

Abstract
Levodopa is the most commonly prescribed drug for Parkinson's disease (PD). Although levodopa improves PD symptoms in the initial stages of the disease, its long-term use is limited by development of side effects, including abnormal involuntary movements (dyskinesias) and psychiatric complications. The endocannabinoid system is emerging as an important modulator of basal ganglia functions and its pharmacologic manipulation represents a promising therapy to alleviate levodopa-induced dyskinesias. Rats with 6-OHDA lesions that are chronically treated with levodopa develop increasingly severe axial, limb, locomotor and oro-facial abnormal involuntary movements (AIMs). Administration of the cannabinoid agonist WIN 55,212−2 attenuated levodopa-induced axial, limb and oral AIMs dose-dependently via a CB1–mediated mechanism, whereas it had no effect on locomotive AIMs. By contrast, systemic administration of URB597, a potent FAAH inhibitor, did not affect AIMs scoring despite its ability to increase anandamide concentration throughout the basal ganglia. Unlike WIN, anandamide can also bind and activate transient receptor potential vanilloid type-1 (TRPV1) receptors, which have been implicated in the modulation of dopamine transmission in the basal ganglia. Interestingly, URB597 significantly decreased all AIMs subtypes only if co-administered with the TRPV1 antagonist capsazepine. Our data indicate that pharmacological blockade of TRPV1 receptors unmasks the anti-dyskinetic effects of FAAH inhibitors and that CB1 and TRPV1 receptors play opposite roles in levodopa-induced dyskinesias.
Keywords: anandamide, endocannabinoid, vanilloid, levodopa, dyskinesias, 6-OHDA, basal ganglia
INTRODUCTION
Since its introduction, L-3,4-dihydroxyphenilalanine (levodopa) has remained the mainstay treatment for PD. Although levodopa alleviates parkinsonsian symptoms, its long-term administration is accompanied by fluctuations in its duration of action and disabling motor complications (dyskinesias) (Obeso et al., 2004). Levodopa-induced dyskinesias (LID) are characterized by choreiform and dystonic movements and are classified according to their temporal profile as “peak-dose” (occurring at peak levodopa concentration in the brain), “diphasic” (at the beginning and end of dosing) and “off” dyskinesias (when levodopa concentration is low) (Fahn, 2000; Nutt et al., 1992). Multiple factors have been shown to contribute to development of LID, including pulsatile stimulation of postsynaptic dopamine receptors (Westin et al., 2006), maladaptive changes in synaptic plasticity (Cenci and Lundblad, 2006; Picconi et al., 2003), neurochemical disturbances and fluctuations of levodopa/dopamine levels (Carta et al., 2006; de la Fuente-Fernandez et al., 2004; Meissner et al., 2006) and altered trafficking of NMDA receptor subunits (Fiorentini et al., 2006; Gardoni et al., 2006). In rodents, LID can be modeled via intracerebral injection of the neurotoxin 6-OHDA - which damages the nigro-striatal pathway - followed by chronic administration of low doses of levodopa, which causes characteristic AIMs and dyskinesias-associated cellular responses (Lundblad et al., 2004). This model has been pharmacologically validated and represents a cost-efficient alternative to non-human primates for screening drugs with potential anti-dyskinetic properties (Lundblad et al., 2002).
Experimental evidence points to the endocannabinoid system as a novel pharmacological target to treat levodopa-associated motor disturbances (Ferrer et al., 2003; Sieradzan et al., 2001; van der Stelt et al., 2005). This system consists of a family of signaling lipids (endocannabinoids) and their allied cannabinoid receptors (Mackie, 2005; Piomelli et al., 2000). In particular, CB1 cannabinoid receptors are highly expressed in brain areas regulating motor functions, including the basal ganglia, cerebellum and sensori-motor cortex (Mackie, 2005). In rodents, activation of dopamine receptors is accompanied by release of the endocannabinoid anandamide (AEA) throughout the basal ganglia (Ferrer et al., 2003; Giuffrida et al., 1999). Dopamine-dependent AEA elevation may serve as an inhibitory feedback to counter dopamine-mediated motor behaviors (Giuffrida et al., 1999; Beltramo et al., 2000) and is disrupted after damaging the nigro-striatal pathway with 6-OHDA (Ferrer et al., 2003), suggesting that alterations in endocannabinoid transmission may affect the dopamine-endocannabinoid crosstalk and eventually result in motor disturbances. In keeping with this hypothesis, administration of the cannabinoid agonist WIN55212−2 (WIN) to rats with 6-OHDA lesions ameliorates levodopa-induced oral AIMs via a CB1-dependent mechanism (Ferrer et al., 2003).
In addition to CB1 receptors, AEA can bind, although with low affinity, to the ionotropic transient receptor potential vanilloid subtype 1 (TRPV1) (Caterina et al., 1997; Ross, 2003). These receptors are co-expressed with CB1 receptors in the striatum and globus pallidus (GP) (Cristino et al., 2006; Toth et al., 2005), and functional interactions between these two receptor types have been reported in vitro (Hermann et al., 2003), and in vivo (Kim et al., 2005).
In this study, we investigated the effects of the cannabinoid agonist WIN on levodopa-induced AIMs in rats with 6-OHDA lesions and tested whether AEA elevation - via pharmacological blockade of its catabolism - produced anti-dyskinetic effects similar to those observed with WIN via CB1- and/or TRPV1-mediated mechanisms.
MATERIALS and METHODS
Chemicals
Fatty acyl chlorides (5,8,11,14-eicosatetraenoylchloride, hexadecanoylchloride and 9-cis-octadecenoylchloride) were from Nu-Check Prep (Elysian, MN). [2-H4]-labeled ethanolamine (98% isotopic atom enrichment), Cambridge Isotope Laboratories (Andover, MA). [2-H5]-labeled 2-AG (98% isotopic atom enrichment), AM251 and URB597 from Cayman Chemical (Ann Arbor, MI). N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) from Supelco (Bellefonte, PA). Halothane from Halocarbon (River Edge, NJ). All organic solvents from Honeywell/Burdick and Jackson (Muskegon, MI). Desipramine hydrochloride, levodopa methyl ester, 6-hydroxydopamine (6-OHDA) hydrochloride and S-(-) carbidopa from Sigma Chemicals Co. (St. Louis, MO); WIN 55−212,2 mesylate and capsazepine from Tocris bioscience, (Ellisville, MI).
Animals and 6-OHDA lesion
Animal care and experiments were conducted in accordance with the National Institutes of Health “Guide for the Care and Use of Laboratory Animals” and approved by the Institutional Animal Care and Use Committee of the University of Texas Health Science Center at San Antonio.
Male Wistar rats (225−250 g; Charles River Laboratories, Wilmington, MA) were housed on a 12-h dark-light cycle, at 22±1 °C with food and water available ad libitum. Animals were habituated to the housing conditions for 1 week before the experiments.
DA-denervating lesions were performed by unilateral injection of 6-OHDA into the left medial forebrain bundle (MFB) as previously reported (Lundblad et al., 2002). Briefly, after intraperitoneal (i.p.) administration of desipramine (25mg/kg, 30 min before surgery), rats were anesthetized with an injection of a cocktail (0.85 ml/kg, i.p.) containing ketamine (100mg/ml), xylazine (100mg/ml) and acepromazine (10mg/ml) in saline solution and positioned in a stereotaxic frame (Kopf Instruments, Tujunga, CA). 6-OHDA (4 μg/μl) was dissolved in 0.2% ascorbate saline. The 6-OHDA solution (2 μl) or a corresponding volume of saline (sham lesion) were injected into the left MFB at a flow rate of 0.5 μl/min using a 10 μl Hamilton microsyringe with a 30-gauge needle at the following coordinates: AP −4.3, ML +1.6, DV −8.3 tooth bar −2.4 (relative to bregma and midline, in mm) (Paxinos and Watson, 1998). Two weeks after the lesion, the rats were screened for apomorphine-induced (0.5 mg/kg, s.c.) contralateral rotation to assess the efficacy of the lesion. Net contralateral turns were calculated by subtracting the number of ipsilateral from contralateral rotations. Only rats displaying more than 300 rotations per 30 min (corresponding to about 90% depletion of tyrosine hydroxylase (TH) positive neurons in the SNc (Ferrer et al., 2003) were included in the study. Two weeks after the apomorphine challenge, animals were treated with a daily injection of levodopa (6 mg/kg, i.p.) plus carbidopa (12 mg/kg, i.p.) for up to 12 days. Chronic administration of this dose of levodopa has been shown to induce a gradual development of dyskinetic-like movements in the majority of rats with 6-OHDA lesions (Lundblad et al., 2002). About 37% of these animals did not develop dyskinesias and were not included in the study.
Behavioral Assays
Catalepsy
WIN-induced catalepsy was determined using the bar test. Catalepsy was assessed by placing both forepaws of the animal on a horizontal bar, 13 cm above the surface, and by measuring the latency to initiate movement using a cut-off time of 60 sec. On the day of the experiment, rats were administered vehicle (saline/PEG/Tween-80, 90/5/5 v/v/v, i.p.) or increasing doses of WIN (0.5, 1, 2.5 mg/kg, i.p.) 15 min before testing. Animals were tested every 30 min for a total time of 120 min following a Latin square design.
AIMs recordings
Abnormal Involuntary Movements (AIMs), consisting of axial, limb, locomotor and orofacial dyskinesia, were measured daily between 10:00 a.m. and 4:00 p.m., over a 2 h period after levodopa administration. The effects of cannabinoid drugs on levodopa-induced AIMs were measured at 60 and 100 minutes after levodopa injection. Animals were placed individually in a Plexiglas box and all observations were carried out for 10 min by a trained researcher blind to the treatment schedule. AIMs were scored on a severity scale ranging from 0 to 4 (modified from (Lundblad et al., 2002). For axial dyskinesias: 0 = absent; 1 = sustained deviation of the head and neck at 30°−60° angle for less than half of the observation time; 2 = as in 1 but for more than half of the observation time; 3 = sustained torsion of the upper trunk at 60°−90° angle for the entire observation time that can be interrupted by a strong noise; 4 = sustained torsion of the upper trunk at 90° angle (causing the rat to lose balance) for all the observation time and that cannot be interrupted by a strong noise. For limb dyskinesias: 0 = absent; 1 = small or low amplitude movements around a fixed position or visible translocation of the distal limb for less than half of the observation time; 2 = low amplitude movements accompanied by notable translocation of the whole limb for more than half of the observation time; 3 = notable translocation and vigorous movement of the whole limb for the entire observation time that can be interrupted by a strong noise; 4 = vigorous limb movements of high amplitude and speed with conspicuous contraction of proximal limb and extensor muscles for the entire observation time that cannot be interrupted by a strong noise. For oral dyskinesias: 0 = absent; 1 = chewing movements present for less than half of the observation time; 2 = chewing movements present for more than half of the observation time; 3 = chewing movements as described for score 2 and accompanied by tongue protrusions; 4 = oral movements present all the time and sometimes accompanied by self-biting. For locomotor dyskinesias: 0 = absent; 1 = rotational behavior present for less than half of the observation time; 2 = rotational behavior present for more than half of the observation time; 3 = rotational behavior present for the entire observation time that can be interrupted by a strong noise; 4 = rotational behavior present for the entire observation time that cannot be interrupted by a strong noise. All behaviors falling within two scoring categories were scored by assigning the lowest score + 0.5. For dose-response experiments, the AIM total score was calculated at day 10 of levodopa administration by adding the score of each AIM subtype (maximum value of 16).
The following drugs (or a combination of them) were tested on levodopa-induced AIMs: the cannabinoid agonist WIN, the FAAH inhibitor URB597, the CB1 antagonists AM251 and SR141716A and the selective TRPV1 antagonist capsazepine (Table 1). WIN, URB597 or capsazepine were administered from day 9 thru day 11 of levodopa treatment, 15 min before levodopa. On day 12, AM251 or SR141716A were given 15 min before co-administration of WIN (or URB597, or URB597+capsazepine) and levodopa.
Table 1.
| |
Property |
Timing (days of levodopa) |
|---|---|---|
| WIN 55,212-2 | Cannabinoid agonist | From day 9 to 12 |
| URB597 | FAAH inhibitor | From day 9 to 12 |
| AM251 | CB1 antagonist | Day 12 |
| SR141617A | CB1 antagonist | Day 12 |
| Capsazepine | TRPV1 antagonist | From day 9 to 12* |
in combination with URB597
In some experiments, rats were killed on day 12, 60 min after levodopa or URB597 or vehicle administration, and the brains rapidly collected to quantify endocannabinoids by GC/MS (see below).
Endocannabinoid Measurements
Animals were anesthetized with halothane and sacrificed by decapitation 60 min after the last injection of cannabinoid drugs or vehicle. Brains were rapidly collected, snap-frozen in cold 2-methylbutane (−50°C), placed on an ice-cold stainless steel mould (Roboz; Rockville, MD) and cut into 1 mm coronal slices using razor blades. Tissue punches (ranging between 5 and 20 mg) were excised from dorsal striatum, globus pallidus (GP) and substantia nigra (SN). The punches were thawed in 1 ml of methanol containing 50 pmol of [2H4]-anandamide and [2H5]-2-arachidonyl glycerol (2-AG) as internal standards. Samples were homogenized with a PowerGen 125 homogenizer (Fisher Scientific; Pittsburgh, PA) and lipids were extracted by adding chloroform and water to yield a methanol/chloroform/water ratio of 1:2:1 (v/v/v) and centrifuged at 800 × g for 5 min at room temperature to allow for phase separation. The lower organic layer (2 ml) was further purified by solid phase extraction using C18 Bond Elut cartridges (100 mg, Varian, Harbor City, CA) as previously described (Hardison et al., 2006). Endocannabinoid-containing fractions were derivitized with 30 μl of BSTFA at room temperature for 30 min, dried under nitrogen, resuspended in 5 μl of hexane and analyzed by GC/chemical ionization mass spectrometry with positive ion detection (PICI) using a TraceDSQ (Thermo Electron; San Jose, CA) equipped with an Rtx-5MS column (15 m × 0.25 mm; Restek; Bellefonte, PA). Quantification of endocannabinoids was carried out using a previously published isotope dilution procedure (Hardison et al., 2006).
Statistical analyses
Data on dyskinesias were expressed as median scores and analyzed using the Kruskal-Wallis test followed by Dunn's multiple comparison test (inter-group analyses) and Friedman test followed by Dunn's multiple comparison test (intra-group analyses). All other data were expressed as the mean±s.e.m. of n experiments. The significance of differences among groups was determined by ANOVA followed by Dunnett's or Bonferroni's test for multiple comparisons, as appropriate. The effects of levodopa and URB597 on endocannabinoid levels were analyzed by two-way ANOVA followed by Bonferroni's posthoc. Rotational responses induced by apomorphine were analyzed by Student's t test. The correlation between apomorphine-induced contralateral rotations and the severity of dyskinesias was calculated by use of a nonparametric tie-corrected Spearman's rank correlation.
The threshold for statistical significance was set at p<0.05.
RESULTS
WIN reduces levodopa-induced AIMs via activation of CB1 receptors
Rats with unilateral 6-OHDA lesion chronically treated with levodopa (6mg/kg, i.p. plus carbidopa, 12 mg/kg, i.p., 1 injection per day for 12 days) developed AIMs which increased over time. No AIMs were observed in either sham-operated or intact animals after chronic levodopa (data not shown). There was no correlation between the number of apomorphine-induced (0.5 mg/kg, s.c.) contralateral rotations, measured 2 weeks after the 6-OHDA lesion, and the severity of dyskinesias (expressed as AIM total score on day 8) (rs=0.0722, p=0.82, n=12).
The intra-day time course of WIN effect on levodopa-induced AIMs (sum of axial, limb, oro-facial and locomotive at day 10 of levodopa administration) showed that WIN had no significant effect within the first 50 min after its administration (Fig. 1A). Therefore, in all subsequent measurements, we chose 60 and 100 min as representative time points of WIN action.
Figure 1.
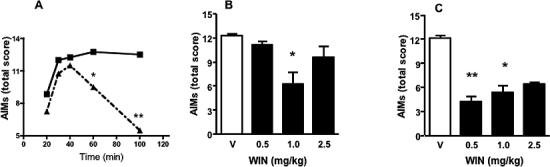
A. Time course of the effect of the cannabinoid agonist WIN (1 mg/kg, i.p., filled triangles) on levodopa-induced AIMs (total score at day 10 of levodopa administration). Filled squares, controls (levodopa + vehicle). B and C. Effects of sub-chronic i.p. administration (day 9−11) of WIN (filled bars) on levodopa-induced dyskinesias (AIMs total score at day 11) measured at 75 (B) and 115 min (C) after WIN injection (corresponding to 60 and 100 min after levodopa administration, respectively). Open bars, vehicle (5%PEG+5%Tween-80 in saline). *p<0.05; **p<0.01, compared to vehicle controls (Kruskal-Wallis followed by Dunn's multiple comparison test. Values represent median scores (n=6). The interquartile ranges have been omitted for clarity).
Systemic administration of increasing doses of WIN (from day 9 to day 11, n=6) significantly decreased levodopa-induced AIMs (measured as total score at day 11). This occurred with the dose of 1 mg/kg at 60 min after levodopa (p<0.05) (Fig.1B), and with the doses of 0.5 (p<0.01) and 1.0 mg/kg (p<0.05) at 100 min after levodopa (Fig. 1C). The highest dose of 2.5 mg/kg produced no anti-dyskinetic effect at 60 min post-levodopa (Fig. 1B), but decreased levodopa-induced AIMs at 100 min, although this trend did not reach statistical significance (Fig. 1C). The anti-dyskinetic effects of WIN were not due to a generalized suppression of motor activity, as none of the doses tested elicited catalepsy 1 h after administration [Time latency (sec): veh, 6.7± 2.5; WIN 0.5, 7.4±2.8; WIN 1.0, 13.0±3.6; WIN 2.5, 12.4±4.2 (repeated measures ANOVA followed by the Dunnett's test. p>0.05, mean±s.e.m., n=6). At 2 h, only the largest dose of WIN (2.5 mg/kg), which did not reduce levodopa-induced AIMs, produced a significant cataleptic effect (veh, 7.9±2.7; WIN 0.5, 6.7±1.9; WIN 1.0, 12.5±3.4; WIN 2.5, 20.9±5.6) (p<0.05, n=6). Furthermore, WIN (1 mg/kg, i.p.) did not alter the rotational response induced by apomorphine (0.05 mg/kg, s.c.) when administered two weeks after the 6-OHDA lesion (apomorphine, 95.8±19.2; apomorphine+WIN, 107±23.4; mean±s.e.m.; Student's t test, p>0.05, n=6).
To test the effect of WIN on each AIM subtype, we selected the lowest dose producing an anti-dyskinetic response (1 mg/kg, i.p.) at 60 and 100 min after levodopa injection. Administration of WIN from day 9 to day 11 significantly reduced axial, limb and oro-facial AIMs at both time points (Fig. 2 and 3), whereas it had no effect on locomotive AIMs (Fig. 2D and Fig. 3D). Post-hoc comparisons revealed a small but significant difference between levodopa- and levodopa+WIN-treated rats at days 10−11, 60 min after levodopa administration (Fig. 2A,B,C), and a more pronounced effect of WIN at days 9−11, 100 min after levodopa (Fig. 3A,B,C). Within-group analysis also revealed a significant effect of WIN at day 10 and 11 compared to day 8 of levodopa at both 60 and 100 min time points (p<0.05). The anti-dyskinetic effect of WIN was reversed by the CB1 antagonist AM251 (1 mg/kg, i.p., 20 min before WIN) on the last day of levodopa+WIN treatment (day 12) (Fig. 2 and 3). The same dose of AM251 had no effect on levodopa-induced AIMs when applied alone (data not shown).
Figure 2.
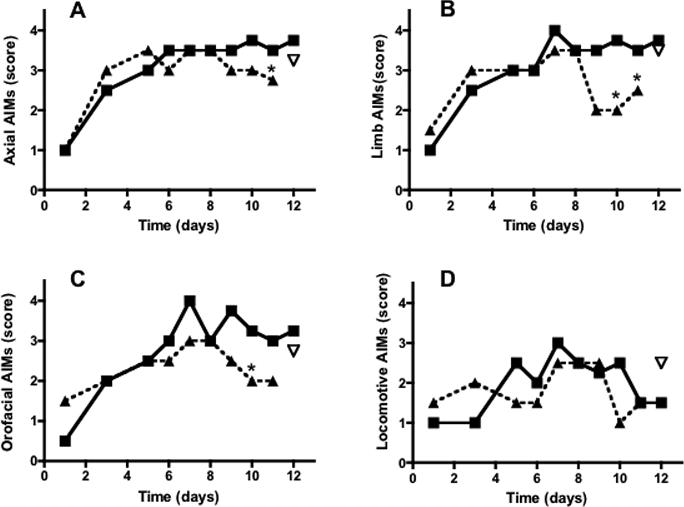
Time course of the effects of systemic administration of levodopa (filled squares, 6 mg/kg, 1 injection per day, n=6) in rats with unilateral 6-OHDA lesions on different types of abnormal involuntary movements (AIMs): Axial (A), Limb (B), orofacial (C) and locomotive (D), measured at 60 min after levodopa injection. A different group of rats (n=6), chronically treated with levodopa as described above, received the cannabinoid agonist WIN (triangles, 1 mg/kg., i.p.) from day 9 to day 11, 15 min before levodopa, whereas rats treated with levodopa only (filled squares) received an injection of vehicle (5%PEG+5%Tween-80 in saline) during the same days. The CB1 antagonist AM251 (inverted triangle, 1 mg/kg., i.p. 15 min before WIN) was administered on day 12 of levodopa treatment. *p<0.05 compared to rats treated with levodopa+vehicle (Kruskal-Wallis followed by Dunn's multiple comparison test. AM251 effect was analyzed by use of Friedman test followed by Dunn's multiple comparison test. Values represent median score. The interquartile ranges have been omitted for clarity).
Figure 3.
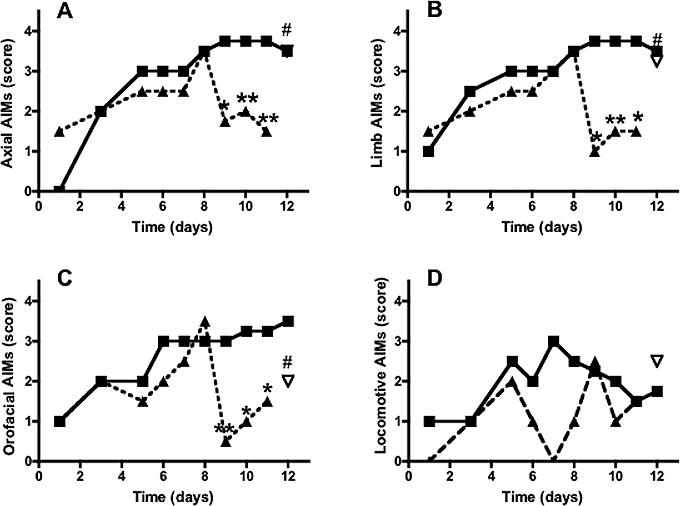
Time course of the effects of systemic administration of levodopa (filled squares, 6 mg/kg, 1 injection per day, n=6) and levodopa+WIN (triangles, 1 mg/kg, i.p.) to rats with unilateral 6-OHDA lesions (n=6) on AIMs (as described in figure 2) measured at 100 min after levodopa injection (115 min after WIN injection). The CB1 antagonist AM251 (inverted triangle, 1 mg/kg., i.p. 15 min before WIN) was administered on day 12 of levodopa treatment. *p<0.05, **p<0.01 compared to rats treated with levodopa+vehicle; #p<0.05 compared to rats treated with levodopa+WIN (Kruskal-Wallis followed by Dunn's multiple comparison test. AM251 effect was analyzed by use of Friedman test followed by Dunn's multiple comparison test. Values represent median score. The interquartile ranges have been omitted for clarity).
Effects of FAAH blockade on endocannabinoid levels
To test whether the anti-dyskinetic effects of WIN could be mimicked by potentiation of AEA action at CB1 receptors, a group of rats with 6-OHDA (n=6) or sham (n=6) lesions undergoing chronic levodopa administration were treated with the FAAH inhibitor, URB597 at a dose known to increase AEA concentrations in rat brain (0.3mg/kg, i.p.,15 min before levodopa from day 9 to day 11) (Fegley et al., 2005). As expected, URB597 significantly increased AEA levels [F(1,19)=53,3 p<0.0001] throughout the basal ganglia of rats with 6-OHDA lesions (Fig. 4), whereas it did not have any effect on 2-AG levels (in nmol/g. Striatum: levodopa, 5.47±0.3; levodopa+URB, 6.62±1.58. GP: levodopa, 5.61±0.65; levodopa+URB, 7.29±0.87. SN: levodopa, 16.58±3.6; levodopa+URB, 19.81±1.67). Systemic administration of the same dose of URB597 to sham-operated animals chronically-treated with levodopa produced a remarkable elevation of AEA [F(1,20=41.08, p<0.0001] (Fig. 4) without affecting 2-AG levels (in nmol/g. Striatum: levodopa, 7.07±0.79; levodopa+URB, 5.58±0.68. GP: levodopa, 7.64±0.28; levodopa+URB, 8.65±1.41. SN: levodopa, 15.22±0.53; levodopa+URB, 11.6±2.65). Chronic treatment with levodopa per se did not elevate AEA (Fig. 4) or 2-AG levels (data not shown) in either group.
Figure 4.
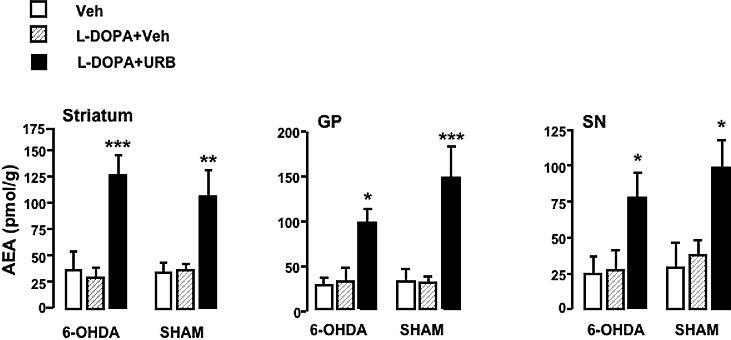
Effects of chronic levodopa (hatched bars, 6 mg/kg, i.p., 1 injection per day for 11 days) or co-administration of chronic levodopa + URB (filled bars, 0.3 mg/kg, i.p., day 9−11) on AEA levels in rats with 6-OHDA and sham lesions, 1 h after the last drug injection. Vehicle (Veh, open bars). GP, globus pallidus; SN, substantia nigra. *p<0.05; **p<0.01; ***p<0.001 compared to levodopa+vehicle (Two-way ANOVA followed by Bonferroni's test. Mean±s.e.m, n=5).
The AEA elevation induced by URB597 in lesioned rats was comparable to that observed after a similar dose of URB597 (0.3 mg/kg, i.p. from day 9 to day 12) in intact animals (in pmol/g. Striatum, 84±11.5; GP, 135.4±62.1; SN, 128±37; mean±s.e.m, n=5).
Effects of FAAH blockade on levodopa-induced AIMs
Systemic administration of URB597 to rats with 6-OHDA lesion at the same dose able to increase AEA levels did not produce any significant effect on levodopa-induced AIMs at either 60 or 100 min time points (data not shown and Fig. 5, respectively).
Figure 5.
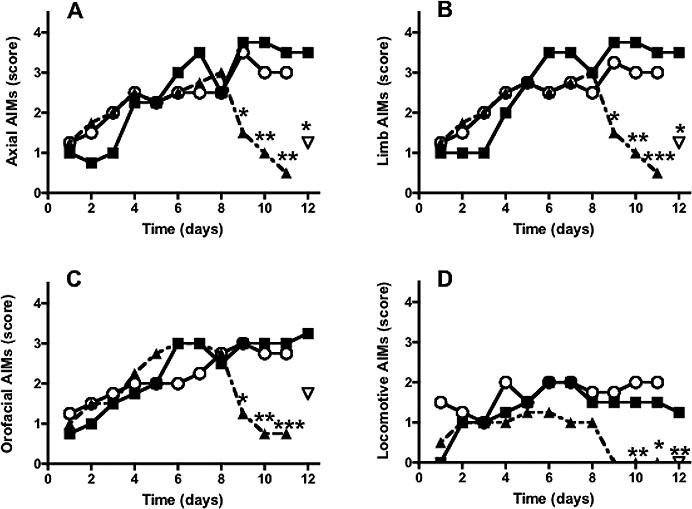
Time course of the effects of systemic administration of levodopa (filled squares, 6 mg/kg, 1 injection per day, n=6) to rats with unilateral 6-OHDA lesion on AIMs (as described in figure 2) measured 100 min after levodopa injection (115 min after URB or capsazepine injection). The FAAH inhibitor, URB597 (open circles, 0.3 mg/kg, i.p.) or URB597 + the TRPV1 antagonist capsazepine (triangles, 0.3+10 mg/kg., i.p., respectively) were administered to distinct groups of rats (n=6) from day 9 to day 11, 15 min before levodopa, whereas rats treated with levodopa only (filled squares) received an injection of vehicle (5%PEG+5%Tween-80 in saline) during the same days. The CB1 antagonist AM251 (inverted triangle, 1 mg/kg., i.p. 15 min before URB+capsazepine) was administered on day 12 of levodopa treatment. *p<0.05, **p<0.01, ***p<0.0001 (Kruskal-Wallis followed by Dunn's multiple comparison test. AM251 effect was analyzed by use of Friedman test followed by Dunn's multiple comparison test. Values represent median score. The interquartile ranges have been omitted for clarity).
As URB597-induced inhibition of FAAH caused a sustained AEA elevation throughout the basal ganglia, we hypothesized that the lack of anti-dyskinetic effect of URB597 might depend on the simultaneous activation of both CB1 and TRPV1 receptors by AEA. To test this hypothesis, URB597 (0.3 mg/kg, i.p.) and the selective TRPV1 antagonist, capsazepine (10 mg/kg i.p.) were co-administered to rats with 6-OHDA lesions, undergoing chronic levodopa treatment, from day 9 to 11, 15 min before levodopa. URB597+capsazepine significantly reduced all AIMs subtypes 100 min after levodopa (Fig. 5), whereas it had no effect during the first hour (data not shown). Within-group analysis also revealed a significant effect of URB597+capsazepine at days 9 (p<0.05) and 10−11 (p<0.01) compared to day 8 of levodopa. Capsazepine per se (10 mg/kg, i.p.) had no effect on levodopa-induced AIMs (data not shown). The anti-dyskinetic effect of URB597+capsazepine on axial, limb and locomotive AIMs was not reversed by the CB1 antagonists SR141716A (1 mg/kg i.p., data not shown) or AM251 (1 mg/kg, i.p.) (Fig. 5A,B,D); this antagonist, however, produced a partial blockade of oro-facial AIMs (Fig 5C).
DISCUSSION
In this study, we showed that: (1) direct activation of CB1 receptors significantly attenuated levodopa-induced AIMs; (2) elevation of brain AEA via pharmacological blockade of its catabolism produced an anti-dyskinetic effect only in the presence of a TRPV1 antagonist.
Our investigations were carried out in rats with unilateral 6-OHDA lesions of the nigro-striatal pathway. In these animals, chronic treatment with levodopa elicited characteristic AIMs, which are distinguishable from stereotypic behavior and model the pathophysiology and pharmacology of levodopa-induced dyskinesias in PD patients (Cenci et al., 2002; Lundblad et al., 2002).
We found no correlation between the extension of the 6-OHDA lesion, as indicated by the number of apomorphine-induced contralateral rotations (Sakai Gash, 1994), and the severity of dyskinesias. As contralateral rotations induced by dopaminergic agonists depend on stimulation of supersensitive post-synaptic dopamine receptors in the injured striatum (Deumens et al., 2002; Marin et al., 2006), our data suggest that the development of AIMs is not related to dopamine receptor supersensitivity and/or to the severity of dopamine depletion, but rather to maladaptive changes in the plasticity of the basal ganglia circuitry following the nigrostriatal lesion (Cenci and Lundblad, 2006).
WIN reduced levodopa-induced AIMs (expressed as total AIM score) at low (0.5−1 mg/kg) but not at high doses (2.5 mg/kg), suggesting that non-specific actions of WIN, when used at doses over 1 mg/kg, may mask its anti-dyskinetic properties. Thus, the possibility of dose-related contrasting effects should be taken into account when testing cannabinoid agonists as anti-dyskinetic agents, and may explain the conflicting results obtained in clinical trials (Sieradzan et al., 2001; Carroll et al., 2004).
Our data indicate that WIN reduced axial, limb and oro-facial AIMs 60 min after levodopa administration and that this effect was more pronounced 100 min post-levodopa. These observations suggest that WIN might be less efficacious in reducing peak-dose dyskinesias, which manifest within 1 h after levodopa administration (Fahn, 2000), whereas it may exert stronger therapeutic benefits at later time points, possibly during diphasic or off states. In keeping with this hypothesis, the cannabinoid agonist nabilone was shown to be particularly effective on diphasic and off-dose dyskinesias in PD patients (Sieradzan et al., 2001). Although a later double-blind, placebo-controlled trial failed to recognize any effect of cannabis on off-dose dyskinesias (Carroll et al., 2004), this assessment was based on diary data from patients, which are limited by incorrect labeling of dyskinetic symptoms (Vitale et al., 2001). Thus, additional studies are necessary to further test this hypothesis.
The anti-dyskinetic effect of WIN was not caused by generalized suppression of motor activity, as the dose of 1 mg/kg had no effect on locomotion (Anderson et al., 1995), nor it elicited catalepsy, nor affected apomorphine-induced rotational behavior.
The CB1 antagonist AM251 reversed the effect of WIN and confirmed our previous study showing that WIN reduced oral dyskinesias via a CB1-mediated mechanism even after chronic administration of larger doses of levodopa (50 mg/kg, i.p.) (Ferrer et al., 2003). The beneficial action of cannabinoid agonists may depend on the ability of these drugs to compensate for the reduced endocannabinoid transmission observed in the striatum of 6-OHDA-treated rats (Ferrer et al., 2003). Although other groups have reported elevated endocannabinoid levels in rat striatum following dopamine depletion (Di Marzo et al., 2000; Gubellini et al., 2002), there is no physiological evidence supporting an increased endocannabinoid tone in 6-OHDA-treated rats (Kreitzer and Malenka, 2007). Thus, these discrepancies may depend on the use of different models (reserpine- versus 6-OHDA-treated rats), time elapsed from the lesion, and/or different analytical methods for the detection and quantification of endocannabinoid substances. The latter point is particularly critical as, for example, the use of silica-gel chromatography for endocannabinoid purification can produce misleading results (Hardison et al., 2006). Unlike in intact rats, levodopa failed to elevate AEA in the basal ganglia of 6-OHDA-treated rats (Ferrer et al., 2003). As AEA counteracts dopamine-dependent hypermotility via activation of CB1 receptors (Giuffrida et al., 1999; Romero et al., 1995), it is plausible to hypothesize that chronic levodopa may produce a “hyperdopaminergic state” in 6-OHDA-treated rats that is not counter-balanced by concomitant dopamine-induced AEA elevation, which in turn may contribute to the development of dyskinesias. In addition, cannabinoid agonists may exert anti-dyskinetic effects by modulating the glutamatergic input from cortico-striatal afferents (Gubellini et al., 2002) and/or by “rescuing” endocannabinoid-mediated synaptic plasticity in the striatum (Kreitzer and Malenka, 2007), which are both dysfunctional following 6-OHDA lesion (Picconi et al., 2003; Kreitzer and Malenka, 2007).
Locomotive AIMs, which consist of levodopa-triggered contralateral rotations, were not attenuated by WIN at any time point. This lack of effect may be ascribed to several factors. First, the rotational response depends on the dose and number of levodopa injections administered per day (Henry et al., 1998). Therefore, the low, single dose used in our paradigm, which triggers modest rotational behavior, may not be sufficient to unmask a possible effect of WIN. Second, locomotive AIMs, unlike other AIMs subtypes, undergo little sensitization, most likely as consequence of the dramatic increase in dystonic postures that limit the expression of rotational behavior. Finally, whether or not levodopa-induced rotations can model dyskinetic movements is controversial (Lane et al., 2006; Marin et al., 2006). Indeed, the doses of levodopa eliciting a stable rotational response are much larger than those producing dyskinesias (Marin et al., 2006); also, drugs that trigger little dyskinesia in the clinic (i.e., ropinirole or bromocriptine) produce full rotational responses in rodents (Lundblad et al., 2002; Ravenscroft et al., 2004). Thus, the inability of WIN to decrease locomotive AIMs may depend on the fact that this behavioral phenotype is not a good measure of dyskinesias.
Previous studies have shown that FAAH inhibitors can reduce hyperdopaminergia-related hyperactivity in mice (Tzavara et al., 2006) and hyperkinesia in a rat model of Huntington's disease (Lastres-Becker et al., 2003) by elevating endocannabinoid levels in the brain. To investigate whether indirect stimulation of CB1 receptors via potentiation of the endogenous ligands could mimic the anti-dyskinetic effect of WIN, we tested the FAAH inhibitor URB597. We found that URB597 elevated AEA throughout the basal ganglia of 6-OHDA-treated rats to an extent comparable to that observed in intact animals. As previously reported (Kathuria et al., 2003), URB597 did not increase 2-AG levels in either animal group. As 2-AG is preferentially metabolized by MAG lipase (Dinh et al., 2002), we were not able to assess if the selective elevation of 2-AG affected levodopa-induced AIMs, as there are no MAG lipase inhibitors currently available for in vivo testing.
Although the unilateral 6-OHDA lesion does not prevent URB597 from elevating brain AEA, URB597 failed to reduce levodopa-induced AIMs when administered alone, suggesting that AEA elevation is not sufficient to attenuate dyskinesias. The contrasting outcomes observed with AEA and WIN may depend on the different pharmacological actions of these compounds. Unlike WIN, which acts as full agonist at CB1 receptors and inhibits TRPV1 receptors via a calcineurin-mediated mechanism (Patwardhan et al., 2006), AEA is a partial agonist at both CB1 and TRPV1 receptors (Ross, 2003). TRPV1 receptors are expressed in selected areas of the central nervous system, such as the striatum, GP and SN (Cristino et al., 2006; Mezey et al., 2000), and regulate basal ganglia functions by affecting the excitability of nigrostriatal dopaminergic neurons (Marinelli et al., 2003). Although the low intrinsic efficacy of AEA at TRPV1 receptors have raised questions whether this lipid might serve as a physiological ligand (Szolcsanyi, 2000; Zygmunt et al., 2000), recent studies indicate that AEA efficacy and potency at TRPV1 receptors can be enhanced by several factors, including pharmacological blockade of FAAH (De Petrocellis et al., 2001; Ross, 2003). Thus, URB597-induced AEA elevation may lead to concomitant stimulation of CB1 and TRPV1 receptors in the basal ganglia.
In our animal model, application of the TRPV1 antagonist capsazepine unmasked an anti-dyskinetic effect of URB597 on all AIMs subtypes. These data suggest that the stimulation of CB1 and TRPV1 receptors by AEA may have opposite effects on levodopa-induced AIMs so that, only when TRPV1 receptors are blocked, AEA exerts its anti-dyskinetic action, possibly via CB1 receptors. These findings differ from those reported by Lee et al. (2006), showing that URB597 alone or stimulation of TRPV1 receptors by capsaicin can attenuate L-DOPA-induced hyperactivity in reserpine-treated rats (Lee et al., 2006). Although in the study of Lee et al. no attempts were made to potentiate (or reverse) the effect of URB597 by co-administration of capsazepine, the discrepancy with our data may be due to the different animal model used and/or the type of behavior measured (vertical motor activity), which models stereotypies rather than dyskinesias (Cenci et al., 2002).
Several studies have shown that stimulation of CB1 and TRPV1 often produce opposite effects in various experimental settings, including changes in intracellular Ca2+ concentrations (Szallasi and Di Marzo, 2000) and glutamate release in the SNc (Marinelli et al., 2003). Nevertheless, whether the anti-dyskinetic effect of AEA unmasked by capsazepine is mediated via activation of CB1 receptors fits our observations only partially. Indeed, although the CB1 antagonist AM251 reversed URB597+capasazepine-induced suppression of oro-facial dyskinesias, it had no effect on other AIMs subtypes. Similarly, SR141716A, which antagonizes AEA effects mediated by a not-yet identified CB receptor different from CB1 and CB2 (Begg et al., 2005), was unable to reverse URB597+capasazepine-induced suppression of AIMs. Thus, targets other than CB1 receptors are likely to play a role in this response.
Under conditions in which FAAH activity is blocked, oxygenase enzymes, such as cyclooxygenase-2 and lipoxygenase, can metabolize AEA (Kozak and Marnett, 2002; Ross et al., 2002), and convert it into prostaglandin- and/or leukotriene-like derivatives. Therefore, we cannot rule out that these metabolites, rather than AEA, might be responsible for the anti-dyskinetic effects of URB597+capsazepine. Alternatively, endocannabinoids may differentially affect distinct AIMs phenotypes, depending on the specific anatomical substrates and/or changes in plasticity regulating each AIM subtype.
In summary, our study shows that AEA elevation via pharmacological blockade of its enzymatic degradation produces a significant anti-dyskinetic effect only if accompanied by the concomitant blockade of TRPV1 receptors. This observation points to TRPV1 receptors as a new, important component in the pathophysiology of levodopa-associated dyskinesias, and opens new pharmacological strategies for the treatments of these disorders.
Acknowledgements
This work was supported by a grant from the National Institute of Neurological Disorders and Stroke (NS 050401 to A.G.) and by a grant from the Italian Ministry of University and Research (FIRB 2006 to V.C.)
We thank Drs. A. Seillier, S. Gaetani, D. Price and A. Frazer for critically reading the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Anderson LA, Anderson JJ, Chase TN, Walters JR. The cannabinoid agonist Win 55,212−2 and CP 55,940 attenuate rotational behavior induced by a dopamine D1 but not a D2 agonist in rats with unilateral lesions of the nigrostriatal pathway. Brain Res. 1995;691:106–114. doi: 10.1016/0006-8993(95)00645-7. [DOI] [PubMed] [Google Scholar]
- Begg M, Pacher P, Batkai S, Osei-Hyiaman D, Offertaler L, Mo FM, Liu J, Kunos G. Evidence for novel cannabinoid receptors. Pharmacol. Ther. 2005;106:133–45. doi: 10.1016/j.pharmthera.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Beltramo M, Rodriguez de Fonseca F, Navarro M, Calignano A, Gorriti MA, Grammatikopoulos G, Sadile AG, Giuffrida A, Piomelli D. Reversal of dopamine D2-receptor responses by an anandamide transport inhibitor. J. Neurosci. 2000;20:3401–3407. doi: 10.1523/JNEUROSCI.20-09-03401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CB, Bain PG, Teare L, Liu X, Joint C, Wroath BA, Parkin SG, Fox P, Wright D, Hobart J, Zajicek JP. Cannabis for dyskinesia in Parkinson disease. Neurology. 2004;63:1245–1250. doi: 10.1212/01.wnl.0000140288.48796.8e. [DOI] [PubMed] [Google Scholar]
- Carta M, Lindgren HS, Lundblad M, Stancampiano R, Fadda F, Cenci MA. Role of striatal L-DOPA in the production of dyskinesia in 6-hydroxydopamine lesioned rats. J. Neurochem. 2006;96:1718–27. doi: 10.1111/j.1471-4159.2006.03696.x. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Lundblad M. Post- versus presynaptic plasticity in L-DOPA-induced dyskinesia. J. Neurochem. 2006;99:381–92. doi: 10.1111/j.1471-4159.2006.04124.x. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Whishaw IQ, Schallert T. Animal models of neurological deficits: how relevant is the rat? Nat. Rev. Neurosci. 2002;3:574–579. doi: 10.1038/nrn877. [DOI] [PubMed] [Google Scholar]
- Cristino L, de Petrocellis L, Pryce G, Baker D, Guglielmotti V, Di Marzo V. Immunohistochemical localization of cannabinoid type 1 and vanilloid transient receptor potential vanilloid type 1 receptors in the mouse brain. Neuroscience. 2006;139:1405–15. doi: 10.1016/j.neuroscience.2006.02.074. [DOI] [PubMed] [Google Scholar]
- de la Fuente-Fernandez R, Sossi V, Huang Z, Furtado S, Lu JQ, Calne DB, Ruth TJ, Stoessl AJ. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson's disease: implications for dyskinesias. Brain. 2004;127:2747–54. doi: 10.1093/brain/awh290. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Bisogno T, Maccarrone M, Davis JB, Finazzi-Agro A, Di Marzo V. The activity of anandamide at vanilloid VR1 receptors requires facilitated transport across the cell membrane and is limited by intracellular metabolism. J. Biol. Chem. 2001;276:12856–63. doi: 10.1074/jbc.M008555200. [DOI] [PubMed] [Google Scholar]
- Deumens R, Blokland A, Prickaerts J. Modeling Parkinson's disease in rats: an evaluation of 6-OHDA lesions of the nigrostriatal pathway. Exp. Neurol. 2002;175:303–17. doi: 10.1006/exnr.2002.7891. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Hill MP, Bisogno T, Crossman AR, Brotchie JM. Enhanced levels of endogenous cannabinoids in the globus pallidus are associated with a reduction in movement in an animal model of Parkinson's disease. FASEB J. 2000;14:1432–1438. doi: 10.1096/fj.14.10.1432. [DOI] [PubMed] [Google Scholar]
- Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahn S. The spectrum of levodopa-induced dyskinesias. Ann. Neurol. 2000;47:S9–11. [PubMed] [Google Scholar]
- Fegley D, Gaetani S, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3'-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. J. Pharmacol. Exp. Ther. 2005;313:352–8. doi: 10.1124/jpet.104.078980. [DOI] [PubMed] [Google Scholar]
- Ferrer B, Asbrock N, Kathuria S, Piomelli D, Giuffrida A. Effects of levodopa on endocannabinoid levels in rat basal ganglia: implications for the treatment of levodopa-induced dyskenesias. Eur. J. Neurosci. 2003;18:1607–1614. doi: 10.1046/j.1460-9568.2003.02896.x. [DOI] [PubMed] [Google Scholar]
- Fiorentini C, Rizzetti MC, Busi C, Bontempi S, Collo G, Spano P, Missale C. Loss of synaptic D1 dopamine/N-methyl-D-aspartate glutamate receptor complexes in L-DOPA-induced dyskinesia in the rat. Mol. Pharmacol. 2006;69:805–12. doi: 10.1124/mol.105.016667. [DOI] [PubMed] [Google Scholar]
- Gardoni F, Picconi B, Ghiglieri V, Polli F, Bagetta V, Bernardi G, Cattabeni F, Di Luca M, Calabresi P. A critical interaction between NR2B and MAGUK in L-DOPA induced dyskinesia. J. Neurosci. 2006;26:2914–22. doi: 10.1523/JNEUROSCI.5326-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida A, Parsons LH, Kerr TM, Rodríguez de Fonseca F, Navarro M, Piomelli D. Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat. Neurosci. 1999;2:358–363. doi: 10.1038/7268. [DOI] [PubMed] [Google Scholar]
- Gubellini P, Picconi B, Bari M, Battista N, Calabresi P, Centonze D, Bernanrdi G, Finazzi-Agro A, Maccarrone M. Experimental parkinsonism alters endocannabinoid degradation: implications for striatal glutamatergic transmission. J. Neurosci. 2002;22:6900–6907. doi: 10.1523/JNEUROSCI.22-16-06900.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardison S, Weintraub ST, Giuffrida A. Quantification of endocannabinoids in rat biological samples by GC/MS: technical and theoretical considerations. Prostaglandins Lip. Med. 2006;81:106–112. doi: 10.1016/j.prostaglandins.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Henry B, Crossman AR, Brotchie JM. Characterization of enhanced behavioral responses to L-DOPA following repeated administration in the 6-hydroxydopamine-lesioned rat model of Parkinson's disease. Exp. Neurol. 1998;151:334–342. doi: 10.1006/exnr.1998.6819. [DOI] [PubMed] [Google Scholar]
- Hermann H, De Petrocellis L, Bisogno T, Schiano Moriello A, Lutz B, Di Marzo V. Dual effect of cannabinoid CB1 receptor stimulation on a vanilloid VR1 receptor-mediated response. Cell Mol. Life Sci. 2003;60:607–16. doi: 10.1007/s000180300052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 2003;1:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Kim SR, Lee DY, Chung ES, Oh UT, Kim SU, Jin BK. Transient receptor potential vanilloid subtype 1 mediates cell death of mesencephalic dopaminergic neurons in vivo and in vitro. J. Neurosci. 2005;25:662–71. doi: 10.1523/JNEUROSCI.4166-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak KR, Marnett LJ. Oxidative metabolism of endocannabinoids. Prostaglandins Leukotr. Essent. Fatty Acids. 2002;66:211–220. doi: 10.1054/plef.2001.0359. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature. 2007;445:643–7. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Lane EL, Cheetham SC, Jenner P. Does contraversive circling in the 6-OHDA-lesioned rat indicate an ability to induce motor complications as well as therapeutic effects in Parkinson's disease? Exp. Neurol. 2006;197:284–90. doi: 10.1016/j.expneurol.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Lastres-Becker I, de Miguel R, De Petrocellis L, Makriyannis A, Di Marzo V, Fernandez-Ruiz J. Compounds acting at the endocannabinoid and/or endovanilloid systems reduce hyperkinesia in a rat model of Huntington's disease. J. Neurochem. 2003;84:1097–1109. doi: 10.1046/j.1471-4159.2003.01595.x. [DOI] [PubMed] [Google Scholar]
- Lee J, Di Marzo V, Brotchie JM. A role for vanilloid receptor 1 (TRPV1) and endocannabinnoid signalling in the regulation of spontaneous and L-DOPA induced locomotion in normal and reserpine-treated rats. Neuropharmacology. 2006;51:557–65. doi: 10.1016/j.neuropharm.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Lundblad M, Andersson M, Winkler C, Kirik D, Wierup N, Cenci MA. Pharmacological validation of behavioral measures of akinesia and dyskinesia in a rat model of Parkinson's disease. Eur. J. Neurosci. 2002;15:120–132. doi: 10.1046/j.0953-816x.2001.01843.x. [DOI] [PubMed] [Google Scholar]
- Lundblad M, Picconi B, Lindgren H, Cenci MA. A model of L-DOPA-induced dyskinesia in 6-hydroxydopamine lesioned mice:relation to motor and cellular parameters of nigrostriatal function. Neurobiol. Dis. 2004;16:110–123. doi: 10.1016/j.nbd.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Mackie K. Distribution of cannabinoid receptors in the central and peripheral nervous system. Handb. Exp. Pharmacol. 2005:299–325. doi: 10.1007/3-540-26573-2_10. [DOI] [PubMed] [Google Scholar]
- Marin C, Rodriguez-Oroz MC, Obeso JA. Motor complications in Parkinson's disease and the clinical significance of rotational behavior in the rat: have we wasted our time? Exp. Neurol. 2006;197:269–74. doi: 10.1016/j.expneurol.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Marinelli S, Di Marzo V, Berretta N, Matias I, Maccarrone M, Bernardi G, Mercuri NB. Presynaptic facilitation of glutamatergic synapses to dopaminergic neurons of the rat substantia nigra by endogenous stimulation of vanilloid receptors. J. Neurosci. 2003;23:3136–44. doi: 10.1523/JNEUROSCI.23-08-03136.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner W, Ravenscroft P, Reese R, Harnack D, Morgenstern R, Kupsch A, Klitgaard H, Bioulac B, Gross CE, Bezard E, Boraud T. Increased slow oscillatory activity in substantia nigra pars reticulata triggers abnormal involuntary movements in the 6-OHDA-lesioned rat in the presence of excessive extracellular striatal dopamine. Neurobiol. Dis. 2006;22:586–98. doi: 10.1016/j.nbd.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Mezey E, Tóth ZE, Cortright DN, Arzubi MK, Krause JE, Elde R, A., G., Blumberg PM, Szallasi A. Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1-like immunoreactivity, in the central nervous system of the rat and human. Proc. Natl. Acad. Sci. 2000;97:3655–3660. doi: 10.1073/pnas.060496197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt JG, Woodward WR, Carter JH, Gancher ST. Effect of long-term therapy on the pharmacodynamics of levodopa. Relation to on-off phenomenon. Arch. Neurol. 1992;49:1123–30. doi: 10.1001/archneur.1992.00530350037016. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Rodriguez-Oroz M, Marin C, Alonso F, Zamarbide I, Lanciego JL, Rodriguez-Diaz M. The origin of motor fluctuations in Parkinson's disease: importance of dopaminergic innervation and basal ganglia circuits. Neurology. 2004;62:S17–30. doi: 10.1212/wnl.62.1_suppl_1.s17. [DOI] [PubMed] [Google Scholar]
- Patwardhan AM, Jeske NA, Price TJ, Gamper N, Akopian AN, Hargreaves KM. The cannabinoid WIN 55,212−2 inhibits transient receptor potential vanilloid 1 (TRPV1) and evokes peripheral antihyperalgesia via calcineurin. Proc. Natl. Acad. Sci. 2006;103:11393–8. doi: 10.1073/pnas.0603861103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson CRR. The Rat Brain in Stereotaxic Coordinates. 1998. [DOI] [PubMed]
- Picconi B, Centonze D, Hakansson K, Bernardi G, Greengard P, Fisone G, Cenci MA, Calabresi P. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat. Neurosci. 2003;6:501–506. doi: 10.1038/nn1040. [DOI] [PubMed] [Google Scholar]
- Piomelli D, Giuffrida A, Calignano A, Rodríguez de Fonseca F. The endocannabinoid system as a target for therapeutic drugs. Trends Pharmacol. Sci. 2000;21:218–224. doi: 10.1016/s0165-6147(00)01482-6. [DOI] [PubMed] [Google Scholar]
- Ravenscroft P, Chalon S, Brotchie JM, Crossman AR. Ropinirole versus L-DOPA effects on striatal opioid peptide precursors in a rodent model of Parkinson's disease: implications for dyskinesia. Exp. Neurol. 2004;185:36–46. doi: 10.1016/j.expneurol.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Romero J, Garcia L, Cebeira M, Zadrozny D, Fernandez-Ruiz JJ, Ramos JA. The endogenous cannabinoid receptor ligand, anandamide, inhibits the motor behaviour: role of nigrostriatal dopaminergic neurons. Life Sci. 1995;56:2033–2040. doi: 10.1016/0024-3205(95)00186-a. [DOI] [PubMed] [Google Scholar]
- Ross RA. Anandamide and vanilloid TRPV1 receptors. Br. J. Pharmacol. 2003;140:790–801. doi: 10.1038/sj.bjp.0705467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RA, Craib SJ, Stevenson LA, Pertwee RG, Henderson A, Toole J, Ellington HC. Pharmacological characterization of the anandamide cyclooxygenase metabolite: prostaglandin E2 ethanolamide. J. Pharmacol. Exp. Ther. 2002;301:900–7. doi: 10.1124/jpet.301.3.900. [DOI] [PubMed] [Google Scholar]
- Sakai K, Gash DM. Effect of bilateral 6-OHDA lesions of the substantia nigra on locomotor activity in the rat. Brain Res. 1994;633:144–50. doi: 10.1016/0006-8993(94)91533-4. [DOI] [PubMed] [Google Scholar]
- Sieradzan KA, Fox SH, Hill M, Dick JPR, Crossman AR, Brotchie JM. Cannabinoids reduce levodopa-induced dyskinesia in Parkinson's disease: a pilot study. Neurology. 2001;57:2108–2111. doi: 10.1212/wnl.57.11.2108. [DOI] [PubMed] [Google Scholar]
- Szallasi A, Di Marzo V. New perspectives on enigmatic vanilloid receptors. Trends Neurosci. 2000;23:491–7. doi: 10.1016/s0166-2236(00)01630-1. [DOI] [PubMed] [Google Scholar]
- Szolcsanyi I. Are cannabinoids endogenous ligands for the VR1 capsaicin receptor. Trends Pharmacol. Sci. 2000;21:41–42. doi: 10.1016/s0165-6147(99)01436-4. [DOI] [PubMed] [Google Scholar]
- Toth A, Boczan J, Kedei N, Lizanecz E, Bagi Z, Papp Z, Edes I, Csiba L, Blumberg PM. Expression and distribution of vanilloid receptor 1 (TRPV1) in the adult rat brain. Brain Res. Mol. Brain Res. 2005;135:162–8. doi: 10.1016/j.molbrainres.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Tzavara ET, Li DL, Moutsimilli L, Bisogno T, Di Marzo V, Phebus LA, Nomikos GG, Giros B. Endocannabinoids activate transient receptor potential vanilloid 1 receptors to reduce hyperdopaminergia-related hyperactivity: therapeutic implications. Biol. Psychiatry. 2006;59:508–15. doi: 10.1016/j.biopsych.2005.08.019. [DOI] [PubMed] [Google Scholar]
- van der Stelt M, Fox SH, Hill M, Crossman AR, Petrosino S, Di Marzo V, Brotchie JM. A role for endocannabinoids in the generation of parkinsonism and levodopa-induced dyskinesia in MPTP-lesioned non-human primate models of Parkinson's disease. FASEB J. 2005;19:1140–2. doi: 10.1096/fj.04-3010fje. [DOI] [PubMed] [Google Scholar]
- Vitale C, Pellecchia MT, Grossi D, Fragassi N, Cuomo T, Di Maio L, Barone P. Unawareness of dyskinesias in Parkinson's and Huntington's diseases. Neurol. Sci. 2001;22:105–6. doi: 10.1007/s100720170066. [DOI] [PubMed] [Google Scholar]
- Westin JE, Lindgren HS, Gardi J, Nyengaard JR, Brundin P, Mohapel P, Cenci MA. Endothelial proliferation and increased blood-brain barrier permeability in the basal ganglia in a rat model of 3,4-dihydroxyphenyl-L-alanine-induced dyskinesia. J. Neurosci. 2006;26:9448–61. doi: 10.1523/JNEUROSCI.0944-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygmunt PM, Julius I, Di Marzo V, Hogestatt ED. Anandamide - the other side of the coin. Trends Pharmacol. Sci. 2000;21:43–44. doi: 10.1016/s0165-6147(99)01430-3. [DOI] [PubMed] [Google Scholar]