

Abstract
Background
Post-hemorrhagic shock mesenteric lymph (PSML) has been shown to activate pulmonary endothelial cells, and cause lung injury. Although multiple mediators may be involved, most of these effects are mediated by NF-κB activation. Degradation of the inhibitor of kappa B (IκB) is a key regulatory step in the activation of NF-κB. We therefore hypothesized that PSML would cause IκB degradation with subsequent NF-κB phosphorylation and nuclear translocation.
Methods
Mesenteric lymph was collected from male rats before shock and each hour after shock for up to 3 hours. (n=5) Buffer (control), buffer + 10% (v/v) lymph, or buffer + TNF (10ng/ml) was incubated with human pulmonary endothelial cells for 30 minutes and then lysed. Immunoblots of lysates were probed for IκB and phospho-p65. Immunohistochemistry was performed on cells grown on glass slides, and then treated as above with the 3rd PSML sample. Cells were fixed and then probed for p65. Statistical analysis was performed with student’s t-test and analysis of variance with significance set at p< 0.05.
Results
Western blots of cell lysates for IκB demonstrated a steady decrease in total IκB with each lymph sample. Phosphorylation of NF-κB, p65 component, steadily increased with each PSML sample, with a maximum reached during the 3rd PSML sample, which also significantly increased translocation of NF-κB to the nucleus.
Conclusion
Post-shock mesenteric lymph bioactivity is mediated by pathways which involved IκB degradation. These pathways offer novel targets for clinical intervention to prevent the distal organ injury caused by postinjury hemorrhagic shock.
Keywords: Mesenteric, Lymph, Shock, IκB, NF-κB
Introduction
Gut ischemia and reperfusion (I/R) is central to the pathogenesis of multiple organ failure after hemorrhagic shock. Although early work focused on the portal circulation as the conduit for biological mediators, more recently, mesenteric lymph has been identified as the mechanistic link between gut I/R and neutrophil-mediated acute lung injury.[1-3]
Both in vivo and in vitro work have focused on the role of neutrophils (polymorph nuclear leukocytes [PMNs]) in mediating lung injury after hemorrhagic shock. We have shown that in a rodent model of hemorrhagic shock, PMN adhesion molecule expression and PMN lung accumulation are diminished with lymph diversion.[4]. Further corroborative work in vitro has shown that isolated PMNs exposed to post-shock mesenteric lymph (PSML) increase surface adhesion molecule expression and increase priming for superoxide production.[5]
Although PMNs play a key role in lung injury [6], activation of the pulmonary vascular endothelium is an essential component.[6] Deitch et al. have found that human umbilical vein endothelium (HUVECs) exposed to PSML undergoes cytotoxicitiy and increased permeability.[2, 7] We have shown that pulmonary microvascular endothelial cells (HMVECs) profoundly up regulate surface expression of ICAM after exposure to PSML.[8]
This increase in ICAM-1 has prompted us to examine the role of NF-κB mediated pathways, since NF-κB is a key regulator of the ICAM-1 gene as well as several other pro-inflammatory genes.[9] Components of the active NF-κB are sequestered in the cytosol by specific proteins of the IκB family.[10, 11] Upon phosphorlyation of IκB, NF-κB components are released and IκB is degraded.[12] Liberation of the NF-κB components allows the molecules to translocate to the nucleus, where they dimerize and initiate transcription of key genes, including ICAM-1.[9, 12] Recently, phosphorylation of NF-κB, specifically phosphorylation at the serine 536 site of p65 RelA, by IκK has emerged as a novel mechanism of transcriptional activation.[13]
We therefore hypothesized that endothelial activation by PSML would involve degradation of IκB with subsequent phosphorylation and translocation of NF-κB to the nucleus in HMVECs.
Methods
Animals
Animal experiments were performed under a protocol approved by the Institutional Animal Care and Use Committee at Denver Health Medical Center, Denver, Colorado. Adult male Sprague-Dawley rats (Harlan Laboratories, Madison, WI) weighing 320-370 g were housed under barrier-sustained conditions and allowed free access to chow and water before use. All animals were maintained in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals.
Materials
All materials were purchased from Sigma-Aldrich Corp. (St. Louis, Mo), unless otherwise specified. All tubing was obtained from Fisher Scientific. Pentobarbital sodium was purchased from Abbott Labs (North Chicago, IL) and heparin from American Pharmaceutical Partner Inc. (Schaumburg, IL). Antibodies against IκBα (C-21) and p65 (C-20) were purchased from Santa Cruz Biotechnology, Inc. Antibodies against actin (AC-40) were purchased from Abcam, Inc. Antibodies against phosph-S536-p65 (93H1) were obtained from Cell Signaling Technology, Inc.
Hemorrhagic shock model
Rats were anesthetized with pentobarbital 50 mg/kg intraperitoneally. The right femoral artery and vein were cannulated with PE-50 tubing. Blood pressure was continuously monitored with a Pro-Paq device (Protocol Systems, Beaverton, Ore). Core body temperature was measured with a rectal thermometer and maintained at 37°C with a heating lamp. A midline laparotomy was performed to expose the common mesenteric lymphatic duct, which was cannulated with 0.02-in silicone tubing. The tubing was secured with a 7-0 prolene suture and exteriorized via a right flank stab wound. The abdomen and groin were then closed with 4-0 monofilament nylons.
Lymph was collected from rats for 60 minutes before (preshock), during nonlethal hemorrhagic shock (mean arterial pressure 30 mm Hg × 40 min), and for each hour after shock up to 3 hours (1st hour PSML, 2nd hour PSML, 3rd hour PSML). Animals were resuscitated during the initial 2 hours after shock with half shed blood volume + 4x shed blood volume as normal saline. Lymph was centrifuged at 6,000 g × 10 minutes at 4°C to remove all cellular components. The supernatant was removed, aliquoted and snap frozen for subsequent analysis.
Cell Culture
Human microvascular endothelial cells (HMVECs) were obtained from Cambrex Bio Science Inc. (Rockland, ME) and cultured in endothelial growth medium. (Cambrex Bio Science, Inc.) Cells were grown to near confluence at 37°C in 5% CO2. After achieving near confluence, cells were incubated with medium, TNF (10ng/ml), or lymph (10% v/v) for 30 minutes at 37°C. To minimize animal-to-animal variability, all post-shock mesenteric lymph samples were compared to the same animal’s preshock sample. Supernatants were removed, and cells were lysed in digestion buffer containing SDS. Cell lysates were heated at 100 °C for 10 minutes, aliquoted and frozen for immunoblot analysis.
Immunoblot Staining
Equal amounts of whole cell lysates were loaded into 8-16% acrylamide gels (Pierce Biotechnology, Inc.), fractioned with SDS-PAGE, and then electrotransferred onto a nitrocellulose membrane (Bio-Rad Laboratories, Inc.). Membranes were blocked with 5% nonfat milk solution and immunoprobed. Bound antibodies were detected with enhanced chemoluminescence detection system (Pierce Biotechnology, Inc.). Densitometry was performed using Image J software program (National Institutes of Health, Bethesda, MD). All plots were normalized to actin and results expressed as a percent of control.
Immunofluorescent microscopy
HMVECs were grown on Lab-Tek Chamber Slides (Nalgene Nunc International, Rochester, NY, USA) and treated with buffer, TNF (ng/ml) or 3rd hour PSML (10% v/v) for 45 minutes. The slides were fixed and permeabilized with 70% acetone / 30% methanol solution at -20°C. Non-specific antigen sites were blocked with 10% donkey serum (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) in PBS for 60 minutes at room temperature. Slides were then probed with rabbit polyclonal anti-p65 antibody (C-20, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) in 1%BSA/PBS for 1 hour. Negative control slides were incubated with isotype (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Following three PBS washes, the slides were incubated for 1hour at room temperature with donkey anti-rabbit Cy3-conjugated antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) and bis-benzimide nuclear stain (Sigma-Aldrich, St. Louis, MO, USA).
Images were acquired with a Zeiss Axiovert fitted with a Cooke CCD SensiCam using a Chroma Sedat with single excitation and emission filters and a multiple bandpass dichroic and Sutter filter control. The two channel images were then digitally processed using Intelligent Imaging Innovations Slidebook 4.1 software (Intelligent Imaging Innovations, Inc., Denver, CO). Nuclear stains were masked to measure the relative p65 mean fluorescence intensity (MFI) within the nuclei. The MFI of at least 70 cells from each experimental group were analyzed by ANOVA using JMP 5.0.1 software. Statistical significance was accepted as p less than 0.001.
Statistical Analysis
Data were analyzed by repeated measures ANOVA using Proc GLM, followed by the Tukey’s multiple comparisons test (SAS Tutorials for Statistical Data Analysis at http://www.stattutorials.com/SAS/). Contrasts were used to test for linear trends between hours pre/post shock and IκB degradation as well as hours pre/post shock and the phosphorylation state of the p65 component. A p-value of less than 0.05 is considered statistically significant.
Results
IκB Degradation
In the first set of experiments, PSML was incubated with HMVECs to determine its effects on IκB degradation. (Figure 1.) The IκB degradation level varied significantly with the different PSML samples (Repeated measures ANOVA p=0.0092); statistical significant differences were detected between the pre-shock levels and the 2nd, 3rd and combined post-shock PSML sample (Tukey’s multiple comparison test, p<0.05). The 3rd postshock hour demonstrated the maximal degradation of IκB over control levels with approximately 40% reduction in IκB. Interestingly, combining each hour of lymph and then adding this combined PSML (“Postshock”) to HMVECs showed a similar amount of IκB degradation as the 3rd postshock hour. A significant linear trend was detected between time (pre, post 1, post 2, post 3 hours) and the IκB degradation level (contrast for linear trend, p=0.0051).
Figure 1. PSML induces IκB degradation in HMVECs.

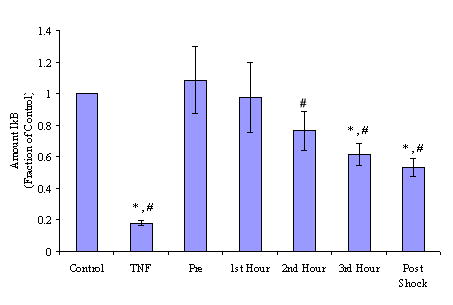
Mesenteric lymph was collected from rats during each hour before and after shock. Lymph was incubated with HMVECs and cells were lysed to determine activation of IκB via IκB degradation. Data are expressed as mean +/- SEM. * p < 0.05 vs. control group. # p < 0.05 vs. preshock group. All PSML induces IκB degradation with maximal degradation occurring after stimulation with lymph from the 3rd hour and combined PSML. a) Representative blot. b) Densitometry analysis of multiple blots.
Phosphorylation of NF-κB
Phosphorylation of NF-κB at serine 536 by IκK is a novel pathway. Phosphorylation at Ser536 profoundly increases its translocation and activation.[13] We therefore measured the phosphorylation of NF-κB by assessing the phosphorylation state of the p65 component. (Figure 2.) TNF stimulation demonstrated a 2-fold increase in p65 phosphorylation over control cells. The p65 phosphorylation level varied significantly with the different PSML samples (Repeated measures ANOVA p=0.0057). The maximal phosphorylation of p65 occurred after stimulation with the 2nd and 3rd hours of PSML (Tukey’s multiple comparison test, p<0.05). In addition, the phosphorylation of p65 observed with the combined PSML sample was significantly higher than the level observed with pre-shock, and the 1st, 2nd and 3rd post-shock hour samples. A significant linear trend was detected between time (pre, 1st, 2nd and 3rd post-shock hour samples) and the phosphorylation of p65 level (contrast for linear trend, p=0.0005).
Figure 2. PSML induces NF-κB phosphorylation.

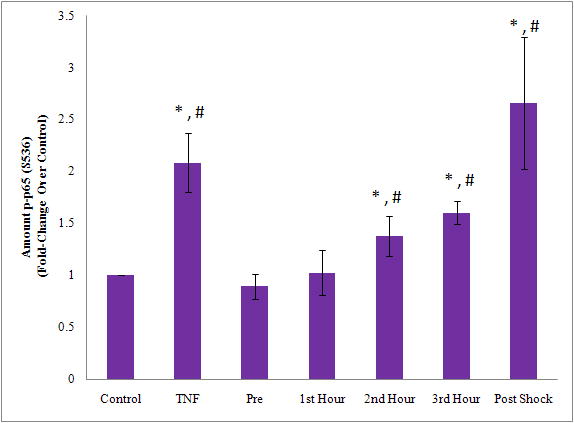
Lymph was incubated with HMVECs for activation. Cells were subsequently lysed and immunoblotted for phospho-p65. * p < 0.05 vs. control group. # p < 0.05 vs. preshock group. PSML increased phosphorylation of NF-κB with maximal phosphorylation occurring after stimulation with 3rd hour PSML and combined PSML. a) Representative blot. b) Densitometry analysis of multiple blots.
NF-κB Translocation
Functional activity of NF-κB is dependent on translocation of NF-κB to the nucleus of cells.[11] Using IHC, we were able to detect the amount of NF-κB in different compartments of the cells. (Figure 3a and 3b.) We chose to focus solely on the 3rd hour PSML hour.
Figure 3. PSML induces nuclear translocation of NF-κB.

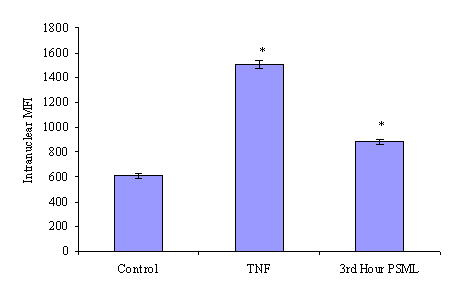
The 3rd hour of PSML was incubated with HMVECs and probed for p65. (a) Immunoflourescence of NF-κB (RelA/p65, red) nuclear translocation. The nuclear stain is removed in the bottom row. (b) Histogram of p65 nuclear mean fluorescence intensity. * p < 0.05 vs. control group. PSML increased translocation of p65 to the nucleus in HMVECs by 30%.
Control cells demonstrated a nuclear mean fluorescence intensity for p65 of 600. TNF stimulated cell demonstrated a marked increase in p65 translocation with an MFI of approximately 1500. The 3rd hour PSML at 10% also induced a significant translocation of p65 to the nucleus, reaching an MFI of approximately 880. This represents a 45% increase in nuclear p65 over control cells.
Discussion
The gut has been invoked as the mechanistic link between post-traumatic shock and ensuing MOF, which remains a major cause of morbidity and mortality after major trauma. Work from the Deitch lab [1, 7, 15-19] as well as work from our group [4, 5, 8, 20, 21] has confirmed that PSML is the mechanistic link between gut I/R and MOF.
Our investigation has focused primarily on the effects of PSML on neutrophils and endothelium. In vitro, PSML primes neutrophils for an increased physiologic response to a second activator.[5, 20] PSML also increases PMN cell surface expression of the adhesion molecule CD11b.[3] Although PMN priming is a critical component in development of lung injury, microvascular endothelial cell activation plays an equally necessary role.[6, 22] To that end, PSML also has a diverse array of proinflammatory effects on endothelial cells. The Deitch lab has demonstrated that in umbilical vein endothelial cells, PSML induces cytotoxicity as well as increased permeability.[2, 7, 19] However, repeated evidence has shown that umbilical vein endothelial physiology is quite different than pulmonary endothelial cells.[23] To that end, endothelial work from our lab has focused on HMVECs. We have previously demonstrated that PSML increases HMVEC ICAM-1 expression, increases in vitro PMN-HMVEC adhesion and increases PMN-mediated HMVEC killing.[8]
Despite multiple attempts to identify the relevant mediators, isolation and identification of the actual mediator remains elusive. Positive results to date suggest that the bioactivity of PSML is likely due to either multiple mediators or an interaction of proteins and lipids. [16][20]. Since the specific bioactive substance remains elusive, the next logical mechanistic targeting site for therapy could be obtained by examining signaling pathways.
Prior work from our lab has demonstrated that PSML in HMVECs induces ICAM-1 expression.[8] In endothelial cells, ICAM-1 is an NF-κB driven gene.[9] We therefore conjectured that signaling would converge onto NK-κB through the IκB proteins. Work from Sasaki et al. has demonstrated that phosphorylation of specific sites of the p50 and p65 component lead to changes in transcriptional activity of NF-κB driven proteins.[13] They have also demonstrated that specific IκK complexes not only phosphorylate IκBs, but also function to specifically phosphorylate NF-κB. The serine 536 residue, for example, is specifically phosphorylated by Iκkα, and is independent of IκB activity.
In this study, we have demonstrated that PSML profoundly induces degradation of IκB. This degradation is followed by translocation of free NF-κB to the nucleus with subsequent gene transcriptional activation. We therefore probed further evidence of IκK activation and found that phosphorylation of NF-κB at the serine 536 site is also enhanced after PSML. This suggests that the signaling pathway of PSML is an noncanonical NF-κB pathway.
In this study, the individual PSML lymph time point with the greatest bioactivity was the 3rd hour. This is congruent with prior work demonstrating that the 3rd postshock hour primed PMNs for an increased response to a second stimulus.[5] This is also congruent with prior work in HUVECs demonstrating the greatest decrease in cell viability after application of 2nd and 3rd hour of PSML.[17] Of interest, combining samples of PSML from each hour has greater bioactivity than any single time point alone. This suggests that there are different mediators acting as the primary inflammatory signal at different time point. Although the specific mediators have not been fully elucidated, this would suggest that different mediators are acting as the primary pro-inflammatory signal during different time points. Previous work has demonstrated that both proteins and lipids act as bioactive mediators in PSML.[16, 20, 21] The tremendous increase seen in the phosphorylation of NF-κB with the combined PSML suggests that there may be a synergistic action by combing lymph samples, such as combining protein carriers for stabilization of the lipid component.
In summary, this study demonstrates that hemorrhagic shock provokes the release of bioactive agents in PSML, which induce degradation of IκB, phosphorylation of RelA/p65 and NF-κB translocation. This study narrows the specific receptors down to those receptors acting via IκK as mediators of the cytotoxicity of lymph.
Acknowledgments
We would like to thank Dr. Angela Sauia, MD, PhD, for her gracious assistance in the statistical analysis of the results.
This study was supported in part by the National Institutes of Health (grant T32GM008315, P50GM49222 and U54GM62119).
Footnotes
Presented at the 2nd Annual Academic Surgical Congress, February 9th, 2007.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Xu D-Z, et al. Trauma-hemorrhagic shock-induced up-regulation of endothelial cell adhesion molecules is blunted by mesenteric lymph duct ligation. Ctitical Care Medicine. 2004;32(3):760–765. doi: 10.1097/01.ccm.0000114815.88622.9d. [DOI] [PubMed] [Google Scholar]
- 2.Magnotti LJ, et al. Gut-Derived Mesenteric Lymph but not Portal Blood Increases Endothelial Cell Permeability and Promotes Lung Injury After Hemorrhagic Shock. Annals of Surgery. 1998;228(4):518–527. doi: 10.1097/00000658-199810000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caruso JM, et al. Factors in Intestinal Lymph after Shock Increase Neutrophil Adhesion Molecule Expression and Pulmonary Leukosequestration. The Journal of Trauma Injury, Infection, and Critical Care. 2003;55(4):727–733. doi: 10.1097/01.TA.0000037410.85492.77. [DOI] [PubMed] [Google Scholar]
- 4.Gonzales RJ, et al. Mesenteric Lymph is Responsible for Post-Hemorrhagic Shock Systemic Neutrophil Priming. The Journal of Trauma Injury, Infection, and Critical Care. 2001;51:1069–1072. doi: 10.1097/00005373-200112000-00008. [DOI] [PubMed] [Google Scholar]
- 5.Masuno T, et al. Bioactivity of postshcok mesenteric lymph depends on the depth and duration of hemorrhagic shock. Shock. 2006;26(3):285–289. doi: 10.1097/01.shk.0000223132.72135.52. [DOI] [PubMed] [Google Scholar]
- 6.Ware LB. Pathophysiologcy of Acute Lung Injury and the Acute Respiratory Distress Syndrome. Sermin Respir Crit Care Med. 2006;27:337–349. doi: 10.1055/s-2006-948288. [DOI] [PubMed] [Google Scholar]
- 7.Lu Q, et al. Hemorrhagic shock induces endothelial cell apoptosis, which is mediatedby factors contained in mesenteric lymph. Critical Care Medicine. 2004;32:2464–2470. doi: 10.1097/01.ccm.0000147833.51214.03. [DOI] [PubMed] [Google Scholar]
- 8.Gonzales RJ, et al. Post-hemorrhagic shock mesenteric lymph activates human pulmonary microvascular endothelium for in vitro neutrophil-mediated injury: the role of intercellular adhesion molecule-1. The Journal of Trauma Injury, Infection, and Critical Care. 2003;54(2):219–223. doi: 10.1097/01.TA.0000047807.12644.95. [DOI] [PubMed] [Google Scholar]
- 9.Ledebur HC, Parks TP. Transcriptional Regulation of the Intercellular Adhesion Molecule-1 Gene by Inflammatory Cytokines in Human Endothelial Cells. The Journal of Biological Chemistry. 1995;270(2):933–943. doi: 10.1074/jbc.270.2.933. [DOI] [PubMed] [Google Scholar]
- 10.Ghosh S, May MJ, Kopp E. NF-kB and REL Proteins: Evolutionarily Conserved Mediators of Immune Responses. Annual Review of Immunology. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 11.Hayden MS, Ghosh S. Signaling to NF-kB. Genes & Development. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 12.Moynagh PN. The NF-κB pathway. Journal of Cell Science. 2005;118(20):4389–4392. doi: 10.1242/jcs.02579. [DOI] [PubMed] [Google Scholar]
- 13.Sasaki C, et al. Phosphorylation of RelA/p65 on S536 Defines an IkB-a-independent NF-kB pathway. Journal of Biological Chemistry. 2001;280(41):34538–34547. doi: 10.1074/jbc.M504943200. [DOI] [PubMed] [Google Scholar]
- 14.Deitch EA. Role of the gut lymphatic system in multiple organ failure. Current Opinion in Critical Care. 2001;7:92–98. doi: 10.1097/00075198-200104000-00007. [DOI] [PubMed] [Google Scholar]
- 15.Zaets S, et al. Mesenteric Lymph Duct Ligation Prevents Shock-Induced RBC Deformability and Shape Changes. Journal of Surgical Research. 2003;109:51–56. doi: 10.1016/s0022-4804(02)00024-0. [DOI] [PubMed] [Google Scholar]
- 16.Dayal SD, et al. Shock mesenteric lymph-induced rat polymorphonuclear neutrophil activation and endothelial cell injury is mediated by aqueous factors. The Journal of Trauma Injury, Infection, and Critical Care. 2002;52:1048–1055. doi: 10.1097/00005373-200206000-00005. [DOI] [PubMed] [Google Scholar]
- 17.Davidson MT, et al. A study of the biologic activity of trauma-hemorrhagic shock mesenteric lymph over time and the relative role of cytokines. Surgery. 2004;136(1):32–41. doi: 10.1016/j.surg.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 18.Dayal SD, et al. Trauma/Hemorrhagic Shock Mesenteric Lymph Upregulates Adhesion Molecule Expression and IL-6 Production in Human Umbilical Vein Endothelial Cells. Shock. 2002;17(6):491–495. doi: 10.1097/00024382-200206000-00009. [DOI] [PubMed] [Google Scholar]
- 19.Davidson MT, et al. Trauma-Hemorrhagic Shock Mesenteric Lymph Induces Endothelial Apoptosis That Involves Both Caspase-Dependent and Caspase-Independent Mechanisms. Annals of Surgery. 2004;240(1):123–131. doi: 10.1097/01.sla.0000129341.94219.cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gonzales RJ, et al. Phospholipase A2-derived neutral lipids from posthemorrhagic shock mesenteric lymph prime the neutrophil oxidative burst. Surgery. 2001;130:192–203. doi: 10.1067/msy.2001.115824. [DOI] [PubMed] [Google Scholar]
- 21.Sarin EL, et al. Systemic neutrohpil priming by lipid mediators in post-shock mesenteric lymph exists across species. The Journal of Trauma Injury, Infection, and Critical Care. 2004;57(5):950–954. doi: 10.1097/01.ta.0000149493.95859.6c. [DOI] [PubMed] [Google Scholar]
- 22.Silliman CC, McLaughlin NJ. Transfusion-related acute lung injury. Blood Reviews. 2006;20:139–159. doi: 10.1016/j.blre.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Choi J, et al. T Lymphocyte-Endothelial Cell Interactions. Annu Rev Immunol. 2004;22:683–709. doi: 10.1146/annurev.immunol.22.012703.104639. [DOI] [PubMed] [Google Scholar]
- 24.Bates PW, Miyamoto S. Expanded Nuclear Roles for IκBs. Sci STKE. 2004;254:pe48. doi: 10.1126/stke.2542004pe48. [DOI] [PubMed] [Google Scholar]