

Abstract
Objectives
Several reports suggest that acute hyperglycemia affects male and female vascular beds differently. However, little is known about the interactions between hyperglycemia and gender in the vasculature. The objectives of our study were to investigate if there is a gender-based difference in the relaxation response of rat aorta after acute exposure to high glucose concentration, and the potential role of protein kinase C-beta (PKCβ), superoxide, and Rho kinase in the gender-specific effect of acute high glucose on the relaxation response.
Methods
Endothelium-dependent dilator responses to acetylcholine (ACh, 10−8 to 10−5 M) were obtained before and after 3 h treatment with Krebs’ solution containing high glucose (46 mM) in aortic rings pre-contracted with phenylephrine (2 μM) taken from female and male Sprague-Dawley rats. Similar experiments were generated in the presence of 1μM LY379196, a selective PKCβ inhibitor, 25 μM MnTMPyP, a superoxide dismutase mimetic, or 1 μM Fasudil, a Rho kinase inhibitor. Furthermore, protein expression of PKCβ isoforms was measured by Western blotting.
Results
We demonstrated that a 3 h incubation with elevated level of glucose impairs ACh responses only in the female rat aortic rings. Inhibition of PKCβ or superoxide production but notRho kinase prevents the high glucose-induced impairment of endothelium-dependent relaxation of female rat aorta. In addition, PKCβ2 expression is significantly higher in the female rat aorta than that in male rat aorta.
Conclusion
These results suggest that the gender difference in the impairment of endothelium-dependent vasodilation after acute exposure to high glucose in rat aorta is partly due to differences in PKCβ2 expression.
Keywords: Endothelial function, Diabetes, Gender, Nitric Oxide, Protein kinase C
Introduction
Cardiovascular disease (CVD) is the leading cause of mortality and morbidity in diabetic patients. There is a considerable body of evidence that early, repeated episodes of asymptomatic hyperglycemia increase the risk of CVD even in absence of overt clinical symptoms of diabetes [1, 2]. Hyperglycemia brings about several changes in vascular homeostasis, and one of the hallmarks of hyperglycemia-induced vascular disease is endothelial cell dysfunction, characterized by diminished nitric oxide (NO)-dependent vasodilation [3]. Endothelium dependent vasodilation (EDV) is generally used as a measure of stimulated release of NO and is a reproducible parameter to probe endothelial function in different pathological conditions. Impaired EDV has been described in diabetes and the degree of impairment of relaxation was shown to be correlated with glycemic control [4].
Several reports suggest that acute hyperglycemia affects male and female vascular beds differently [5]. Clinically these differences are manifested by a stronger association of CVD and diabetes in women than in men [5–7]. Furthermore, premenopausal diabetic women loose their gender-based cardiovascular protection [3, 8, 9]. Nonetheless, there is insufficient evidence to establish the timeline or the mechanism(s) underlying the loss of premenopausal female specific cardiovascular protection in diabetes. Thus, the initial aim of our study was to determine whether there is a gender difference in the development of abnormal endothelium-dependent responses in rat aorta following exposure to an acute hyperglycemic state.
The second aim of this study was to explore a potential mechanism by which hyperglycemia impairs vasodilator function in animals. The mechanism by which glucose impairs vascular dilatation is not clearly defined, but increased de novo synthesis of diacylglycerol (DAG) from glucose has been proposed [10]. DAG is a known activator of protein kinase C (PKC). Recent evidence implies a prominent role for PKC in that the various isoforms such as α, β1, and β2 are upregulated and activated in hyperglycemia and diabetes [11]. PKC activation causes generation of reactive oxygen species (ROS) such as superoxide (.O2) [12] furthering the cyclic effects of high-glucose induced alteration of vascular relaxation [13] by leading to a reduction in NO viability and loss of NO-dependent vasodilation. Activation of PKCβ isoform also leads to decreased NO production in human [13], mice [14] and rat [15] vasculature. Thus, experiments were carried out to examine the role of PKCβ and superoxide in the abnormal vascular responses to acute hyperglycemia in rat aorta. Specifically, we sought to determine whether inhibiting PKCβ activity or scavenging superoxide would reverse the impairment of endothelium-dependent vasodilation. Furthermore, estrogen is known to up-regulate various isoforms of PKC mRNA in different cell types, such as pituitary cells [16], breast cancer cells [17] and ovarian granulosa cells [18]. Therefore, we tested the hypothesis that aortic PKCβ expression in rat is gender-dependent.
In addition to the possible role of PKCβ and superoxide, several reports demonstrated that the Rho A/Rho kinase (ROCK) pathway plays a role in suppression of endothelial NO production by down regulating of endothelial nitric oxide synthase (eNOS) in human endothelial cells [19, 20]. This prompted us to investigate whether ROCK is also involved in acute hyperglycemia mediated impairment of vascular responses in the rat.
2. Methods
2.1 Experimental Animals
Adult male and female Sprague-Dawley rats weighing 225–250 g were used (Simonsen Laboratories, CA). All animal protocols were approved by the Animal Care Committee of the University of the Pacific and complied with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). Rats were sacrificed using a CO2 gas chamber.
2.2. Tissue collection and measurement of arterial tension
The thoracic aorta was cleaned of adhering tissue and cut into 3–4 mm rings in oxygenated Krebs’ solution of following composition (in mM) NaCl 118.3, KCl 4.7, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25.0, EDTA 0.023, and glucose 6.0 at 37 °C.
Aortic rings were suspended horizontally between two stainless steel hooks for measurement of isometric tension in organ baths containing 20 ml of Krebs’ solution at 37°C, bubbled with 95% O2/5% CO2. The isometric tension of aortic rings was monitored with a computer-based data acquisition system (ADI Instrument, CO). Rings were equilibrated for 45 min under a resting tension of 2 g to allow development of a stable basal tone. Stimulation of rings with 80 mM KCl was repeated every 15 min until contractile responses were stable. Acetylcholine (ACh, 10 μM) induced relaxation of the phenylephrine (PE, 2 μM) pre-contracted vessels was taken as evidence for the preservation of an intact endothelium.
2.2.1. Dilator concentration response curves to ACh
Aortic rings were contracted with PE (2 μM), which produced about 80% of the maximal contraction. The first concentration-response curve (CRC) was obtained in Krebs containing normal glucose (NG, 6 mM) by the addition of increasing concentrations of ACh (10−8 to 10−5 M). Tissues were then washed with Krebs’ solution for 1 h to allow the rings to reach the basal tone. Rings were again pre-contracted with PE and the second CRC to ACh was generated in Krebs containing NG.
2.2.2. Effects of high glucose or PMA on endothelium-dependent relaxation
At this point, the tissues taken from each animal were designated for 2 sets of experiments. One set was incubated with Krebs containing NG (6 mM) and the other set was incubated with Krebs containing high glucose (HG, 46 mM) for 3 h or 0.1 μM phorbol 12-myristate 13-acetate (PMA), a PKC activator, for 3 h. A third dilator CRC to ACh was then obtained in pre-contracted aortic rings.
In a second set of experiments, we determined the role of PKCβ, PKC-alpha (PKCα), PKC-delta (PKCδ), superoxide or ROCK in HG- or PMA-induced blunting of ACh relaxation. Aortic rings were incubated with 1 μM of LY379196 (a gift of Lilly Research Laboratories, Indianapolis, IN), a selective inhibitor of PKCβ, 9 nM of Ro-32-0432, a selective PKCα inhibitor [21] (Calbiochem, CA), 6 μM of Rottlerin, a selective PKCδ inhibitor [22] (Calbiochem, CA), 25 μM of MnTMPyP, a membrane permeant mimetic of superoxide dismutase (SOD) (Sigma, MO), or 1 μM of fasudil, an inhibitor of ROCK (Sigma, MO), for 20 min prior to the addition of HG (46 mM) or 0.1 μM PMA into the buffer. Incubation was continued for additional 3 h and the third CRC to ACh was then performed. The concentration of LY379196 was identical to concentration of LY333531, a selective inhibitor of PKCβ, used in the similar in vitro vascular study as reported previously [23]. To investigate the effects of the above compounds alone on the ACh–induced relaxation, in some tissues the second CRC were generated after pretreatment with 1 μM LY379196, 25 μM MnTMPyP or 1 μM fasudil for 20 min. Tissues were then washed with Krebs solution for 1 h to allow a return to basal tone.
2.2.3. Effects of high glucose on endothelium-independent relaxation
Effects of HG on sodium nitroprusside (SNP, 10−9–10−6 M), a NO-donor, induced relaxation of pre-contracted aortic rings with PE (2 μM), was investigated following a 3 h incubation of the aortic ring with HG (46 mM).
2.2.4. Effects of high glucose on bradykinin-induced endothelium-dependent relaxation
Effects of HG on bradykinin (BK, 10−8–10−3 M), a BK receptor-mediated endothelium-dependent vasodilator, induced relaxation of pre-contracted aortic rings with PE (2 μM), was investigated following a 3 h incubation of the aortic ring with HG (46 mM).
PE, ACh, BK, SNP, Fasudil and MnTMPyP were dissolved in double-distilled H2O, and all other drugs were dissolved in DMSO. In each case, the vehicle had no effect on vascular reactivity.
2.3. Western analysis
Protein samples from aortas taken from 5 female and 5 male rats were prepared as described previously by us [24]. 50 μg of protein from each sample was loaded on 8% SDS-polyacrylamide gels, and transferred to Nitrocellulose membranes. Membranes were blocked for 1 h in Tris-buffered saline plus Tween-20 (0.1%; TBS-T) containing 5% fat free milk powder. Blots were incubated with Rabbit polyclonal antibodies against PKCβ1 and PKCβ2 (1:1000, Santa Cruz Biotechnology Inc., CA,) at 4°C overnight. After the washes, blots were incubated with the secondary antibody Bovine anti-rabbit linked to horseradish peroxidase (1:5000, Santa Cruz Biotechnology., CA) for 1 h at room temperature. Enhanced chemiluminescence was performed (Pierce Biotechnology, Rockford, IL) just before developing the film. GAPDH was used for internal standard (1:2500, Cell Signaling Danvers, MA).
2.4. Data analysis
The maximum relaxation (Emax) to ACh is expressed as the percent decreased from maximum PE-induced contraction. The EC50 values were used to calculate pD2 values (-log EC50), which are normally distributed. Western blot gels were analyzed densitometrically and normalized to expression of GAPDH protein levels respectively. Data are reported as the mean ± standard error of the mean (S.E.M.). Data were analyzed by paired t-test for comparisons of two group means in a pre/post-test format or unpaired t-test for comparisons of two group means. A probability value of less than 5% (P < 0.05) was considered significant.
3. Results
3.1. Analysis of relaxation responses to ACh
To examine if there is a gender difference in EDV in aortic rings of rats exposed to HG, the CRC to ACh in the Krebs containing HG (46 mM) was compared to the second CRC to ACh generated in Krebs containing NG (6 mM) in PE pre-contracted aortic rings of male and female animals. In rat aorta, PE-induced contraction was significantly (P < 0.05) decreased by incubation of tissues with HG in both female and male animals. PE-induced tensions in aortic rings taken from female rats (n=9) were 0.72 ± 0.07 g and 0.61 ± 0.09 g for NG and HG, respectively. PE-induced tensions in aortic rings taken from male rats (n=8) were 0.75 ± 0.01 g and 0.57 ± 0.07 g for NG and HG, respectively.
ACh (10−8 to 10−5 M) relaxed aortic ring pre-constricted with PE (2 μM) in a concentration-dependent manner. No significant differences in response to ACh occurred among the first, second and third CRCs generated in Krebs containing NG (Tables 1 & 2).
Table 1.
Values of sensitivity to ACh as expressed by pD2 in experimental groups (CRC; concentration-response curves; NG; Normal Glucose, HG; High Glucose).
| (A) Males | |||
|---|---|---|---|
| 1st CRC in | 2nd CRC in | 3rd CRC in | |
|
Group
n = 8 |
NG
6.8 ± 0.05 |
NG
6.9 ± 0.07 |
NG
6.7 ± 0.09 |
|
Group
n = 8 |
NG
6.71 ± 0.09 |
NG
6.64 ± 0.23 |
HG
6.67 ± 0.18 |
|
Group
n = 5 |
NG
6.64 ± 0.32 |
NG
6.66 ± 0.21 |
PMA
6.70 ± 0.25 |
| (B) Females | |||
|
Group
n = 9 |
NG
6.67 ± 0.16 |
NG
6.65 ± 0.13 |
NG
6.65 ± 0.17 |
|
Group
n = 9 |
NG
6.68 ± 0.18 |
NG
6.64 ± 0.11 |
HG
6.70 ± 0.15 |
|
Group
n = 5 |
NG
6.85 ± 0.18 |
LY379196 + NG
6.83 ± 0.13 |
LY379196 + HG
6.89 ± 0.10 |
|
Group
n = 5 |
NG
6.89 ± 0.12 |
Fasudil + NG
6.87 ± 0.10 |
Fasudil + HG
6.70 ± 0.14 |
|
Group
n = 5 |
NG
7.06 ± 0.19 |
Ro-32-0432+ NG
7.03 ± 0.14 |
Ro-32-0432 + HG
7.14 ± 0.22 |
|
Group
n = 5 |
NG
6.94 ± 0.22 |
Rottlerin + NG
6.87 ± 0.21 |
Rottlerin+ HG
6.78 ± 0.33 |
|
Group
n = 5 |
NG
6.87 ± 0.13 |
LY341684 + NG
6.88 ± 0.18 |
LY341684 + HG
7.06 ± 0.16 |
|
Group
n = 5 |
NG
6.64 ± 0.12 |
MnTMPyP + NG
6.62 ± 0.18 |
MnTMPyP + HG
6.66 ± 0.17 |
|
Group
n = 5 |
NG
6.81 ± 0.16 |
NG
6.82 ± 0.15 |
PMA
6.83 ± 0.16 |
|
Group
n = 5 |
NG
6.87 ± 0.16 |
LY379196 + NG
6.80 ± 0.12 |
LY379196 + PMA
6.99 ± 0.17 |
|
Group
n = 5 |
NG
6.78 ± 0.03 |
Fasudil + NG
6.74 ± 0.25 |
Fasudil + PMA
6.78 ± 0.11 |
|
Group
n = 5 |
NG
7.02 ± 0.18 |
Ro-32-0432+ NG
6.99 ± 0.15 |
Ro-32-0432 + PMA
7.15 ± 0.18 |
|
Group
n = 5 |
NG
6.96 ± 0.22 |
Rottlerin + NG
6.89 ± 0.19 |
Rottlerin+ PMA
6.93 ± 0.25 |
|
Group
n = 5 |
NG
6.86 ± 0.16 |
LY341684 + NG
6.80 ± 0.21 |
LY341684 + PMA
7.10 ± 0.15 |
|
Group
n = 5 |
NG
6.75 ± 0.18 |
MnTMPyP + NG
6.78 ± 0.16 |
MnTMPyP + PMA
6.71 ± 0.13 |
Data are expressed as Mean ± S.E.M.
Table 2.
Maximal response (Emax) to Ach in experimental groups (CRC; concentration-response curves; NG; Normal Glucose, HG; High Glucose
| (A) Males | |||
|---|---|---|---|
| 1st CRC in | 2nd CRC in | 3rd CRC in | |
|
Group
n= 8 |
NG
77.8 ± 3.4 |
NG
76.9 ± 3.4 |
NG
73 ± 3.5 |
|
Group
n = 8 |
NG
75.7 ± 5.6 |
NG
77.4 ± 5.2 |
HG
67.5 ± 5.6 |
|
Group
n = 5 |
NG
72.2 ± 8.7 |
NG
75.4 ± 6.7 |
PMA
64.3 ± 8.8 |
| (B) Females | |||
|
Group
n = 9 |
NG
84.5 ± 4.4 |
NG
86.9 ± 3.6 |
NG
83.4 ± 8.4 |
|
Group
n = 9 |
NG
79.0 ± 4.0 |
NG
77.8 ± 4.6 |
HG
56.2 ± 4.3* |
|
Group
n = 5 |
NG
81.9 ± 4.0 |
LY379196 + NG
80.9 ± 4.2 |
LY379196 + HG
78.3 ± 5.4 |
|
Group
n = 5 |
NG
80.7 ± 4.9 |
Fasudil + NG
81.0 ± 3.4 |
Fasudil + HG
57.5 ± 6.1* |
|
Group
n = 5 |
NG
96 ± 2.0 |
Ro-32-0432+ NG
93.3 ± 2.1 |
Ro-32-0432 + HG
73.4 ± 10.3* |
|
Group
n = 5 |
NG
93.7 ± 2.2 |
Rottlerin + NG
90.1 ± 3.5 |
Rottlerin+ HG
67.2 ± 3.9* |
|
Group
n = 5 |
NG
90.1 ± 3.4 |
LY341684 + NG
87.6 ± 5.7 |
LY341684 + HG
88.9 ± 5.9 |
|
Group
n = 5 |
NG
88 ± 3.2 |
MnTMPyP + NG
87 ± 3.2 |
MnTMPyP+ HG
76 ± 0.6* |
|
Group
n = 5 |
NG
79.1 ± 3.1 |
NG
82.4 ± 2.8 |
PMA
62 ± 3.5* |
|
Group
n = 5 |
NG
81.9 ± 4.0 |
LY379196 + NG
80.9 ± 4.2 |
LY379196 + PMA
78.3 ± 5.4 |
|
Group
n = 5 |
NG
78.0 ± 6.2 |
Fasudil + NG
79.8 ± 5.9 |
Fasudil + PMA
57.9 ± 5.8* |
|
Group
n = 5 |
NG
98.3 ± 2.1 |
Ro-32-0432 + NG
93.2 ± 4.1 |
Ro-32-0432 + PMA
73.0 ± 6.5* |
|
Group
n = 5 |
NG
96.3 ± 2.0 |
Rottlerin + NG
93.3 ± 4.0 |
Rottlerin + PMA
73.5 ± 6.5* |
|
Group
n = 5 |
NG
88.0 ± 5.2 |
LY341684 + NG
87.5 ± 4.2 |
LY341684 +PMA
88.6 ± 4.9 |
|
Group
n = 5 |
NG
89.5 ± 3.1 |
MnTMPyP + NG
90.4 ± 4.6 |
MnTMPyP+ PMA
79.6 ± 2.9* |
Data are expressed as Mean ± SEM (*P < 0.05 vs 2nd CRC, paired t-test).
Incubation of aortic rings taken from female rats for 3 h in Krebs containing HG attenuated ACh (10−6 to 10−5 M) -induced relaxation compared to the second CRC in NG (Figure 1B, P < 0.05; n = 9). The Emax to ACh was 77.8 ± 4.6% (before HG) and 56.2 ± 4.3% after incubation with HG (Table 2B). At ACh concentrations of less than 10−6 M, there was no significant effect of HG. Sensitivity to ACh as assessed by −logEC50 (pD2) was the similar for NG and HG (Table 1B). In contrast, ACh induced relaxation was not affected by HG in aortic rings taken from male rats (Figure 1A), as also indicated by no significant differences at the level of 5% in pD2 and Emax values (Tables 1A–2A).
Figure 1. Effect of high glucose on the relaxation response to cumulative concentrations of acetylcholine (ACh) in intact aortic rings pre-contracted with phenylephrine (PE; 2 μM) taken from male (A) and female (B) rats.
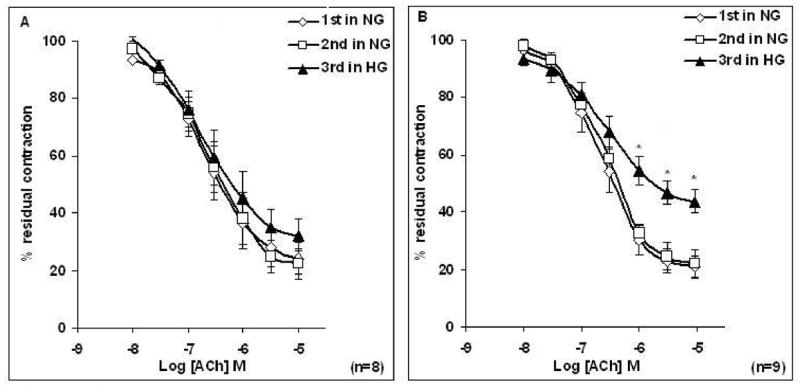
Relaxation to ACh is expressed as a percentage of PE induced maximum contraction. Data are presented as the mean ± S.E.M.
Rings were incubated with Krebs containing normal glucose (NG, 6mM) and/or Krebs containing high glucose (HG, 46 mM) for 3 h before the concentration response curve (CRC) to ACh was determined. The 3rd CRC at higher concentration of ACh was reduced when compared with the 1st and 2nd CRC only in aortic rings taken from female rats (*P < 0.05, paired t-test).
3.1.1. Effects of PKC and ROCK activation on the relaxation responses to ACh
To examine the role of PKCβ and ROCK as potential downstream effectors of HG-induced endothelial impairment, aortic rings from female rats were incubated for 20 min with either 1 μM LY379196 (a PKCβ inhibitor) or 1μM fasudil (a ROCK inhibitor) prior to 3 h incubation in Krebs containing HG. Pretreatment of aortic rings taken from female rats with LY379196, but not fasudil prior to the 3 h incubation of tissues in Krebs containing HG prevented the HG-induced attenuation of ACh-induced relaxation of the aortic ring (Figures 2A–2B). Emax to ACh was significantly reduced after HG treatment of female rat aorta (Table 2B); prior treatment with LY379196 restored full ACh response. LY379196 or fasudil alone did not alter ACh response in NG (Table 2B). To rule out the possible involvement of other PKC isoforms, such as PKCα or PKCδ, the same experiment was performed in the presence of 9 nM Ro-32-0432 (a PKCα inhibitor) or 6 μM Rottlerin (a PKCδ inhibitor). Prior treatment of tissues with Ro-32-0432 or Rottlerin did not prevent the impairment of EDV induced by HG in female aortic rings (Figures 2C–2D).
Figure 2. Effects of LY379196, Fasudil, Ro-32-0432, or Rottlerin on relaxations induced by acetylcholine (ACh) in female aortic rings in normal glucose and high glucose.

Responses to ACh were determined in rings incubated with Krebs containing normal glucose (NG, 6mM) as 1st response, Krebs containing LY379196 (1 μM), Fasudil (1μM), Ro-32-0432 (9 nM), or Rottlerin (6 μM) for 20 min as 2nd response, and Krebs containing LY379196, Fasudil, Ro-32-0432, or Rottlerin, in high glucose (HG, 46 mM) for 3 h as 3rd response. (A) LY379196 restored the abnormal ACh relaxation of female rat aorta incubated in HG. (B-D) Fasudil, Ro-32-0432 or Rottlerin did not prevent the HG-induced impairment of responses to ACh. Data are presented as the mean ± S.EM. (*P < 0.05, paired t-test).
Similarly acute treatment of tissues with Krebs containing PMA (0.1 μM), a DAG analogue, for 3 h significantly attenuated ACh-induced relaxation of aortic rings taken from female animals only (P < 0.05, n=5) (Figure 3B). Inhibition of PKCβ but not of PKCα, PKCδ or ROCK prior to the 3 h incubation in PMA prevented PMA-induced impairment of EDV of female aortic rings (Figures 3C–3F). Finally, LY341684 (salt form of LY333531, a gift of Lilly Research Laboratories) was used to confirm findings with LY379196. As with LY379196, 1 μM LY341684 restored responses to ACh in female rat aortic rings exposed to high glucose or PMA (Figures 4A–4B).
Figure 3. Effect of activation of PKC by PMA on the relaxation response to cumulative concentrations of acetylcholine (ACh) in intact aortic rings from male (A) and female (B) rats, and the effect of pretreatment of rings with LY379196, Fasudil, Ro-32-0432, or Rottlerin.
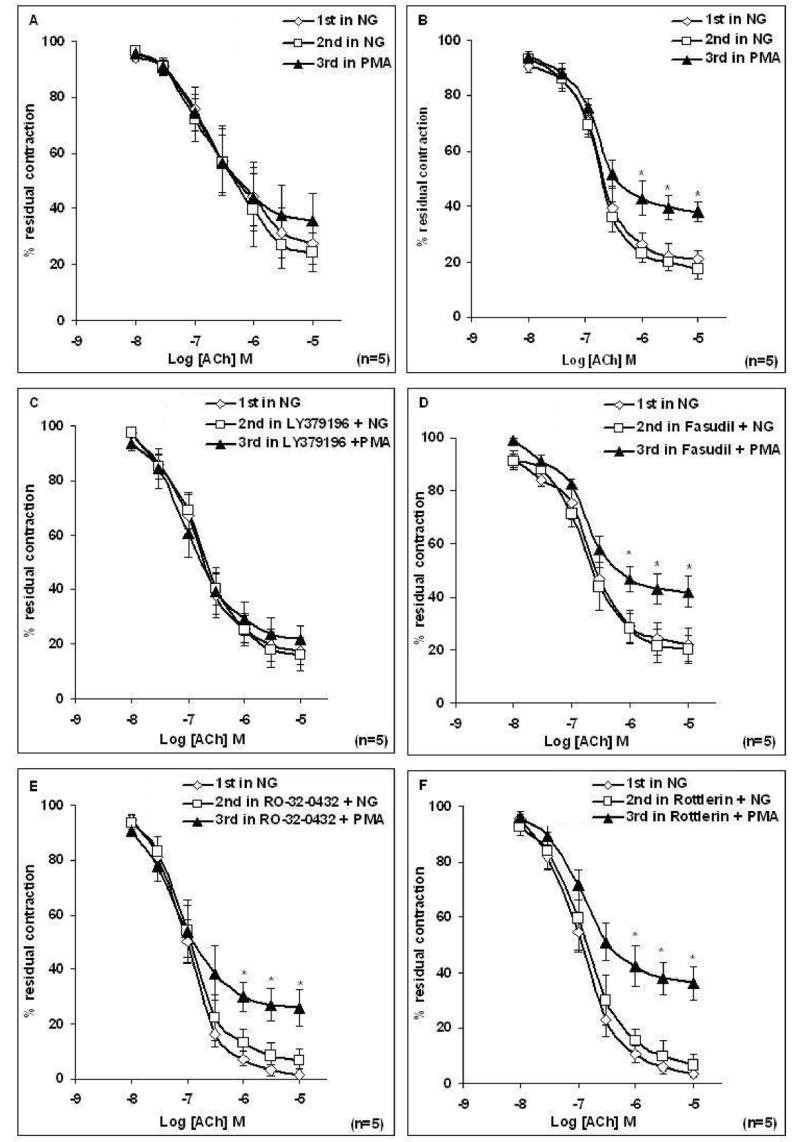
(A–B) Rings were incubated with Krebs containing normal glucose (NG, 6mM) and/or Krebs containing PMA (0.1 μM) for 3 h before concentration response curve (CRC) to ACh was determined. Treatment of aortic rings with PMA for 3 h caused a significant reduction in maximal relaxation to ACh in females. (C–F) Rings from female rats were incubated with Krebs containing NG or Krebs containing LY379196 (1 μM), Fasudil (1μM), Ro-32-0432 (9 nM), or Rottlerin (6 μM) for 20 min or Krebs containing LY379196, Fasudil, Ro-32-0432 or Rottlerin and PMA (0.1 μM) for 3 h before the CRC to ACh was determined. Treatment of aorta with LY379196, but not Fasudil, Ro-32-0432 or Rottlerin prior to PMA restored the abnormal ACh relaxation of aorta. Data are presented as the mean ± S.EM. (*P < 0.05 paired t-test).
Figure 4. Effect of LY341684 on high glucose (A) or PMA (B) induced attenuation of ACh relaxation of aortic rings taken from female rats.

Rings were incubated with Krebs containing normal glucose (NG, 6 mM) or Krebs containing LY341684 (1 μM) for 20 min or Krebs containing LY341684 and high glucose (HG, 46mM) (A) or LY341684 and PMA (0.1 μM) (B) for 3 h before responses to ACh were determined. Pretreatment of aorta with LY341684 prior to HG or PMA restored the abnormal ACh relaxation of aorta. Data are presented as the mean ± S.EM. (*P < 0.05 paired t-test).
3.1.2. Effect of superoxide generation
Figures 5C–5D show that acute pretreatment of female rat aortic rings with 25 μM MnTMPyP, a SOD mimetic which acts as superoxide anion scavenger, for 20 min prior to the 3 h incubation in Krebs containing HG (46 mM) or PMA (0.1 μM) partially but significantly improved the altered ACh response (P < 0.05 vs CRCs to ACh in HG or PMA, unpaired t-test). However, prior treatment with MnTMPyP did not fully restore ACh response; the Emax to ACh was 87 ± 3.2% and 90 ± 4.6% (before HG and PMA, respectively) and 76 ± 0.6% and 79.6 ± 2.9% after incubation with HG and PMA, respectively (P < 0.05, paired t-test, n=5).
Figure 5. Effects of MnTMPyP on high Glucose (A) or PMA (B) induced attenuation of ACh relaxation of aortic rings taken from female rats.

Rings were incubated with Krebs containing normal glucose (NG, 6 mM), or Krebs containing MnTMPyP (25 μM) for 20 min or Krebs containing MnTMPyP and high glucose (HG, 46mM) for 3 h (A) or MnTMPyP and PMA (0.1 μM) for 3 h (B) before concentration response curve (CRC) to ACh was determined. Data are presented as the mean ± S.E.M. Pretreatment of aorta with MnTMPyP prior to HG or PMA partially but significantly improved altered ACh responses in HG or PMA (#P < 0.05 vs ACh CRCs in HG or PMA (dotted lines), unpaired t-test). However, prior treatment with MnTMPyP did not fully restore ACh responses in HG or PMA (*P < 0.05 vs 2nd CRC in NG, paired t-test).
3.2. Analysis of relaxation responses to sodium nitroprusside (SNP)
Unlike the effects on ACh-induced relaxation, incubation of aortic rings taken from female animals with Krebs containing HG (46 mM) for 3 h had no effect on SNP-induced relaxation of PE pre-contracted rings (Figure 6).
Figure 6. Effects of high Glucose on the relaxation response to cumulative concentrations of sodium nitroprusside (SNP) in intact aortic rings pre-contracted with phenylephrine (PE; 2 μM) taken from female rats.
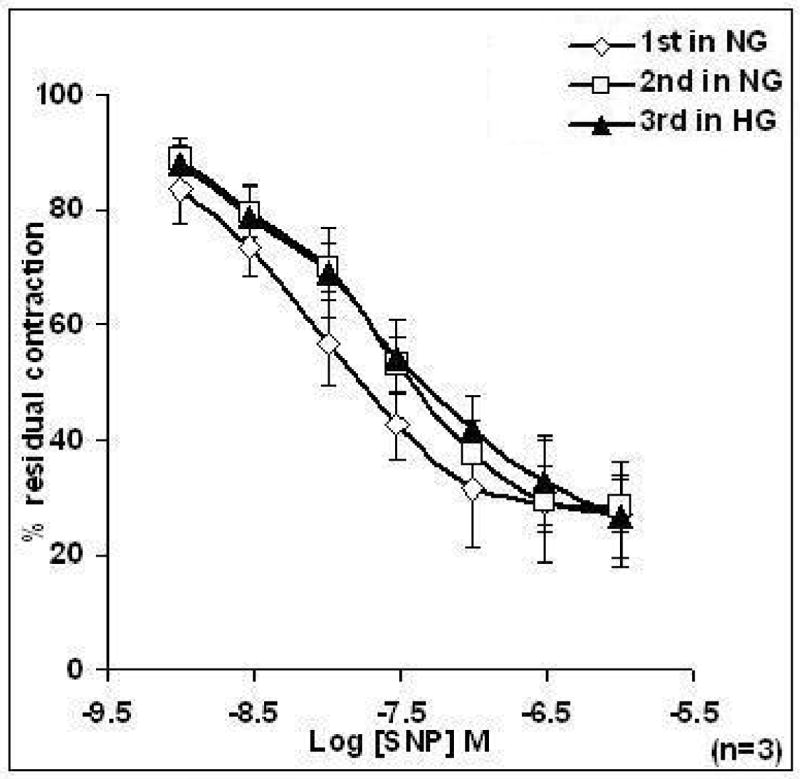
Rings were incubated with Krebs containing normal glucose (NG, 6 mM) and or Krebs containing high glucose (HG, 46 mM) for 3 h before concentration response curve (CRC) to SNP was determined. The 1st and 2nd CRCs to SNP of aorta in Krebs containing NG were not significantly different from the 3rd CRC in HG. Data are presented as the mean ± S.E.M.
3.3. Analysis of relaxation responses to bradykinin (BK)
BK (10−8 to 10−3 M) relaxed aortic ring pre-constricted with PE (2 μM) in a concentration-dependent manner. Incubation of aortic rings taken from female rats for 3 h in Krebs containing HG significantly attenuated BK (10−4 to 10−3 M) -induced relaxation compared to the CRC in NG (P < 0.05; n=3) (data not shown).
3.4. Analysis of PKCβ expression
As shown in Figure 7, there was no difference in PKCβ1 expression in female and male rat aorta. However, level of protein expression for PKCβ2 was significantly higher in aorta taken from female rats compared to those observed in male rats (P < 0.05, unpaired t-test, n=5 per each group).
Figure 7. PKCβ protein expression in female and male rat aorta.

(A) Representative Western blot for PKCβ isoform expression detected in aorta from three males and three females. (B) PKCβ1 and PKCβ2 protein levels in aorta from male (open bar) and female (slash bar) rats normalized to GAPDH protein (internal control). The PKCβ2 protein expression is significantly higher in aorta taken from female rats compared to that in male rats. Each bar represents the mean ± S.E.M. of 5 animals per each group (* P < 0.05 vs. Males, unpaired t-test).
4. Discussion
The novel findings of this investigation are that 1) acute exposure to HG reveals a gender difference in the development of impaired EDV in rat aorta, and 2) PKC β2 protein expression is higher in female rat aorta than in male rat aorta. To our knowledge, this is the first report showing that a 3 h incubation of aortic rings with Krebs containing HG impairs ACh-induced EDV only in aorta from female rats but not in aorta from male rats.
Many of the vascular complications in diabetes have been attributed to repeated episodes of acute hyperglycemia that result in vascular injury. It is now known that cardiovascular complications start early as disease progresses from normal glucose tolerance to impaired glucose tolerance and then to overt type 2 diabetes. In line with previous reports that demonstrate compromised EDV in different vascular beds under hyperglycemic conditions [13, 15, 25, 26], we also found that HG impairs EDV in rat aorta. However, we noted a significant gender based difference in the vascular response to an acute hyperglycemic condition. This contrast with the findings of Wang et al [26], who reported that a 6 h exposure to HG (44 mM) resulted in an impairment of EDV in aortic rings from male rats. In fact, we also obtained similar findings by incubating male aorta with HG for 6 h (data not shown). Thus, in male rats longer exposures to HG would lead to the impairment of vascular response similar to those observed in female rats. These data support the hypothesis that in hyperglycemia there is a predisposition of female cardiovascular system to earlier vascular injury leading to an increased risk for the development of CVDs.
Epidemiological evidence suggests that hyperglycemia/diabetes overcomes gender-based protective effects on the cardiovascular system in premenopausal female. This study suggests a potential underlying mechanism for those observations. Activation of the PKCβ signaling pathway in hyperglycemia has been increasingly recognized as an early event leading to cardiovascular dysfunction both in humans [13] and animals [26, 27]. We sought to investigate the role PKCβ activation in mediating the selective impairment of EDV in female aortic rings following exposure to HG.
The hyperglycemia induced impairment of EDV in female rat aorta was prevented by prior incubation of the aortic rings with PKCβ blockers, LY379196 or LY341684 (Figures 2A & 4A), but not with PKCα and PKCδ inhibitors. This suggests that impairment of EDV under acute hyperglycemic conditions in female aortic rings was partly due to an increase in the activity of PKCβ. This hypothesis is further supported by the impairment of ACh-induced relaxation in female aortic rings exposed to Krebs containing PMA, a PKC activator. Also consistent with our working hypothesis are the data demonstrating that the attenuation of EDV by PMA only occurs in females, and is completely prevented following prior treatment of tissues with selective PKCβ inhibitors. Modulation of ACh-induced relaxation can occur at several points; namely, bioavailability of NO, stimulation of release, and NO-interaction with smooth muscle. The effects of hyperglycemia on vascular smooth muscle cell function are less clear and not the focus of our current study. Nevertheless, in the current study, acute HG resulted in decreased contractile responses to PE in male and female rat aorta. Published data demonstrate both an increase, decrease, and no change in vasoconstriction under conditions of hyperglycemia [25, 28], but altered sensitivity to vasoconstriction does not appear to contribute significantly to the impairment of EDV in human diabetic patients or in healthy human volunteers exposed to hyperglycemia [29]. Furthermore, in our study, SNP-induced aortic relaxation in female animals was not affected by HG. SNP is a NO donor, leading to a rise of cGMP-mediated endothelium-independent relaxation in smooth muscle cells. This suggests that the responsiveness of female vascular smooth muscle to NO is not affected by acute hyperglycemia. Lastly, relaxation of aortic rings by bradykinin, another type of receptor-mediated endothelium-dependent vasodilator, was also affected by HG in females (data not shown). These data suggest that the impaired response to ACh is representative a general phenomenon of endothelial dysfunction.
Three hours of hyperglycemic condition did not impair EDV in male animals (Figure 1B), suggesting that PKCβ activity was less affected in male aortic rings exposed to HG than that in female tissues. The enhanced activity of PKCβ in female rat aortas exposed to HG may partly result from an elevated level of PKCβ expression. Several studies have shown that estrogen up-regulates various isoforms of PKC mRNA expression in cell types like pituitary cells [16], breast cancer cells [17] and ovarian granulosa cells [18]. Therefore, we tested the hypothesis that there may be a gender difference in aortic PKCβ1 and PKCβ2 expression in rat. Interestingly, we observed that PKCβ2, but not PKCβ1, expression was higher in aorta taken from female rats compared to those in male rats. Clearly, the precise location, endothelium or smooth muscle, of enhanced PKCβ2 expression needs to be resolved. In the hyperglycemic condition, due to the de novo synthesis of DAG, a PKC activator [10], the higher basal level of PKCβ2 could lead to a greater increase in total activity of PKCβ2 in female aortic rings than in male aortic rings. PKC activation has been reported to cause generation of ROS furthering the cyclic effects of HG-induced alteration of EDV [12]. To test the hypothesis that superoxide underlies the vascular dysfunction observed in HG, we investigated the role of superoxide production in HG- or PMA-induced alteration of vascular responses to ACh in the female rat aorta. Prior incubation of female rat aorta with a SOD mimetic agent partially but significantly improved the altered ACh responses in the presence of HG or PMA suggesting that impairment of EDV is due, in part, to augmented superoxide production, but superoxide alone is insufficient to account for all of the endothelial dysfunction observed in female rat aorta exposed to HG or PMA.
Although the functional consequences of an elevated basal expression of PKCβ2 in female aorta under normal conditions are unclear, these data are consistent with studies showing that estrogen up-regulates various isoforms of PKC mRNA expression. Additional studies will be needed to document the intracellular and functional effects of elevated basal expression of PKCβ2 in female aorta. It is also important to note that, despite the presence of higher expression of basal PKCβ2 in female aorta, under normoglycemic conditions, the eNOS and NO release are not affected in female aorta. In fact, we previously reported that eNOS expression [30] and NO release [31] is higher in aorta taken from female rats than those in male rats. This lack of effect of high level of PKCβ expression on NO production may be attributed to the availability of DAG which is the rate limiting step for activation of PKCβ.
Finally, it has been demonstrated that ROCK mediates endothelial dysfunction [19, 20, 32]. We therefore sought to investigate whether ROCK is involved in HG-induced endothelial dysfunction in female aorta. Inhibition of ROCK by fasudil failed to prevent the HG- or PMA-induced impairment of EDV in female rat aortic rings suggesting that impairment of ACh-induced relaxation by acute hyperglycemic condition is not mediated by ROCK activation. These findings differ from those of Rikitake et al [33] who showed that 16 h exposure of human endothelial cells to a hyperglycemic condition caused activation of ROCK through activation of PKC. However, these investigators made their observations in an endothelial cell line that may differ significantly from our setting. These data also suggest that the duration of exposure to hyperglycemia may play a role in the activation of ROCK.
In summary, this is the first report showing that after a 3 h exposure of rat aorta to elevated glucose, there is a selective impairment of stimulated release of NO in female animals. A role for PKC and superoxide was suggested by the facts that a PKCβ inhibitor or a superoxide scavenger could significantly correct the effect of HG, and PKC activation could mimic the action of glucose in tissues taken from females. Furthermore, we found that there is a gender difference in endothelial PKCβII expression in rat aorta. We therefore speculate that differences in levels of PKCβ may contribute to the factors responsible for the overcoming of gender based cardiovascular protection in premenopausal hyperglycemic female leading to cardiovascular dysfunction.
Acknowledgments
This work was supported by grant from the National Heart, Lung, and Blood Institute (NHLBI). R15HL71520, and the National Institute for Dental and Craniofacial Research (NIDCR), DE016587.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de Vegt F, Dekker JM, Ruhe HG, Stehouwer CD, Nijpels G, Bouter LM, et al. Hyperglycaemia is associated with all-cause and cardiovascular mortality in the Hoorn population: the Hoorn Study. Diabetologia. 1999;42:926–31. doi: 10.1007/s001250051249. [DOI] [PubMed] [Google Scholar]
- 2.Group DS. Glucose tolerance and cardiovascular mortality: comparison of fasting and 2-hour diagnostic criteria. Arch Intern Med. 2001;161:397–405. doi: 10.1001/archinte.161.3.397. [DOI] [PubMed] [Google Scholar]
- 3.Steinberg HO, Paradisi G, Cronin J, Crowde K, Hempfling A, Hook G, et al. Type II diabetes abrogates sex differences in endothelial function in premenopausal women. Circulation. 2000;101:2040–6. doi: 10.1161/01.cir.101.17.2040. [DOI] [PubMed] [Google Scholar]
- 4.Stettler C, Allemann S, Juni P, Cull CA, Holman RR, Egger M, et al. Glycemic control and macrovascular disease in types 1 and 2 diabetes mellitus: Meta-analysis of randomized trials. Am Heart J. 2006;152:27–38. doi: 10.1016/j.ahj.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 5.Hu G. Gender difference in all-cause and cardiovascular mortality related to hyperglycaemia and newly-diagnosed diabetes. Diabetologia. 2003;46:608–17. doi: 10.1007/s00125-003-1096-6. [DOI] [PubMed] [Google Scholar]
- 6.Onat A, Hergenc G, Keles I, Dogan Y, Turkmen S, Sansoy V. Sex difference in development of diabetes and cardiovascular disease on the way from obesity and metabolic syndrome. Metabolism. 2005;54:800–8. doi: 10.1016/j.metabol.2005.01.025. [DOI] [PubMed] [Google Scholar]
- 7.Janghorbani M, Jones RB, Gilmour WH, Hedley AJ, Zhianpour M. A prospective population based study of gender differential in mortality from cardiovascular disease and “all causes” in asymptomatic hyperglycaemics. J Clin Epidemiol. 1994;47:397–405. doi: 10.1016/0895-4356(94)90161-9. [DOI] [PubMed] [Google Scholar]
- 8.Kannel WB. Metabolic risk factors for coronary heart disease in women: perspective from the Framingham Study. Am Heart J. 1987;114:413–9. doi: 10.1016/0002-8703(87)90511-4. [DOI] [PubMed] [Google Scholar]
- 9.Di Carli MF, Afonso L, Campisi R, Ramappa P, Bianco-Batlles D, Grunberger G, et al. Coronary vascular dysfunction in premenopausal women with diabetes mellitus. Am Heart J. 2002;144:711–8. [PubMed] [Google Scholar]
- 10.Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci U S A. 1992;89:11059–63. doi: 10.1073/pnas.89.22.11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo M, Wu MH, Korompai F, Yuan SY. Upregulation of PKC genes and isozymes in cardiovascular tissues during early stages of experimental diabetes. Physiol Genomics. 2003;12:139–46. doi: 10.1152/physiolgenomics.00125.2002. [DOI] [PubMed] [Google Scholar]
- 12.Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–45. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 13.Beckman JA, Goldfine AB, Gordon MB, Garrett LA, Creager MA. Inhibition of protein kinase Cbeta prevents impaired endothelium-dependent vasodilation caused by hyperglycemia in humans. Circ Res. 2002;90:107–11. doi: 10.1161/hh0102.102359. [DOI] [PubMed] [Google Scholar]
- 14.Nangle MR, Cotter MA, Cameron NE. Protein kinase C beta inhibition and aorta and corpus cavernosum function in streptozotocin-diabetic mice. Eur J Pharmacol. 2003;475:99–106. doi: 10.1016/s0014-2999(03)02113-7. [DOI] [PubMed] [Google Scholar]
- 15.Cotter MA, Jack AM, Cameron NE. Effects of the protein kinase C beta inhibitor LY333531 on neural and vascular function in rats with streptozotocin-induced diabetes. Clin Sci (Lond) 2002;103:311–21. doi: 10.1042/cs1030311. [DOI] [PubMed] [Google Scholar]
- 16.Maeda T, Lloyd RV. Protein kinase C activity and messenger RNA modulation by estrogen in normal and neoplastic rat pituitary tissue. Lab Invest. 1993;68:472–80. [PubMed] [Google Scholar]
- 17.Karp G, Maissel A, Livneh E. Hormonal regulation of PKC: Estrogen up-regulates PKCeta expression in estrogen-responsive breast cancer cells. Cancer Lett. 2006 doi: 10.1016/j.canlet.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 18.Peters CA, Cutler RE, Maizels ET, Robertson MC, Shiu RP, Fields P, et al. Regulation of PKC delta expression by estrogen and rat placental lactogen-1 in luteinized rat ovarian granulosa cells. Mol Cell Endocrinol. 2000;162:181–91. doi: 10.1016/s0303-7207(00)00193-3. [DOI] [PubMed] [Google Scholar]
- 19.Eto M, Barandier C, Rathgeb L, Kozai T, Joch H, Yang Z, et al. Thrombin suppresses endothelial nitric oxide synthase and upregulates endothelin-converting enzyme-1 expression by distinct pathways: role of Rho/ROCK and mitogen-activated protein kinase. Circ Res. 2001;89:583–90. doi: 10.1161/hh1901.097084. [DOI] [PubMed] [Google Scholar]
- 20.Ming XF, Viswambharan H, Barandier C, Ruffieux J, Kaibuchi K, Rusconi S, et al. Rho GTPase/Rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Mol Cell Biol. 2002;22:8467–77. doi: 10.1128/MCB.22.24.8467-8477.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilkinson SE, Parker PJ, Nixon JS. Isoenzyme specificity of bisindolylmaleimides, selective inhibitors of protein kinase C. Biochem J. 1993;294(Pt 2):335–7. doi: 10.1042/bj2940335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, et al. Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun. 1994;199:93–8. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- 23.Lagaud GJ, Masih-Khan E, Kai S, van Breemen C, Dube GP. Influence of type II diabetes on arterial tone and endothelial function in murine mesenteric resistance arteries. J Vasc Res. 2001;38:578–89. doi: 10.1159/000051094. [DOI] [PubMed] [Google Scholar]
- 24.Zhang YM, Bo J, Taffet GE, Chang J, Shi J, Reddy AK, et al. Targeted deletion of ROCK1 protects the heart against pressure overload by inhibiting reactive fibrosis. Faseb J. 2006;20:916–25. doi: 10.1096/fj.05-5129com. [DOI] [PubMed] [Google Scholar]
- 25.Giugliano D, Marfella R, Coppola L, Verrazzo G, Acampora R, Giunta R, et al. Vascular effects of acute hyperglycemia in humans are reversed by L-arginine. Evidence for reduced availability of nitric oxide during hyperglycemia. Circulation. 1997;95:1783–90. doi: 10.1161/01.cir.95.7.1783. [DOI] [PubMed] [Google Scholar]
- 26.Wang SX, Xiong XM, Song T, Liu LY. Protective effects of cariporide on endothelial dysfunction induced by high glucose. Acta Pharmacol Sin. 2005;26:329–33. doi: 10.1111/j.1745-7254.2005.00042.x. [DOI] [PubMed] [Google Scholar]
- 27.Bohlen HG, Nase GP. Obesity lowers hyperglycemic threshold for impaired in vivo endothelial nitric oxide function. Am J Physiol Heart Circ Physiol. 2002;283:H391–7. doi: 10.1152/ajpheart.00019.2002. [DOI] [PubMed] [Google Scholar]
- 28.Lott ME, Hogeman C, Herr M, Gabbay R, Sinoway LI. Effects of an Oral Glucose Tolerance Test on the Myogenic Response in Healthy Individuals. Am J Physiol Heart Circ Physiol. 2006 doi: 10.1152/ajpheart.00940.2005. [DOI] [PubMed] [Google Scholar]
- 29.Johnstone MT, Creager SJ, Scales KM, Cusco JA, Lee BK, Creager MA. Impaired endothelium-dependent vasodilation in patients with insulin-dependent diabetes mellitus. Circulation. 1993;88:2510–6. doi: 10.1161/01.cir.88.6.2510. [DOI] [PubMed] [Google Scholar]
- 30.Rahimian R, Dube GP, Toma W, Dos Santos N, McManus BM, van Breemen C. Raloxifene enhances nitric oxide release in rat aorta via increasing endothelial nitric oxide mRNA expression. Eur J Pharmacol. 2002;434:141–9. doi: 10.1016/s0014-2999(01)01546-1. [DOI] [PubMed] [Google Scholar]
- 31.Rahimian R, Laher I, Dube G, van Breemen C. Estrogen and selective estrogen receptor modulator LY117018 enhance release of nitric oxide in rat aorta. J Pharmacol Exp Ther. 1997;283:116–22. [PubMed] [Google Scholar]
- 32.Wolfrum S, Dendorfer A, Rikitake Y, Stalker TJ, Gong Y, Scalia R, et al. Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arterioscler Thromb Vasc Biol. 2004;24:1842–7. doi: 10.1161/01.ATV.0000142813.33538.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rikitake Y, Liao JK. Rho-kinase mediates hyperglycemia-induced plasminogen activator inhibitor-1 expression in vascular endothelial cells. Circulation. 2005;111:3261–8. doi: 10.1161/CIRCULATIONAHA.105.534024. [DOI] [PMC free article] [PubMed] [Google Scholar]