

Abstract
Estrogen can significantly influence CD16 expression and alter monocytic cytokine release upon CD16 receptor activation. However, the function of the estrogen receptor (ER) α and β in this response is unclear. To test whether estrogen binds ERα and/or ERβ to affect CD16 expression, monocytic cells were treated with and without physiological levels of 17 β-estradiol and various doses of the ERα and ERβ antagonist fulvestrant followed by measurement of CD16 transcript levels. To determine how estrogen induced changes in TNF-α and IL-1β release due to CD16 activation we quantitated the amount of cytokines after treatment with estrogen, fulvestrant and antibodies that specifically bind and activate the CD16 receptor. Interaction of ERα and the CD16 promoter was then determined by chromatin immunoprecipitation. Furthermore, specific promoter elements utilized by estrogen to control CD16 expression were mutated and expression from a luciferase reporter quantitated after transfection. Using the luciferase reporter construct containing a wild type CD16 promoter, the role of ERα and ERβ in the estrogen response was tested by treating transfected monocytes with an ERα specific agonist or an ERβ specific agonist and measuring expression. Our results show that CD16 transcript levels significantly decreased in monocytic cells due to estrogen and that the observed decrease in message was blocked by the antagonist fulvestrant. Estrogen reduced CD16 expression and decreased TNF-α and IL-1β release upon CD16 activation but the administration of fulvestrant blocked this decrease. ERα was found to interact with a region 5′ of the CD16 gene in the presence of estrogen, and site-directed mutational analysis of this region indicated the necessity for an estrogen response element in modulating estrogen effects on CD16 expression. Moreover, both an ERα and an ERβ agonist reduced expression of the CD16 reporter construct suggesting both receptors can play a role in CD16 regulation. In conclusion, CD16 expression can be altered by the activity of ERα or ERβ and our results also show that ERα can associate with a region within the CD16 promoter that is important in production of transcript.
Keywords: CD16, estrogen receptor, promoter, expression, auto-immune, luciferase, chromatin immunoprecipitation, estradiol
Introduction
CD16, also termed Fc gamma receptor III-A, is expressed on monocytes and macrophages. CD16 binds auto-antigens that are important in the onset and maintenance of auto-immune diseases (Edwards and Cambridge, 1998). Binding and cross-linking or aggregation of the CD16 receptor induces cytokine release that can promote cartilage reabsorbtion, inhibit the synthesis of proteoglycans and cause inflammation, which are all major symptoms of auto-immune disorders (Saklatvala, 1986; Pettipher et al., 1986; Henderson and Pettipher, 1989; Wooley et al., 1993; Abrahams et al., 2000; Kramer et al., 2004). Estrogen can decrease the expression of CD16 on monocytes modulating cytokine release (Kramer et al., 2004). Changes in the systemic output of estrogen is a potential reason why women have a higher incidence of auto-immune disorders, such as systemic lupus erythematuosus, scleroderma, SjÖgrens syndrome, Graves disease and rheumatoid arthritis (Ansar et al., 1985; Felten, 1993). For example, rheumatoid arthritis animal models indicate that low estrogen concentrations, such as those observed post-partum, leads to greater onset of arthritis (Mattsson et al., 1991) and estrogen treatment ameliorates collagen induced arthritic symptoms (Holmdahl et al., 1987). Epidemiological evidence in humans confirms animal model data indicating that during pregnancy (high estrogen) rheumatoid arthritis symptoms are ameliorated but post-partum and at menopause (low estrogen) arthritis onset and symptoms increase (Hench, 1938; Persellin, 1976; Ostensen et al., 1983; McHugh, 1990). One pathway by which estrogen could impact these immune disorders is by modulating CD16 expression.
Cytokine production and release from monocytes and macrophages can be induced by antigen binding and cross-linking the CD16 receptor (Abrahams et al., 2000). The functional CD16 receptor in monocytes consists of a α subunit that spans the cell membrane to bind antigens within the blood. As well as, an internal portion of the receptor, the γ subunit (FcεRIγ), which is necessary for signal transduction (Wirthmueller et al., 1992). After antigen binding the CD16 receptor will cross-link or aggregate initiating a change in the receptor that triggers the internal FcεRI portion to signal multiple events, such as, autophosphorylation and tyrosine kinase activation (Oliver et al., 1988; Benhamou and Siraganian, 1992; Beaven and Metzger, 1993; Paolini et al., 1994). These internal signaling events trigger changes in cytokine release. Importantly, CD16 binding is preferential for binding small IgG dimer or trimer complexes that include IgG anti-IgG antibody complexes. These complexes are important components of auto-antigens and rheumatoid factors that potentially trigger the onset or maintenance of auto-immune diseases such as rheumatoid arthritis (Pope et al., 1974; Nardella et al., 1981; Klaassen et al., 1988; Edwards and Cambridge, 1998). Furthermore, the expression of CD16 on monocytes/macrophages is restricted to tissues (e.g., synovial tissue, pericardium) impacted by rheumatoid arthritis (Bhatia et al., 1998). Previous studies indicated that Fc gamma receptors I and II have a higher level of expression on splenic-macrophages due to estrogen treatment that contributes to the clearance of IgG-sensitized cells (Friedman et al., 1985; Gomez et al., 2001). Our own studies have contributed to this contemporary body of knowledge by showing that estrogen can significantly influence CD16 expression. Changes in the level of CD16 present on monocytes and macrophages leads to altered cytokine release upon receptor cross-linking (Kramer et al., 2004).
Estrogen receptor α and β transcripts (i.e., ERα and ERβ, respectively) have been detected in primary macrophages and in the monocytic leukemia cell line THP-1 (Vegeto et al., 2001; Cutolo et al., 2001). Additional data showed that ERβ protein is present in primary macrophages (Kramer et al., 2004), suggesting that ERs function in transducing the estrogen response. As a direct result, studies as to the mechanisms by which estrogen regulates cytokine production and release from macrophages was pursued, which led to the conclusion that estrogen regulates IL-1β indirectly through post-translational processes involving caspase-1 cleavage (Krieg et al., 2001). TNF- α production is potentially regulated though kinase signaling (Srivastava et al., 1999) and IL-6 can be directly regulated by estrogen (Pottratz et al., 1994). The reports discussed above suggest that changes in estrogen concentrations initiate or exacerbate autoimmune disease symptoms as a result of estrogen modulating the macrophage cytokine production. In addition, these reports demonstrate that the mechanism by which estrogen impacts cytokine production can be indirect (such as signal transduction cascades) or direct, in which the ligand-bound (ER) α and β acts at the promoter to affect transcription.
Although our previous work provided evidence that estrogen decreased CD16 expression and reduced cytokine release from monocytes activated by cross-linking the CD16 receptor, the precise mechanism by which this occurs is unknown. In this study, we show that estrogen modulates CD16 expression in monocytes due to ERα interaction with the CD16 promoter region and that the change in CD16 expression, resulting from estrogen exposure, can be due to both ERα and ERβ.
Materials and Methods
THP-1 cell culture and treatments
THP-1 cells (leukemic, monocytic cell line -ATCC) were cultured in RMPI 1640 minus phenol red, 2% fetal bovine serum (FBS) and antibiotics to a density of 5 × 105 cells/ml. THP-1 cells are a proven model for primary macrophages (Fenton et al., 1988; Auwerx, 1991) and for ER expression (Cutolo et al., 2001). THP-1 cells, concentrated to 2.5 × 106 cells/ml, were treated with 15 ng/ml of phorbol 12-myristate 13-acetate (PMA, Sigma) to induce differentiation into a macrophage-like THP-1 cell (Kramer and Wray, 2002). Forty-eight hours after PMA treatment, the cells were rinsed in PBS three times and placed in media consisting of RMPI 1640 (minus phenol red), 100 mg/ml sodium pyruvate, MEM vitamins, MEM non-essential amino acids, G-418 (all components from Gibco/BRL, Rockville, MD) and 2% charcoal-stripped FBS. The THP-1 cell line can be cultured in serum-free media for over a week without any detectable change (data not shown). Twenty-four hours later, the cultures were divided such that the cells were untreated or treated with 1 × 10−8 17-β-estradiol, unless specified that the cells were treated with 1 × 10−9 M 17-β-estradiol. We utilized a dose of 1 × 10−8 M 17-β-estradiol, or approximately 10,000 pmol/L, to treat the monocyte cultures because this value falls near the range of estrogen detected in human serum at the preovulatory high of 2393 ± 356 pmol/L (Polan et al., 1990). Moreover, previously published reports describe this concentration of estrogen (i.e., 1 × 10−8 M 17-β-estradiol) as being a physiological dose (Cutolo et al., 1995). After 24 hours, the estrogen-treated cultures were rinsed three times in PBS and split again such that half were placed in media with estrogen and half without; 10nM, 100nM or 1000nM of fulvestrant was added to a portion of these cultures. Fulvestrant has been identified as “Faslodex” or ICI 182, 780 (Tocris Cookson, Inc., Ballwin, MO) and has been shown to be an antagonist for both the ERα and ERβ (Tremblay et al., 1997; Kuiper et al., 1997; Wakeling, 2000). After 0, 12, 24 and 48 hours, the cells were harvested, and real-time RT-PCR was performed.
Primary monocyte isolation and culture
Primary monocyte isolation occurred in 3 stages. First, buffy coats from healthy female volunteers between the ages of 20 and 45 years were obtained from the Carter Blood Care Center following IRB-approved protocols. Second, the blood products were diluted in 0.15 M NaCl/ 2 mM EDTA; 31 ml of this mixture was layered on 18 ml of Ficoll-hypaque (Sigma) solution in a 50 ml polypropylene tube and separated by density gradient centrifugation. The banded peripheral blood mononuclear cell fraction (PBMC) (monocytes, T and B cells) was removed from the gradient, red blood cells and polymorphonuclear cells form a pellet that is discarded. Approximately 7×109 PBMCs were supplied from each 50 ml sample obtained from the blood bank. Third, the PBMC fraction was suspended in RPMI media with 10% fetal bovine serum (FBS) and 10% DMSO and frozen in liquid nitrogen (1×106 cells/vial).
Frozen vials of human female PBMCs were thawed quickly at 37°C, added to 8 ml of media (RPMI 1640 minus phenol red, plus 2% charcoal-stripped FBS), centrifuged, the supernate discarded, and the monocytes re-plated in fresh media. Monocytes will adhere to the plate while T and B cells remain suspended. After 45 minutes, the plates were gently washed with three rinses of prewarmed PBS, and the cells were resuspended in media containing 10 ng/ml transforming growth factor-β1 for further experimentation. Functional CD16 expression can be increased on peripheral blood monocytes with TGF β (Welch et al., 1990).
Monocyte activation and treatment
Monocytes from one frozen vial were resuspended in 24 mls and divided into 1 ml aliquots. These 24 aliquots were treated with ±1 × 10−8 M 17 β-estradiol and with ±100 nM of fulvestrant. A portion of the cultures was isolated for RNA analysis at 0, 12 and 24 hours following treatment. After 48 hours in culture, 25 μg/ml of anti-CD16 anti-human monoclonal antibody (clone 3G8, Ancell Immunology Research Products) or 25 μg/ml of an IgG1 isotype control anti-human antibody (Sigma, St. Louis, MS) was added to a portion of the cultures. Antibody clone #3G8 specifically cross-links the CD16 receptor, inducing TNF-α and IL-1 cytokine release (Abrahams et al., 2000).
Real time RT-PCR analysis of gene transcript
To analyze the transcript levels, total RNA was isolated from the treated cells using the RNeasy RNA isolation kit (Qiagen, Valencia, CA). Total RNA was quantitated, and the RNA quality was determined using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA) following the manufacturer's directions. Reverse transcription was completed using the RETROscript First Strand Synthesis Kit (Ambion, Austin, TX) according to the manufacturer's directions by isolating the total RNA from the cells as a template. Quantitation of gene expression was completed using real-time PCR by incorporating Syber green I dye. The real time PCR reaction mixture contained 1X Brilliant SYBR Green QPCR Master Mix (Stratagene, La Jolla, CA), 0.3 μM of the CD16 primers or 18S primers (Table 1) and 2 μl of the reverse transcriptase reaction. PCR reactions were completed on a MX4000 multiplex quantitative PCR system (Stratagene) and consisted of a 10 min denaturation step at 95°C, followed by 40 cycles of 95°C for 30 seconds, 56°C for 1 minute and 72°C for 1.5 minutes and finally, a 10-minute 72°C extension. After this set of cycles was completed, a melting curve was generated by a 1-minute 95°C denaturation step and then by 81 cycles that increased by 0.5°C per cycle starting from 55°C. The data were gathered with MX4000 software (version 4). Control RNA samples that contained a mock reverse transcription reaction (no reverse transcriptase added) were used to verify the absence of contamination in the solutions. Standard curves were generated for both the gene-specific product and the 18S transcripts using increasing concentrations of the reverse transcriptase reaction from the calibrator samples. Calibrator samples (i.e., control samples) were untreated cells grown on Petri plates for 0 days and thus, were at basal conditions. Ct (threshold cycle) values obtained from the MX4000 software (Stratagene, La Jolla, CA) using an amplification-based threshold determination based on a fixed amplification position of 32.5% of the maximum first derivative were used in further calculations to determine relative gene expression. The maximum first derivative is set at 100%, a point near the end of the exponential amplification phase. Relative CD16 expression was calculated according to published methods (Pfaffl, 2001), Ct values were plotted on the y-axis and the amount of input cDNA on the x-axis to produce a standard curve and generate a slope value for both the gene-specific product and 18S genes was determined. PCR efficiency (E) for both genes was determined from the slope of the standard curve calculated as 10 (−1/slope). Relative gene expression was calculated using the formula below:
where Egene is the real-time PCR efficiency of the gene of interest transcript (i.e., CD16), and E18S is the real-time PCR efficiency of the 18S gene transcript. The Ct values were generated from a known quantity of the template obtained from the calibrator sample using the gene-specific and 18S primers. The Ct values from the calibrator cell samples above were subtracted from the Ct values generated using the template from the treated cells. The target gene expression was normalized against an unregulated reference gene (the ribosomal 18S gene) by dividing these subtracted Ct values. The assumption was that 18S was transcribed in such large amounts relative to all mRNA transcripts present in the cell that only global changes in cellular metabolism would appreciably affect the expression of 18S.
Table 1.
RT-PCR primer sets
| Primer ID | Primer sequence |
|---|---|
| Real time RT-PCR | |
| CD16 primers | 5′-gacagtgtgactctgaag-3′, 5′-gcacctgtactctccac-3′ |
| 18S primers | 5′- ccgaagcgtttactttga-3′, 5′-gccgtccctcttaatcat-3′ |
| ChIP analysis | |
| −353 to −50 of the CD16 5′ flanking region |
5′-gtggatccaaatccaggaga-3′, 5′-gggaccaaaccgactagaca-3′ |
| −864 to −257 of the CD16 5′ flanking region |
5′-accagcccagatccagtg-3′, 5′-aggtctgtggctgagcatct-3′ |
| −1113 to −820 of the CD16 5′ flanking region |
5′-gcctcctgggcttttctag-3′, 5′-gtcctgcggatttagctcag-3′ |
| Luciferase reporter constructs | |
| −1833 to + 62 of the CD16 gene | 5′ aaccagggagaccctgactta 3′, 5′ aatgaaaaccccctgaaagc 3′ |
| Sp1(−89) & Sp1(−83) | 5′-gggtgacagagatgtgtggaggtgctggtgaaaggctgtttac-3′ 5′-gtaaacagcctttcaccagcacctccacacatctctgtcaccc-3' |
| 1/2ERE(−251) & Sp1 (−242) | 5′-agatgctcagccacagaactttgagggagtaaagg-3′ 5′-cctttactccctcaaagttctgtggctgagcatct-3′ |
| 1/2ERE(−299) & Sp1 (−300) | 5′-gcaagcatcctgggatagctgaaggcatactctggcagattc-3′ 5′-gaatctgccagagtatgccttcagctatcccaggatgcttgc-3′ |
| ERE(−366) | 5′-atccccagggactcacagtcccattcttgg-3′ 5′-ccaagaatgggactgtgagtccctggggat-3′ |
ELISA analysis of TNF-α and IL-1β
The quantitative measurement of TNF-α and IL-1β was performed using commercially available sandwich ELISA kits and following the manufactures directions (Invitrogen, Carlsbad, CA). TNF-α and IL-1β were assessed 48 hours after the addition of anti-CD16 or the isotype IgG control antibody. The exact amounts of cytokine within the samples were determined by comparing them to standards generated from recombinant TNF-α or IL-1β protein.
Chromatin immunoprecipitation (ChIP) assay
The chromatin immunoprecipitation was performed utilizing the ChIP-IT chromatin Immunoprecipitation kit and shearing kit following the manufacturer's directions (Active Motif, Carlsbad, CA). Briefly, monocytes or U937 cells were cultured in 10% charcoal-stripped FBS, split in half, and treated with or without 1×10−8 M 17 β-estradiol; 24 hours later the ChIP assay was performed. Treated monocytes or U937 cells (2-3 × 107) were pelleted (250 RCF, 5 min), suspended in 1% formaldehyde/1X PBS for 10 minutes and rinsed in 10 ml PBS at 4°C. The cells were then pelleted and suspended in 10 ml of glycine buffer in PBS. Cells were pelleted, suspended in 1.5 ml of ice-cold lysis buffer/protease inhibitor cocktail/PMSF and dounced to release the nuclei. The cell lysates were pelleted (2400 RCF, 10 min, 4°C), resuspended in 350 μl of shearing buffer and sonicated at 4°C with sixteen 15-second pulses at 10% power and ten 15 second pulses at 50% power (Virsonic 60, Virtis Inc., Gardner, NY). Protein G beads were added to the sheared lysate. The samples were mixed for 2 hours at 4°C and then pelleted at 4000 RCF. The supernatants that represented the total chromatin “input DNA” were isolated. Four micrograms of antibody (IgG, Active Motif or ERα, MC-20, Santa Cruz Biotechnologies, Santa Cruz, CA) were added to 170 μl of total chromatin input DNA and incubated overnight at 4°C. Following the overnight incubation, 100 μl of protein G beads were added. The mixture was incubated for 1.5 hours at 4°C, and the beads were rinsed. DNA was eluted from the beads (1% SDS, 50 mM NaHCO3) and digested with RNaseA overnight at 65°C, followed by a proteinase K digestion at 42°C according to the manufacturer's directions. The DNA was purified using the DNA purification mini-columns provided in the kit.
The analysis of the areas where the ER interacts with the 5′ flanking region of the CD16 gene was completed by PCR analysis of three overlapping, consecutive regions using primers that amplified bases −353 to −50, −864 to −257 and bases −1113 to −820 of the CD16 5′ flanking region (Table 1). The PCR required 40 cycles of the following conditions: 94°C for 20 seconds, 55°C for 30 seconds, and 72°C for 30 seconds. The PCR products were resolved on a 1.5% agarose gel, stained with 0.5 μg/ml ethidium bromide and the images captured.
Production of reporter constructs
Construction of the CD16-luciferase reporter plasmid (CD16-Luc) required amplification of an 1895 bp sequence −1833 to + 62 of the 5′ region of the CD16 gene (GenBank accession# Z46222). Amplification was completed using the primers spanning that region (Table 1) and chromosomal DNA isolated from monocytes and 34 cycles of the following conditions: 94°C for 1 minute, 54°C for 1 minute and 72°C for 2 minutes. The PCR product was cloned into the PCR blunt II topo vector (Invitrogen, Carlsbad, CA), which was digested with SacI and XhoI, and the promoter fragment was isolated. To generate the final reporter construct CD16-Luc, the fragment was ligated into a predigested pGL3 basic vector (Promega, Madison, WI) containing the firefly luciferase gene. Potential ER binding sites, such as estrogen response elements or Sp1 consensus sequences within the CD16-Luc construct, were determined using two software packages called alibaba2 (www.gene-regulation.com) and Estrogen Response Element Finder, ERE-V2 (sdmc.lit.org.sg). Based on the binding sites predicted by this software and the chromosomal immunoprecipitation data site, directed mutations were made in four different regions of the sequence upstream of the CD16 gene. Either one, two or three nucleotides were changed simultaneously in each region using the site-directed mutagenesis kit from Stratagene (La Jolla, CA). The site-directed mutatgenesis protocol utilized the CD16-Luc construct as a template and one of four different primer sets (Table 1) to produce the constructs termed Sp1(−89) & Sp1(−83), 1/2ERE(−251) & Sp1 (−242), 1/2ERE(−299) & Sp1 (−300) and ERE(−366)
Transfection of primary monocytes
Cell transfections were completed by first mixing 3×106−1×107 monocytes in 100 μl of Human Monocyte Nucleofector Medium supplemented with 2 mM glutamine and 10% FCS, according to the manufacturer's directions (Amaxa, Gaithersburg, MD). Second, 1 μg of CD16-Luc or mutated CD16-Luc and 1 μg phRL-SV40 (Renilla luciferase plasmid with the SV40 promoter; Promega, Madison ,WI) were added to the solution and mixed. Cells mixed with DNA were transferred to an Amaxa cuvette and electroporated on program Y-001. They were immediately removed from the cuvette and placed in 1 ml of RPMI 1640 plus 2% charcoal-stripped FBS ± 1 × 10−8 M 17 βestradiol or the ERα-specific agonist 4,4′,4″-(4-Propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol (PPT) (Tocris Bioscience, Ellisville, MS) or the ERβ-specific agonist diarylpropionitrile (DPN) at a concentration of 1 × 10−9 M. After a 4-hour incubation at 37°C, the cells were harvested and the luciferase activity measured using the Dual-Luciferase® Reporter Assay System (Promega). Luminescent photon measurements were made on a liquid scintillation analyzer (Tricarb 2900TR, Packard BioScience Company, Meriden, CT) with the coincidence function disabled. Relative firefly luciferase activity was normalized to co-transfected Renilla luciferase activity to obtain luciferase activity plotted in arbitrary units termed “relative light units” (RLU).
Statistics
Statistical analysis was completed using a Mann Whitney, two-tailed t-test. For the real time RT-PCR data two-way analysis of variance (ANOVA) was performed using treatment and time as variables. Significance is indicated if p < 0.05. All values are given as the mean ± SEM. Three independent experiments were completed for all treatment groups.
Results
THP-1 cells have a significantly higher CD16 transcript level after estrogen withdrawal in comparison to monocytes continuously treated with 10−8M and 10−9M 17 β-estradiol for the same amount of time (Fig. 1A and B). Similar results were found in the primary monocytes (Table 2). A withdrawal paradigm more closely simulates the real life scenario in that cells are exposed to estrogen such as during pregnancy or in a female having normal menstral cycles and then the estrogen is withdrawn post-partum and at menopause. Thus, we wanted to simulate the in vivo scenario but from our previous studies (Kramer et al., 2004) it does not appear to make a significant difference as to whether the cells were not treated with estrogen or whether the estrogen was withdrawn. The THP-1 cells treated with the ERα and ERβ antagonist fulvestrant (100 or 1000 nM), concurrent with exposing THP-1 cells to 10−8M 17 β-estradiol did not show a change in the level of CD16 transcript (Fig. 2A). The withdrawal of estrogen increased the CD16 transcript levels, and this increase after withdrawal was observed with antagonist added (Fig. 2B). Increasing the dose of antagonist did not increase the amount of CD16 gene expression further (Fig. 2B). No significant change in the CD16 levels was observed in the monocytes treated with three concentrations of the ERα and ERβ antagonist in media lacking 17 β-estradiol (Fig. 2C).
Figure 1. Estrogen modulates CD16 transcript levels in THP-1 monocytes.
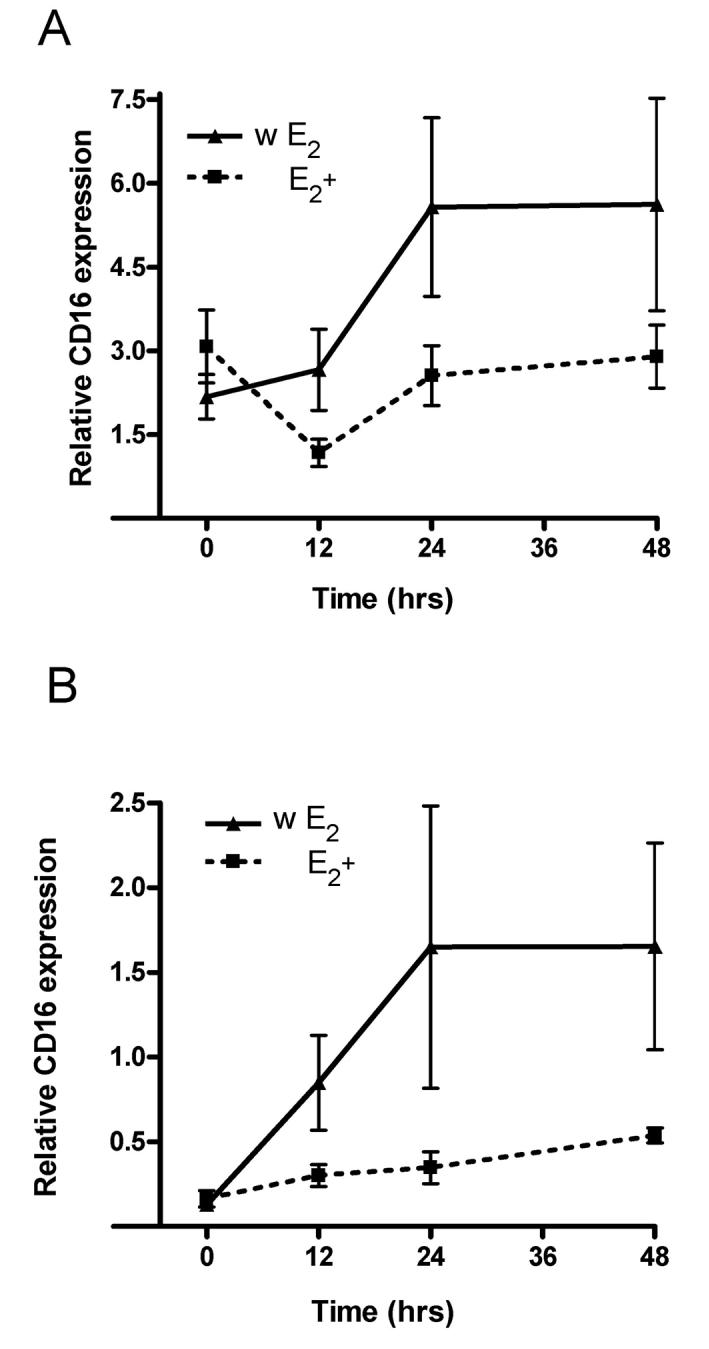
In panel A, cells were treated with 1 × 10−8 M 17 β-estradiol (E2+), and in panel B, cells were treated with 1 × 10−9 M 17 β-estradiol (E2+). In a portion of the cultures treated with 17 β-estradiol, the hormone was withdrawn (w E2). Samples were collected at 0, 12, 24 and 48 hours after hormonal treatment or withdrawal of hormone. Transcript levels for CD16 were analyzed using real-time PCR. Two-way analysis of variance (ANOVA) was completed for the data in panel A and panel B. ANOVA results indicate that the data for the two treatment groups (E2+ and w E2) in panel A and in panel B were significantly different p<0.05. Three experiments were performed for each treatment at each time point. Relative CD16 expression was calculated in reference to the amount of 18S RNA present within the cells. Values are the mean ± SEM.
Table 2.
CD16 transcript levels in monocytes treated with and without 17 β-estradiol
| Time (hrs) | 10−8M 17 β-estradiol | Media without 17 β-estradiol |
|---|---|---|
| 0 | 3.1 ± 0.65 | 5.1 ± 1.4 |
| 12 | 1.2 ± 0.24 | 3.6 ± 0.4 |
| 24 | 2.5 ±.0.53 | 4.6 ± 0.6 |
Cells were collected 0, 12, and 24 hours after estrogen treatment, and CD16 levels were determined by real-time PCR. At least six experiments were performed for each treatment at each time. Relative CD16 expression was calculated in reference to the amount of 18S RNA present within the cells. Values are given as the mean ± SEM.
Figure 2. ER antagonist fulvestrant effects CD16 levels in THP-1 monocytes treated with high physiological levels of estrogen.
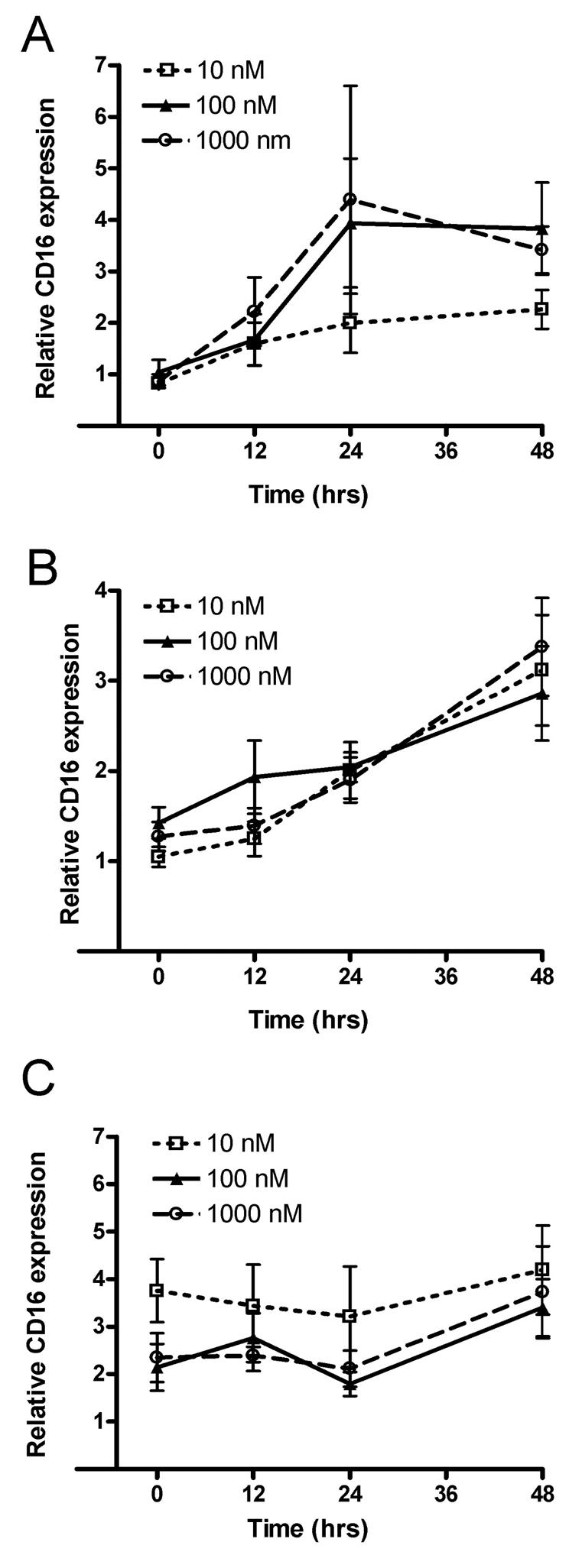
In panel A, monocytes were treated with 1 × 10−8 M 17 β-estradiol and three different concentrations of fulvestrant (i.e., ICI 182,780), starting at time zero. In panel B, monocytes were initially treated with 1 × 10−8 M 17 β-estradiol and then had 17 β-estradiol withdrawn concomitant with addition of fulvestrant. In panel C, monocytes were cultured in media without the addition of 17 β-estradiol, and fulvestrant was added. After the addition of fulvestrant cells were collected at 0, 12, 24 and 48 hours. CD16 transcript levels were determined by real-time PCR. Three experiments were performed for each treatment at each time. Relative CD16 was calculated in reference to the amount of 18S RNA present within the cells. Values are the mean ± SEM.
To determine how estrogen and the estrogen receptor function in monocyte activity, the TNF-α and IL-1β levels were quantitated after activating the monocytes by cross-linking the CD16 receptor. The TNF-α release (Fig. 3A) and production (Fig. 3B) significantly decreased after CD16 cross-linking in the estrogen-treated cultures. The fulvestrant blockade of the estrogen receptor prevented this reduction in TNF-α release (Fig. 3A) and production (Fig. 3B). Studies of IL-1β showed that a significant decrease in production is detected after estrogen treatment and that this decrease can be blocked by fulvestrant administration; no estrogen (29±7 pg/ml); with estrogen (15±2.7 pg/ml); with estrogen and fulvestrant (25±2.6 pg/ml); experimental replicates ≥ 3.
Figure 3. TNF-α expression in activated primary monocytes in the presence of estrogen and fulvestrant.
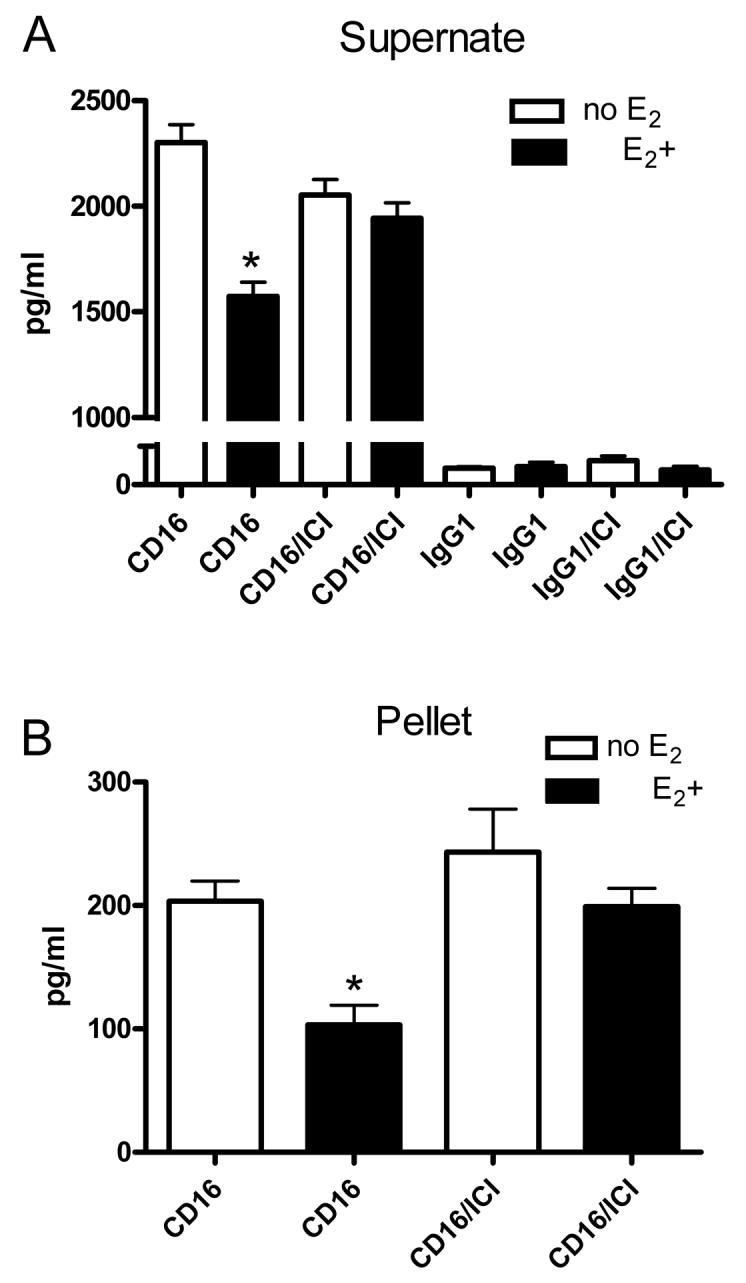
Primary monocytes were cultured without (no E2) or with (E2+) 1 × 10−8 M 17 β-estradiol. A portion of these cultures was treated with 100 nm fulvestrant (i.e., ICI 182, 780). After 48 hours, antibody against CD16 (25 μg/ml) or an IgG1 isotype control antibody (25 μg/ml) was added. The supernate (A) and cell pellets (B) were collected 48 hours after addition of antibody and the amount of TNF-α quantitated in each by ELISA. Values represent the mean amount of TNF-α (pg) from 1 ml of cultured cells. * = P<0.05 comparing estrogen treated to non-estrogen treated cells. Four experiments were performed for each treatment group.
Chromosomal immunoprecipitation of DNA from monocytes showed that in the presence of estrogen, the ERα associated to a greater extent with a CD16 promoter region encompassing bases −353 to −50, as indicated by a greater amount of PCR product from the estrogen-treated monocytes (Fig. 4A, compare lanes 5 and 6). A smear of DNA was detected from the estrogen-treated monocytes after amplification of bases encompassing −864 to −257 (Fig. 4B, compare lanes 9 and 10); a distinct band could not be produced using varied PCR conditions. The promoter sequence further upstream of base −864 did not show any change in the immunoprecipitated product due to estrogen treatment (Fig. 4 C, compare lanes 12 and 13). Multiple consensus elements and the potential transcription factor binding sites known to interact with the ERα or ERβ were found in the 353 to −50 region (Fig. 4D). As a positive control, non-precipitated, input DNA from both estrogen and non-estrogen-treated monocytes was used as a template for PCR. Using this input DNA as a template, PCR produced a distinct band for all three primer sets encompassing the CD16 promoter and, as expected, hormonal treatment did not affect the level of PCR product (Fig. 4, lanes 4, 7, 8, 11, 14 and 15). As a negative control, IgG antibodies did not immunoprecipitate the CD16 promoter region and gave no PCR product (Fig. 4A, lane 1).
Figure 4. Chromatin immunoprecipitation (ChIP) analysis of the CD16 promoter in the monocytic cell line U937 and in primary monocytes.
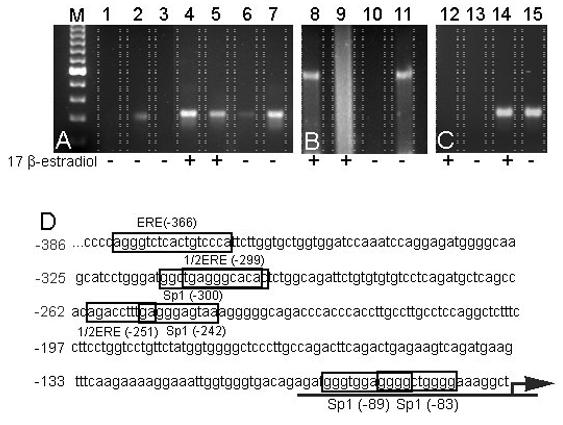
Chromatin was isolated from U937 cells (lanes 1 through 3) and from primary monocytes (lanes 4 through 15). Chromatin was isolated from cells treated for 24 hours with or without 1 × 10−8 M 17 β-estradiol, as indicated by “+” or “−“ below each lane. The first lane shows a 100 base pair ladder (M). In lane 1, chromatin was immunoprecipitated with a non-specific IgG control antibody, and in lane 2, chromatin was immunoprecipitated with an ERα specific antibody followed by PCR amplification of a promoter region of the CD16 gene. Lane 3 represents a negative control PCR reaction using no chromatin as a template. Additional PCR reactions used template from either total chromatin, as a positive control (lanes 4, 7, 8, 11, 14 and 15) or chromatin immunoprecipitated with an ERα specific antibody (lanes 2, 5, 6, 9, 10, 12 and 13). PCR products from the ChIP analysis were resolved on an agarose gel, stained with ethidium bromide and imaged. Images are representative of duplicate experiments. In panel A, the PCR product comprised bases −353 to −50 of the CD16 5′ flanking region. In panel B, the PCR product included bases −864 to −257, and in panel C, the PCR product included bases −1113 to −820 of the CD16 5′ flanking region. Panel D shows the upstream promoter regions of the CD16 gene from bases −386 through −74. Estrogen response elements (ERE) or estrogen response element half-sites (1/2ERE), as well as Sp1 consensus binding sequences, are shown as boxed regions. Arrow indicates the direction of transcription.
To further define which sites in the CD16 promoter are used by estrogen to regulate expression, putative transcription factor binding sites were identified and mutated. These sites had consensus sequences that could potentially bind ERα or ERβ. Mutations were made in the reporter construct containing 1,833 base pairs upstream of the CD16 gene. Mutation of the four potential transcription binding sites in the CD16 promoter included changing one, two or three base pairs at each site (Fig. 5A, B). Transfection of these constructs in the primary monocytes in the presence or absence of estrogen indicated that mutating the 1/2ERE (−299):Sp1 (−300) site encompassing bases −299 to −313 prevented estrogenic action (Fig. 5C). A mutation at the Sp1 sites between bases −83 to −99 lowered the CD16 expression. A mutation at the estrogen response element between bases −366 to −381 increased the CD16 expression but the estrogen continued to significantly lower the CD16 expression (Fig. 5C). A mutation at the remaining sequence had no effect.
Figure 5. Expression of mutated CD16 promoter constructs.

Six different DNA constructs were generated for transfection into the primary monocytes. One construct had an SV40 promoter driving luciferase expression (positive control), one construct had a 1,833 base region of the CD16 promoter driving luciferase expression, and the remaining four constructs had site-directed mutations of the 1,833 base CD16 promoter, as indicated in panel A. Two of the mutated regions have three simultaneous base changes, one has two simultaneous base changes, and one had a single base change. Arrowhead points to the wild type base that was mutated in the sense strand of the CD16 promoter sequence and the bold letter above the arrowhead is the mutated base that was incorporated. Relative positions of these mutations within the 1,833 base CD16 promoter are shown in panel B. Primary monocytes transfected with one of the six constructs shown in panel B were cultured with (E2+) or without (no E2) 1 × 10−8 M 17 β-estradiol. Luciferase expression from each construct in panel B is aligned horizontally with the bar graph in panel C. Letter “a” over each bar in panel C indicates a significant difference (p<0.05) between the luminescence of cells treated with or without 17 β-estradiol after transfection with a particular DNA construct. Letter “b” indicates a significant difference (p<0.05) between the luminescence of cells transfected with the mutant construct and cells transfected with the intact CD16 promoter fragment (−1833 to −1). In panel D the cells were transfected with a reporter construct containing the CD16 promoter driving luciferase expression. After transfection cells were treated with or without 1 × 10−8 M 17 β-estradiol or the ERα-specific agonist PPT or the ERβ-specific agonist DPN at a concentration of 1 × 10−9 M. Comparing treatment groups significant differences are indicated by * = P<0.05, *** = P<0.001. Relative light units (RLU) represent the luminescent signal from the firefly luciferase normalized to an internal control Renilla luciferase construct. Three experiments were performed for each treatment group transfected with the site directed mutagenesis constructs and four experiments were performed for each group treated with the estrogen receptor antagonists.
To verify that estrogen utilizes the CD16 promoter to modulate expression, luciferase activity from a reporter construct containing 1,833 bases of the CD16 promoter was transfected into primary monocytes. The estrogen significantly suppressed the luciferase activity from the CD16 promoter/luciferase construct (Fig. 5). The role that ERα or ERβ have in the estrogenic effect was tested by adding ERα agonist PPT or ERβ agonist DPN to the media after transfection of the CD16 promoter/luciferase construct. At a concentration of 1×10−9 M, both PPT and DPN significantly lowered the luciferase activity compared to the non-estrogen treated monocytes (Fig. 5D). The addition of both PPT and DPN led to an even greater reduction in luciferase activity (Fig. 5D).
Discussion
Estrogen has been found to modulate CD16 expression, causing changes in monocytic cytokine release upon receptor activation (Kramer et al., 2004). For example, the CD16 levels increased after estrogen withdrawal and were consistently high in non-estrogen treated cells. The process by which estrogen affects CD16 expression is unclear, so the aim of this study was to test the role of ERα and ERβ in modulating CD16 expression due to changes in estrogen concentration. We showed that estrogen decreased CD16 expression and cytokine production, which is consistent with previous results, and that the ERα and ERβ antagonist fulvestrant reduced this inhibitory effect. In those cells from which estrogen was withdrawn or were nonestrogen-treated, fulvestrant caused no additional rise in CD16 levels. Treatment with ERα and ERβ agonists PPT and DPN suggest that both ERα and ERβ can suppress CD16 expression A region in the CD16 promoter was associated with ERα to a greater extent after estrogen treatment, consistent with the idea that a decrease in CD16 expression upon estrogen treatment was partly dependent on ERα. Mutation of several putative transcription factor binding sites within in the CD16 promoter indicated that the sequence between bases −299 to −313 was necessary for estrogen to suppress CD16 expression. Interestingly, this region has the potential to interact with ERα and ERβ and convey estrogen signals within this promoter.
Antagonist affects ER function
To determine the role of the ERα and ERβ in transmitting the effect of estrogen on the CD16 expression, we treated monocytes with the antagonist fulvestrant, which competitively inhibits binding of estrogen to ERα and ERβ (Wakeling and Bowler, 1988; Tremblay et al., 1997; Kuiper et al., 1997; Wakeling, 2000). The fulvestrant binding affinity is 89% that of estrogen, while the affinity of tamoxifen is significantly lower (Wakeling and Bowler, 1987). Fulvestrant binding impairs receptor dimerization and blocks nuclear localization of the receptor (Fawell et al., 1990; Dauvois et al., 1993). The remaining receptor that does localize to the nucleus and dimerize will be transcriptionally inactive because both the AF1 and AF2 are disabled due to fulvestrant binding. Fulvestrant-bound receptor is unstable, resulting in accelerated degradation of receptor protein compared with estradiol- or tamoxifen (Nicholson et al., 1995) but the mRNA levels do not decrease. Thus, fulvestrant binds, blocks and accelerates the degradation of receptor protein leading to complete inhibition of estrogen signaling (Wakeling, 1995; Wardley, 2002; Osborne et al., 2004). Estrogen suppressed the CD16 transcript levels and cytokine production from activated monocytes. The addition of the ERα and ERβ antagonist fulvestrant blocked estrogen binding to the receptor, leading to increased levels of CD16 but only at higher concentrations (i.e., 100 nM, 1000 nM), which suggested a dose-dependent effect. A dose of 100 nM fulvestrant also prevented the estrogen from decreasing the cytokine production after cross-linking CD16 receptors, suggesting that estrogen reduces cytokine production and the level of receptors present through an estrogen receptor-dependent mechanism.
Genomic action of estrogen at the CD16 promoter
Previous studies indicated that actinomycin D blockade of de novo transcriptional processes prevented estrogen modulation of CD16 expression (Kramer et al., 2004). Data from our lab also showed that changes in the CD16 transcript levels due to the withdrawal or treatment of estrogen can not be observed until after 24 hours. Nongenomic ER action occurs rapidly, generally in less than an hour also, non-genomic action, such as modulation of signal transduction, does not usually result in a transcriptional event (Sak and Everaus, 2004; Levin, 2005). Because changes in CD16 expression were slow and resulted in a change in transcript levels, estrogen is likely acting through a genomic pathway and not a signal transduction pathway. Further evidence that estrogen modulates transcription through a genomic pathway is that the ERα is associated with the CD16 5′ flanking region between bases −353 to −50.
Estrogen action at the CD16 promoter
Chromatin immunoprecipitation analysis demonstrated an interaction between ERα and the CD16 promoter after estrogen treatment. The promoter sequence contains several estrogen response elements and several Sp1 consensus sequences. Mutational analysis indicated that one particular site from −299 to −313, having homology to both an estrogen response element and a Sp1 binding site, was necessary for estrogen to suppress CD16 expression. Moreover, the treatment of monocytes transfected with a CD16 promoter, luciferase reporter construct demonstrated that both ERα and ERβ agonists significantly reduce luciferase production. Studies have shown that 17 β-estradiol can inhibit Sp1 protein production by utilizing either ERα or ERβ (Ling et al., 2001∼). MCP-1 gene expression was also inhibited when estrogen, bound to ERβ, blocked Sp1 transcriptional activities (Kanda and Watanabe, 2003). Luteinizing hormone receptor is another gene having estrogen response element half-sites in the promoter that can be inhibited by estrogen (Geng et al., 1999); demonstrating further that estrogen bound to the ERα or ERβ can inhibit gene expression through interacting with that genes promoter. Further studies will be needed to determine how the ERα or ERβ interacts with the CD16 promoter. For example, the ER can interact directly with the promoter by binding to the promoter sequence or the ER can act indirectly by affecting the function of other transcription factors.
Estrogen and CD16 linked to disease
The CD16 receptor binds self antigens and rheumatoid factors that are potential triggers for onset or maintenance of auto-immune diseases such as rheumatoid arthritis (Pope et al., 1974; Nardella et al., 1981; Klaassen et al., 1988; Edwards and Cambridge, 1998). The expression of CD16 on monocytes is restricted to tissues impacted by rheumatoid arthritis (Bhatia et al., 1998) and individuals with rheumatoid arthritis have shown higher levels of CD16 expression in these tissues (Kawanaka et al., 2002; Masuda et al., 2003a; Masuda et al., 2003b). Higher levels of CD16 can contribute to greater joint destruction and inflammation (Baeten et al., 2000; Kawanaka et al., 2002; Yano et al., 2007). Moreover, CD16 has been linked to arthritis processes in mice models, including collagen-induced arthritis (CIA) (Kleinau et al., 2000; Diaz et al., 2002; Kagari et al., 2003). Together these studies suggest that estrogen can affect auto-immune disease symptoms by modulating CD16 expression.
To summarize, it has been reported that women have a higher incidence of auto-immune disorders, such as systemic lupus erythematuosus, scleroderma, Sjogrens syndrome, Graves disease and rheumatoid arthritis (Ansar et al., 1985; Felten, 1993) but the mechanisms for this higher incidence are still unclear. Our results show that CD16 expression is reduced by estrogen and that reduced CD16 levels lower the amount of proinflammatory cytokine release from monocytes upon cross-linking the CD16 receptor with antibody. Estrogen modulates CD16 expression by a mechanism that can utilize ERα or ERβ and where ERα interacts with the CD16 promoter. Thus, by estrogen modulating CD16 expression and cytokine release resulting from CD16 cross-linking, changes in estrogen can affect the inflammation and joint degradation processes subsequently affecting the incidence of auto-immune disorders found in women.
Acknowledgements
This work was supported by a grant DE015372-01 from the NIH institutes National Institute of Dental and Craniofacial Research (NIDCR) and Office of Research on Women's Health (ORWH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrahams VM, Cambridge G, Lydyard PM, Edwards JC. Induction of tumor necrosis factor alpha production by adhered human monocytes: a key role for Fcgamma receptor type IIIa in rheumatoid arthritis. Arthritis Rheum. 2000;43:608–616. doi: 10.1002/1529-0131(200003)43:3<608::AID-ANR18>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Ansar AS, Penhale WJ, Talal N. Sex hormones, immune responses, and autoimmune diseases. Mechanisms of sex hormone action. Am. J. Pathol. 1985;121:531–551. [PMC free article] [PubMed] [Google Scholar]
- Auwerx J. The human leukemia cell line, THP-1: a multifacetted model for the study of monocyte-macrophage differentiation. Experientia. 1991;47:22–31. doi: 10.1007/BF02041244. [DOI] [PubMed] [Google Scholar]
- Baeten D, Boots AM, Steenbakkers PG, Elewaut D, Bos E, Verheijden GF, Berheijden G, Miltenburg AM, Rijnders AW, Veys EM, De KF. Human cartilage gp-39+,CD16+ monocytes in peripheral blood and synovium: correlation with joint destruction in rheumatoid arthritis. Arthritis Rheum. 2000;43:1233–1243. doi: 10.1002/1529-0131(200006)43:6<1233::AID-ANR6>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Beaven MA, Metzger H. Signal transduction by Fc receptors: the Fc epsilon RI case. Immunol Today. 1993;14:222–226. doi: 10.1016/0167-5699(93)90167-j. [DOI] [PubMed] [Google Scholar]
- Benhamou M, Siraganian RP. Protein-tyrosine phosphorylation: an essential component of Fc epsilon RI signaling. Immunol Today. 1992;13:195–197. doi: 10.1016/0167-5699(92)90152-w. [DOI] [PubMed] [Google Scholar]
- Bhatia A, Blades S, Cambridge G, Edwards JC. Differential distribution of Fc gamma RIIIa in normal human tissues and co-localization with DAF and fibrillin-1: implications for immunological microenvironments. Immunology. 1998;94:56–63. doi: 10.1046/j.1365-2567.1998.00491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutolo M, Sulli A, Seriolo B, Accardo S, Masi AT. Estrogens, the immune response and autoimmunity. Clin. Exp. Rheumatol. 1995;13:217–226. [PubMed] [Google Scholar]
- Cutolo M, Villaggio B, Bisso A, Sulli A, Coviello D, Dayer JM. Presence of estrogen receptors in human myeloid monocytic cells (THP-1 cell line) Eur Cytokine Netw. 2001;12:368–372. [PubMed] [Google Scholar]
- Dauvois S, White R, Parker MG. The antiestrogen ICI 182780 disrupts estrogen receptor nucleocytoplasmic shuttling. J Cell Sci. 1993;106(Pt 4):1377–1388. doi: 10.1242/jcs.106.4.1377. [DOI] [PubMed] [Google Scholar]
- Diaz d. S., Andren M, Martinsson P, Verbeek JS, Kleinau S. Expression of FcgammaRIII is required for development of collagen-induced arthritis. Eur. J Immunol. 2002;32:2915–2922. doi: 10.1002/1521-4141(2002010)32:10<2915::AID-IMMU2915>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Edwards JC, Cambridge G. Rheumatoid arthritis: the predictable effect of small immune complexes in which antibody is also antigen. Br. J. Rheumatol. 1998;37:126–130. doi: 10.1093/rheumatology/37.2.126. [DOI] [PubMed] [Google Scholar]
- Fawell SE, White R, Hoare S, Sydenham M, Page M, Parker MG. Inhibition of estrogen receptor-DNA binding by the “pure” antiestrogen ICI 164,384 appears to be mediated by impaired receptor dimerization. Proc. Natl. Acad. Sci U. S. A. 1990;87:6883–6887. doi: 10.1073/pnas.87.17.6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felten DL. Epidemiology of the rheumatic diseases. In: McCarty DJ, Koopman WJ, editors. Arthritis and allied conditions. A textbook of rheumatology. Lea & Febiger; Philadelphia: 1993. pp. 17–48. [Google Scholar]
- Fenton MJ, Vermeulen MW, Clark BD, Webb AC, Auron PE. Human pro-IL-1 beta gene expression in monocytic cells is regulated by two distinct pathways. J. Immunol. 1988;140:2267–2273. [PubMed] [Google Scholar]
- Friedman D, Netti F, Schreiber AD. Effect of estradiol and steroid analogues on the clearance of immunoglobulin G-coated erythrocytes. J. Clin. Invest. 1985;75:162–167. doi: 10.1172/JCI111669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y, Tsai-Morris CH, Zhang Y, Dufau ML. The human luteinizing hormone receptor gene promoter: activation by Sp1 and Sp3 and inhibitory regulation. Biochem. Biophys. Res. Commun. 1999;263:366–371. doi: 10.1006/bbrc.1999.1374. [DOI] [PubMed] [Google Scholar]
- Gomez F, Ruiz P, Bernal JA, Escobar M, Garcia-Egido A, Lopez-Saez JJ. Enhancement of splenic-macrophage fcgamma receptor expression by treatment with estrogens. Clin. Diagn. Lab. Immunol. 2001;8:806–810. doi: 10.1128/CDLI.8.4.806-810.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hench PS. The ameliorating effect of pregnancy on chronic atrophi (infectious rheumatoid) arthritis, fibrosis and interim heat hydarthrosis. Mayo Clin.Proc. 1938;13:161–167. Ref Type: Journal (Full) [Google Scholar]
- Henderson B, Pettipher ER. Arthritogenic actions of recombinant IL-1 and tumour necrosis factor alpha in the rabbit: evidence for synergistic interactions between cytokines in vivo. Clin. Exp. Immunol. 1989;75:306–310. [PMC free article] [PubMed] [Google Scholar]
- Holmdahl R, Jansson L, Meyerson B, Klareskog L. Oestrogen induced suppression of collagen arthritis: I. Long term oestradiol treatment of DBA/1 mice reduces severity and incidence of arthritis and decreases the anti type II collagen immune response. Clin. Exp. Immunol. 1987;70:372–378. [PMC free article] [PubMed] [Google Scholar]
- Kagari T, Tanaka D, Doi H, Shimozato T. Essential role of Fc gamma receptors in anti-type II collagen antibody-induced arthritis. J Immunol. 2003;170:4318–4324. doi: 10.4049/jimmunol.170.8.4318. [DOI] [PubMed] [Google Scholar]
- Kanda N, Watanabe S. 17Beta-estradiol inhibits MCP-1 production in human keratinocytes. J Invest Dermatol. 2003;120:1058–1066. doi: 10.1046/j.1523-1747.2003.12255.x. [DOI] [PubMed] [Google Scholar]
- Kawanaka N, Yamamura M, Aita T, Morita Y, Okamoto A, Kawashima M, Iwahashi M, Ueno A, Ohmoto Y, Makino H. CD14+,CD16+ blood monocytes and joint inflammation in rheumatoid arthritis. Arthritis Rheum. 2002;46:2578–2586. doi: 10.1002/art.10545. [DOI] [PubMed] [Google Scholar]
- Klaassen RJ, Goldschmeding R, Tetteroo PA, Von dem Borne AE. The Fc valency of an immune complex is the decisive factor for binding to low-affinity Fc gamma receptors. Eur. J. Immunol. 1988;18:1373–1377. doi: 10.1002/eji.1830180911. [DOI] [PubMed] [Google Scholar]
- Kleinau S, Martinsson P, Heyman B. Induction and suppression of collagen-induced arthritis is dependent on distinct fcgamma receptors. J Exp. Med. 2000;191:1611–1616. doi: 10.1084/jem.191.9.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer PR, Kramer SF, Guan G. 17 beta-estradiol regulates cytokine release through modulation of CD16 expression in monocytes and monocyte-derived macrophages. Arthritis Rheum. 2004;50:1967–1975. doi: 10.1002/art.20309. [DOI] [PubMed] [Google Scholar]
- Kramer PR, Wray S. 17-Beta-estradiol regulates expression of genes that function in macrophage activation and cholesterol homeostasis. J. Steroid Biochem. Mol. Biol. 2002;81:203–216. doi: 10.1016/s0960-0760(02)00065-1. [DOI] [PubMed] [Google Scholar]
- Krieg SA, Krieg AJ, Shapiro DJ. A unique downstream estrogen responsive unit mediates estrogen induction of proteinase inhibitor-9, a cellular inhibitor of IL-1beta- converting enzyme (caspase 1) Mol. Endocrinol. 2001;15:1971–1982. doi: 10.1210/mend.15.11.0719. [DOI] [PubMed] [Google Scholar]
- Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138:863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- Levin ER. Integration of the Extra-nuclear and Nuclear Actions of Estrogen. Mol. Endocrinol. 2005 doi: 10.1210/me.2004-0390. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S, Deng G, Ives HE, Chatterjee K, Rubanyi GM, Komesaroff PA, Sudhir K. Estrogen inhibits mechanical strain-induced mitogenesis in human vascular smooth muscle cells via down-regulation of Sp-1. Cardiovasc. Res. 2001;50:108–114. doi: 10.1016/s0008-6363(01)00200-0. [DOI] [PubMed] [Google Scholar]
- Masuda M, Morimoto T, De Haas M, Nishimura N, Nakamoto K, Okuda K, Komiyama Y, Ogawa R, Takahashi H. Increase of soluble FcgRIIIa derived from natural killer cells and macrophages in plasma from patients with rheumatoid arthritis. J. Rheumatol. 2003a;30:1911–1917. [PubMed] [Google Scholar]
- Masuda M, Morimoto T, Kobatake S, Nishimura N, Nakamoto K, Dong XH, Komiyama Y, Ogawa R, Takahashi H. Measurement of soluble Fcgamma receptor type IIIa derived from macrophages in plasma: increase in patients with rheumatoid arthritis. Clin. Exp. Immunol. 2003b;132:477–484. doi: 10.1046/j.1365-2249.2003.02168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson R, Mattsson A, Holmdahl R, Whyte A, Rook GA. Maintained pregnancy levels of oestrogen afford complete protection from post-partum exacerbation of collagen-induced arthritis. Clin. Exp. Immunol. 1991;85:41–47. doi: 10.1111/j.1365-2249.1991.tb05679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh NJ. Pregnancy loss, menopause, and the onset of rheumatoid arthritis. Ann. Rheum. Dis. 1990;49:817–818. doi: 10.1136/ard.49.10.817-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardella FA, Teller DC, Mannik M. Studies on the antigenic determinants in the self-association of IgG rheumatoid factor. J. Exp. Med. 1981;154:112–125. doi: 10.1084/jem.154.1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson RI, Gee JM, Manning DL, Wakeling AE, Montano MM, Katzenellenbogen BS. Responses to pure antiestrogens (ICI 164384, ICI 182780) in estrogen-sensitive and -resistant experimental and clinical breast cancer. Ann. N. Y. Acad. Sci. 1995;761:148–163. doi: 10.1111/j.1749-6632.1995.tb31376.x. [DOI] [PubMed] [Google Scholar]
- Oliver JM, Seagrave J, Stump RF, Pfeiffer JR, Deanin GG. Signal transduction and cellular response in RBL-2H3 mast cells. Prog. Allergy. 1988;42:185–245. [PubMed] [Google Scholar]
- Osborne CK, Wakeling A, Nicholson RI. Fulvestrant: an oestrogen receptor antagonist with a novel mechanism of action. Br. J Cancer. 2004;90(Suppl 1):S2–S6. doi: 10.1038/sj.bjc.6601629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostensen M, Aune B, Husby G. Effect of pregnancy and hormonal changes on the activity of rheumatoid arthritis. Scand. J. Rheumatol. 1983;12:69–72. doi: 10.3109/03009748309102886. [DOI] [PubMed] [Google Scholar]
- Paolini R, Numerof R, Kinet JP. Kinase activation through the high-affinity receptor for immunoglobulin E. Immunomethods. 1994;4:35–40. doi: 10.1006/immu.1994.1005. [DOI] [PubMed] [Google Scholar]
- Persellin RH. The effect of pregnancy on rheumatoid arthritis. Bull. Rheum. Dis. 1976;27:922–927. [PubMed] [Google Scholar]
- Pettipher ER, Higgs GA, Henderson B. Interleukin 1 induces leukocyte infiltration and cartilage proteoglycan degradation in the synovial joint. Proc. Natl. Acad. Sci. U. S. A. 1986;83:8749–8753. doi: 10.1073/pnas.83.22.8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polan ML, Kuo A, Loukides J, Bottomly K. Cultured human luteal peripheral monocytes secrete increased levels of interleukin-1. J. Clin. Endocrinol. Metab. 1990;70:480–484. doi: 10.1210/jcem-70-2-480. [DOI] [PubMed] [Google Scholar]
- Pope RM, Teller DC, Mannik M. The molecular basis of self-association of antibodies to IgG (rheumatoid factors) in rheumatoid arthritis. Proc. Natl. Acad. Sci. U. S. A. 1974;71:517–521. doi: 10.1073/pnas.71.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottratz ST, Bellido T, Mocharla H, Crabb D, Manolagas SC. 17 beta-Estradiol inhibits expression of human interleukin-6 promoter- reporter constructs by a receptor-dependent mechanism. J. Clin. Invest. 1994;93:944–950. doi: 10.1172/JCI117100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sak K, Everaus H. Nongenomic effects of 17beta-estradiol--diversity of membrane binding sites. J Steroid Biochem. Mol. Biol. 2004;88:323–335. doi: 10.1016/j.jsbmb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Saklatvala J. Tumour necrosis factor alpha stimulates resorption and inhibits synthesis of proteoglycan in cartilage. Nature. 1986;322:547–549. doi: 10.1038/322547a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Weitzmann MN, Cenci S, Ross FP, Adler S, Pacifici R. Estrogen decreases TNF gene expression by blocking JNK activity and the resulting production of c-Jun and JunD. J. Clin. Invest. 1999;104:503–513. doi: 10.1172/JCI7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay GB, Tremblay A, Copeland NG, Gilbert DJ, Jenkins NA, Labrie F, Giguere V. Cloning, chromosomal localization, and functional analysis of the murine estrogen receptor beta. Mol. Endocrinol. 1997;11:353–365. doi: 10.1210/mend.11.3.9902. [DOI] [PubMed] [Google Scholar]
- Vegeto E, Bonincontro C, Pollio G, Sala A, Viappiani S, Nardi F, Brusadelli A, Viviani B, Ciana P, Maggi A. Estrogen prevents the lipopolysaccharide-induced inflammatory response in microglia. J. Neurosci. 2001;21:1809–1818. doi: 10.1523/JNEUROSCI.21-06-01809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeling AE. Use of pure antioestrogens to elucidate the mode of action of oestrogens. Biochem. Pharmacol. 1995;49:1545–1549. doi: 10.1016/0006-2952(94)00528-t. [DOI] [PubMed] [Google Scholar]
- Wakeling AE. Similarities and distinctions in the mode of action of different classes of antioestrogens. Endocr. Relat Cancer. 2000;7:17–28. doi: 10.1677/erc.0.0070017. [DOI] [PubMed] [Google Scholar]
- Wakeling AE, Bowler J. Steroidal pure antioestrogens. J. Endocrinol. 1987;112:R7–10. doi: 10.1677/joe.0.112r007. [DOI] [PubMed] [Google Scholar]
- Wakeling AE, Bowler J. Novel antioestrogens without partial agonist activity. J Steroid Biochem. 1988;31:645–653. doi: 10.1016/0022-4731(88)90014-3. [DOI] [PubMed] [Google Scholar]
- Wardley AM. Fulvestrant: a review of its development, pre-clinical and clinical data. Int. J Clin. Pract. 2002;56:305–309. [PubMed] [Google Scholar]
- Welch GR, Wong HL, Wahl SM. Selective induction of Fc gamma RIII on human monocytes by transforming growth factor-beta. J. Immunol. 1990;144:3444–3448. [PubMed] [Google Scholar]
- Wirthmueller U, Kurosaki T, Murakami MS, Ravetch JV. Signal transduction by Fc gamma RIII (CD16) is mediated through the gamma chain. J. Exp. Med. 1992;175:1381–1390. doi: 10.1084/jem.175.5.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooley PH, Whalen JD, Chapman DL, Berger AE, Richard KA, Aspar DG, Staite ND. The effect of an interleukin-1 receptor antagonist protein on type II collagen-induced arthritis and antigen-induced arthritis in mice. Arthritis Rheum. 1993;36:1305–1314. doi: 10.1002/art.1780360915. [DOI] [PubMed] [Google Scholar]
- Yano R, Yamamura M, Sunahori K, Takasugi K, Yamana J, Kawashima M, Makino H. Recruitment of CD16+ monocytes into synovial tissues is mediated by fractalkine and CX3CR1 in rheumatoid arthritis patients. Acta Med. Okayama. 2007;61:89–98. doi: 10.18926/AMO/32882. [DOI] [PubMed] [Google Scholar]