0264-410x(01)00189-x. [DOI] [PubMed] [Google Scholar]
- 14.Khlebnikov VS, Vetchinin SS, Grechko GK, Averina AA, Golovlev IR, Averin SF, Zhemchugov VE, Konovalov SI, Anisimov GA, Afanas’ev SS. The preventive activity of monoclonal antibodies specific to the lipopolysaccharide of Francisella tularensis. Zh Mikrobiol Epidemiol Immunobiol. 1992:67–70. [PubMed] [Google Scholar]
- 15.Kirimanjeswara GS, Golden JM, Bakshi CS, Metzger DW. Prophylactic and Therapeutic Use of Antibodies for Protection against Respiratory Infection with Francisella tularensis. J Immunol. 2007;179:532–539. doi: 10.4049/jimmunol.179.1.532. [DOI] [PubMed] [Google Scholar]
- 16.Shulman M, Wilde CD, Kohler G. A better cell line for making hybridomas secreting specific antibodies. Nature. 1978;276:269–270. doi: 10.1038/276269a0. [DOI] [PubMed] [Google Scholar]
- 17.Sharon J, Gefter ML, Manser T, Ptashne M. Site-directed mutagenesis of an invariant amino acid residue at the variable-diversity segments junction of an antibody. Proc Natl Acad Sci U S A. 1986;83:2628–2631. doi: 10.1073/pnas.83.8.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herlyn DM, Steplewski Z, Herlyn MF, Koprowski H. Inhibition of growth of colorectal carcinoma in nude mice by monoclonal antibody. Cancer Res. 1980;40:717–721. [PubMed] [Google Scholar]
- 19.Schlimok G, Gottlinger H, Funke I, Swierkot S, Hauser H, Riethmuller G. In vivo and in vitro labelling of epithelial tumor cells with anti 17-1A monoclonal antibodies in bone marrow of cancer patients. Hybridoma. 1986;5(Suppl 1):S163–170. [PubMed] [Google Scholar]
- 20.Khlebnikov VS, Golovlev IR, Tokhtamysheva NV, Averin SF, Kulevatskii DP, Grechko GK, Averina AA, Vetchinin SS. The determination of the antigenic determinant of protective monoclonal antibodies specific to the Francisella tularensis lipopolysaccharide] Zh Mikrobiol Epidemiol Immunobiol. 1993:83–88. [PubMed] [Google Scholar]
- 21.Kugeler KJ, Pappert R, Zhou Y, Petersen JM. Real-time PCR for Francisella tularensis types A and B. Emerg Infect Dis. 2006;12:1799–1801. doi: 10.3201/eid1211.060629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wickstrum JR, Hong KJ, Bokhari S, Reed N, McWilliams N, Horvat RT, Parmely MJ. Coactivating signals for the hepatic lymphocyte gamma interferon response to Francisella tularensis. Infect Immun. 2007;75:1335–1342. doi: 10.1128/IAI.01203-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Westphal O, Luderitz O, Bister F. Uder die extraktion of bakterien mit wasser. Z Naturforsch. 1952;B7:148–155. [Google Scholar]
- 24.Hood AM. Virulence factors of Francisella tularensis. J Hyg (Lond) 1977;79:47–60. doi: 10.1017/s0022172400052840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phillips NJ, Schilling B, McLendon MK, Apicella MA, Gibson BW. Novel modification of lipid A of Francisella tularensis. Infect Immun. 2004;72:5340–5348. doi: 10.1128/IAI.72.9.5340-5348.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davies DH, Liang X, Hernandez JE, Randall A, Hirst S, Mu Y, Romero KM, Nguyen TT, Kalantari-Dehaghi M, Crotty S, Baldi P, Villarreal LP, Felgner PL. Profiling the humoral immune response to infection by using proteome microarrays: high-throughput vaccine and diagnostic antigen discovery. Proc Natl Acad Sci U S A. 2005;102:547–552. doi: 10.1073/pnas.0408782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weil CS. Tables for convenient calculation of median-effective dose (LD50 or ED50) and instruction in their use. Biometrics. 1952;8:249–263. [Google Scholar]
- 28.Fulop MJ, Webber T, Manchee RJ, Kelly DC. Production and characterization of monoclonal antibodies directed against the lipopolysaccharide of Francisella tularensis. J Clin Microbiol. 1991;29:1407–1412. doi: 10.1128/jcm.29.7.1407-1412.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gubbins MJ, Berry JD, Schmidt L, Cabral T, Kabani A, Tsang RS. Production and characterization of a monoclonal antibody to Francisella tularensis lipopolysaccharide. Hybridoma (Larchmt) 2007;26:98–103. doi: 10.1089/hyb.2006.049. [DOI] [PubMed] [Google Scholar]
- 30.Hotta A, Uda A, Fujita O, Tanabayashi K, Yamada A. Preparation of monoclonal antibodies for detection and identification of Francisella tularensis. Clin Vaccine Immunol. 2007;14:81–84. doi: 10.1128/CVI.00057-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Narayanan RB, Drabick JJ, Williams JC, Fortier AH, Meltzer MS, Sadoff JC, Bolt CR, Nacy CA. Immunotherapy of tularemia: characterization of a monoclonal antibody reactive with Francisella tularensis. J Leukoc Biol. 1993;53:112–116. doi: 10.1002/jlb.53.1.112. [DOI] [PubMed] [Google Scholar]
- 32.Ericsson M, Tarnvik A, Kuoppa K, Sandstrom G, Sjostedt A. Increased synthesis of DnaK, GroEL, and GroES homologs by Francisella tularensis LVS in response to heat and hydrogen peroxide. Infect Immun. 1994;62:178–183. doi: 10.1128/iai.62.1.178-183.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ericsson M, Kroca M, Johansson T, Sjostedt A, Tarnvik A. Long-lasting recall response of CD4+ and CD8+ alphabeta T cells, but not gammadelta T cells, to heat shock proteins of Francisella tularensis. Scand J Infect Dis. 2001;33:145–152. doi: 10.1080/003655401750065562. [DOI] [PubMed] [Google Scholar]
- 34.Havlasova J, Hernychova L, Brychta M, Hubalek M, Lenco J, Larsson P, Lundqvist M, Forsman M, Krocova Z, Stulik J, Macela A. Proteomic analysis of anti-Francisella tularensis LVS antibody response in murine model of tularemia. Proteomics. 2005;5:2090–2103. doi: 10.1002/pmic.200401123. [DOI] [PubMed] [Google Scholar]
- 35.Deng K, Blick RJ, Liu W, Hansen EJ. Identification of Francisella tularensis genes affected by iron limitation. Infect Immun. 2006;74:4224–4236. doi: 10.1128/IAI.01975-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee BY, Horwitz MA, Clemens DL. Identification, recombinant expression, immunolocalization in macrophages, and T-cell responsiveness of the major extracellular proteins of Francisella tularensis. Infect Immun. 2006;74:4002–4013. doi: 10.1128/IAI.00257-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Melillo A, Sledjeski DD, Lipski S, Wooten RM, Basrur V, Lafontaine ER. Identification of a Francisella tularensis LVS outer membrane protein that confers adherence to A549 human lung cells. FEMS Microbiol Lett. 2006;263:102–108. doi: 10.1111/j.1574-6968.2006.00413.x. [DOI] [PubMed] [Google Scholar]
- 38.Twine SM, Petit MD, Shen H, Mykytczuk NC, Kelly JF, Conlan JW. Immunoproteomic analysis of the murine antibody response to successful and failed immunization with live anti-Francisella vaccines. Biochem Biophys Res Commun. 2006;346:999–1008. doi: 10.1016/j.bbrc.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 39.Janovska S, Pavkova I, Hubalek M, Lenco J, Macela A, Stulik J. Identification of immunoreactive antigens in membrane proteins enriched fraction from Francisella tularensis LVS. Immunol Lett. 2007;108:151–159. doi: 10.1016/j.imlet.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 40.Hartley MG, Green M, Choules G, Rogers D, Rees DG, Newstead S, Sjostedt A, Titball RW. Protection afforded by heat shock protein 60 from Francisella tularensis is due to copurified lipopolysaccharide. Infect Immun. 2004;72:4109–4113. doi: 10.1128/IAI.72.7.4109-4113.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Samuelson JC, Chen M, Jiang F, Moller I, Wiedmann M, Kuhn A, Phillips GJ, Dalbey RE. YidC mediates membrane protein insertion in bacteria. Nature. 2000;406:637–641. doi: 10.1038/35020586. [DOI] [PubMed] [Google Scholar]
- 42.Luirink J, von Heijne G, Houben E, de Gier JW. Biogenesis of inner membrane proteins in Escherichia coli. Annu Rev Microbiol. 2005;59:329–355. doi: 10.1146/annurev.micro.59.030804.121246. [DOI] [PubMed] [Google Scholar]
- 43.Duckett NS, Olmos S, Durrant DM, Metzger DW. Intranasal interleukin-12 treatment for protection against respiratory infection with the Francisella tularensis live vaccine strain. Infect Immun. 2005;73:2306–2311. doi: 10.1128/IAI.73.4.2306-2311.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Metzger DW, Bakshi CS, Kirimanjeswara G. Mucosal Immunopathogenesis of Francisella tularensis. Ann N Y Acad Sci. 2007 doi: 10.1196/annals.1409.007. [DOI] [PubMed] [Google Scholar]
- 45.Pammit MA, Raulie EK, Lauriano CM, Klose KE, Arulanandam BP. Intranasal vaccination with a defined attenuated Francisella novicida strain induces gamma interferon-dependent antibody-mediated protection against tularemia. Infect Immun. 2006;74:2063–2071. doi: 10.1128/IAI.74.4.2063-2071.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu TH, Hutt JA, Garrison KA, Berliba LS, Zhou Y, Lyons CR. Intranasal vaccination induces protective immunity against intranasal infection with virulent Francisella tularensis biovar A. Infect Immun. 2005;73:2644–2654. doi: 10.1128/IAI.73.5.2644-2654.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johansson A, Farlow J, Larsson P, Dukerich M, Chambers E, Bystrom M, Fox J, Chu M, Forsman M, Sjostedt A, Keim P. Worldwide genetic relationships among Francisella tularensis isolates determined by multiple-locus variable-number tandem repeat analysis. J Bacteriol. 2004;186:5808–5818. doi: 10.1128/JB.186.17.5808-5818.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bruggemann M, Williams GT, Bindon CI, Clark MR, Walker MR, Jefferis R, Waldmann H, Neuberger MS. Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J Exp Med. 1987;166:1351–1361. doi: 10.1084/jem.166.5.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dangl JL, Wensel TG, Morrison SL, Stryer L, Herzenberg LA, Oi VT. Segmental flexibility and complement fixation of genetically engineered chimeric human, rabbit and mouse antibodies. Embo J. 1988;7:1989–1994. doi: 10.1002/j.1460-2075.1988.tb03037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rashid A, Auchincloss H, Jr, Sharon J. Comparison of GK1.5 and chimeric rat/mouse GK1.5 anti-CD4 antibodies for prolongation of skin allograft survival and suppression of alloantibody production in mice. J Immunol. 1992;148:1382–1388. [PubMed] [Google Scholar]
- 51.Janeway CA, Travers P, Walport M, Shlomchik M. Immunobiology: The Immune System in Health and Disease. 6. New York: Garland Science; 2005. [Google Scholar]
- 52.Lugtenberg B, Van Alphen L. Molecular architecture and functioning of the outer membrane of Escherichia coli and other gram-negative bacteria. Biochim Biophys Acta. 1983;737:51–115. doi: 10.1016/0304-4157(83)90014-x. [DOI] [PubMed] [Google Scholar]
- 53.Sharon J, Sompuram SR, Yang CY, Williams BR, Sarantopoulos S. Construction of polyclonal antibody libraries using phage display. Methods Mol Biol. 2002;178:101–112. doi: 10.1385/1-59259-240-6:101. [DOI] [PubMed] [Google Scholar]
- 54.Sharon J, Liebman MA, Williams BR. Recombinant polyclonal antibodies for cancer therapy. J Cell Biochem. 2005;96:305–313. doi: 10.1002/jcb.20536. [DOI] [PubMed] [Google Scholar]
- 55.Sarantopoulos S, Kao CY, Den W, Sharon J. A method for linking VL and VH region genes that allows bulk transfer between vectors for use in generating polyclonal IgG libraries. J Immunol. 1994;152:5344–5351. [PubMed] [Google Scholar]
- 56.Wiberg FC, Rasmussen SK, Frandsen TP, Rasmussen LK, Tengbjerg K, Coljee VW, Sharon J, Yang CY, Bregenholt S, Nielsen LS, Haurum JS, Tolstrup AB. Production of target-specific recombinant human polyclonal antibodies in mammalian cells. Biotechnol Bioeng. 2006;94:396–405. doi: 10.1002/bit.20865. [DOI] [PubMed] [Google Scholar]
- 57.Mirick GR, Bradt BM, Denardo SJ, Denardo GL. A review of human anti-globulin antibody (HAGA, HAMA, HACA, HAHA) responses to monoclonal antibodies. Not four letter words. Q J Nucl Med Mol Imaging. 2004;48:251–257. [PubMed] [Google Scholar]