

SUMMARY
Extracellular serpins such as antithrombin and α1-antitrypsin are the quintessential regulators of proteolytic pathways. In contrast, the biological functions of the intracellular serpins remain obscure. Using live-animal imaging and reverse genetics in C. elegans, the intracellular serpin, SRP-6, exhibited a pro-survival function by blocking necrosis. Minutes after hypotonic shock, srp-6 nulls underwent a catastrophic series of events culminating in lysosomal disruption, cytoplasmic proteolysis and death. This newly defined hypo-osmotic stress lethal (Osl) phenotype was dependent upon calpains and lysosomal cysteine peptidases, two in vitro targets of SRP-6. By protecting against both the induction of, and the lethal effects from, lysosomal injury, SRP-6 also blocked death induced by heat shock, oxidative stress, hypoxia and MEC-4(d). These findings suggested that multiple noxious stimuli converged upon a peptidase-driven, core stress response pathway that, in the absence of serpin regulation, triggered a lysosomal-dependent necrotic cell death routine.
INTRODUCTION
Serpins are the quintessential regulators of extracellular serine peptidase pathways, where they prevent the deleterious effects of excessive peptide bond hydrolysis (Law et al., 2006; Silverman et al., 2001). For example, thrombin, which converts fibrinogen to fibrin, is generated by activated upstream peptidases including coagulation factors X (fXa) and fIXa (Church et al., 2007). Antithrombin prevents excessive thrombosis by inhibiting fXa, fIXa and a thrombin-mediated amplification loop that perpetuates zymogen conversion. Thus, serpins regulate proteolytic pathways by inhibiting multiple peptidases and by employing a unique suicide-substrate-like mechanism to irreversibly terminate enzymatic activity.
Serpins are the largest (n >1500) and most widely dispersed family of peptidase inhibitors, with representatives in Archaea, Prokarya and Eukarya as well as some viruses (Law et al., 2006). The 37 human serpins are divided into 9 clades (A-I) (Silverman et al., 2001). Most of these serpins are secreted and constitute ~10% of the plasma proteins. In contrast, the vertebrate clade B serpins belong to a larger intracellular subfamily (Silverman et al., 2004). The intracellular serpins (serpinsIC) lack cleavable N-terminal signal peptides and dwell within the nucleo-cytosolic compartment. Several serpinsIC also neutralize papain-like lysosomal cysteine peptidases, but the biological significance of this activity is unknown (Silverman et al., 2004). Based on their subcellular location, we postulate that serpinsIC provide an important survival function by limiting the collateral damage induced by endogenous lysosomal cysteine or serine peptidases. If this hypothesis is correct, then the loss of lysosomal integrity must be a common occurrence during cellular stress. However, the role of lysosomes in cellular injury is uncertain. Initially considered ‘suicide-bags’ that provoked programmed cell death and tissue involution, the inability to show a causal relationship between lysosomal fragility and cell death led to the supposition that lysosmes were durable structures, which released their powerful complement of acid hydrolases into the cytosol only during postmortem autolysis (de Duve, 1983). Subsequently, caspase-dependent apoptosis (type I death) was considered the predominant form of programmed cell death with lysosomes relegated to an injurious role only in cases where autophagy precipitated injury (type II death) (Clarke, 1990). More recently, lysosomes were implicated, albeit marginally, in the induction or execution of the three major cell death routines, including necrosis (type III death) (Golstein and Kroemer, 2007; Zong and Thompson, 2006). Indeed, studies suggest that increasing lysosomal membrane permeability by exposure to oxygen free radicals, photo-oxidation products, or calpains, shifts the cell death routine from apoptosis (ordered and programmed) to necrosis (unordered and unprogrammed) (Golstein and Kroemer, 2007; Zong and Thompson, 2006).
Since there is little data suggesting that eukaryotic serpinsIC protect against caspase-dependent apoptosis (Silverman et al., 2004), these inhibitors may play a role by preventing lysosomal peptidases from exacerbating caspase-independent apoptosis or necrosis. Experimental support for this hypothesis, in vivo, has yet to come from targeted deletions of serpinsIC in mice. Although Serpinb9 nulls show increased loss of cytotoxic lymphocytes, Serpinb2 or -b6 nulls show normal development, survival, fertility, wound healing and resistance to infection (Dougherty et al., 1999; Scarff et al., 2004; Zhang et al., 2006). The large number of serpinsIC in the mouse genome (n = 28) and functional overlaps between paralogues may confound this experimental approach (Silverman et al., 2004). Members of the serpinIC family are also conserved in the genomes of simpler multicellular organisms, such as Caenorhabditis elegans. This nematode harbors 9 serpinsIC of which 6 are bona fide peptidase inhibitors (Luke et al., 2006). We sought to determine whether a reverse genetic approach in a model organism with a smaller serpinIC repertoire could yield insight into the biologic function of this evolutionarily well-conserved, yet enigmatic family of proteins. We identified a surprisingly virulent necrotic phenotype (Osl) in animals (srp-6(ok319)) with homozygous loss of the inhibitory-type serpinIC, srp-6. Interestingly SRP-6 protected animals from necrosis by blocking calpain-associated lysosomal lysis and by neutralizing lysosomal cysteine peptidases released from injured organelles. In addition, SRP-6 protected against necrosis induced by heat stress, hypoxia hyperoxia and cation channel hyperactivity. Taken together, these studies showed that massive cellular necrosis was neither unregulated nor unprogrammed, but was triggered by an ordered, SRP-6-regulated, peptidase-driven stress response pathway targeting the lysosomal compartment. Thus, serpinsIC may serve as pro-survival factors by protecting against both the induction and sequelae of lysosomal injury.
RESULTS
srp-6 Nulls Defined the Hypo-osmotic Stress Lethal (Osl) Phenotype
An interstitial deletion encompassing the reactive site loop (RSL) and 85% of the srp-6 coding region occurred in srp-6(ok319) animals (Supplemental Results, Figure S1). srp-6 nulls were similar to wild-type Bristol N2 (srp-6(+)) animals in terms of anatomy, pharyngeal pumping, lifespan, fecundity and movement (not shown). At 20-25 °C, however, the survival of the srp-6(ok319) animals was decreased markedly when they were collected in water rather than isotonic M9 buffer (Figure 1A). DIC microscopy showed that the srp-6(ok319) animals had suffered a grim fate (Figures 1B-1K). srp-6(ok319) larvae thrashed about for a few minutes and became rod-like with multiple vacuoles and refractile cell corpses (Figures 1D-1K; Movies S1A and S1B). Animals were unresponsive to light touch and never recovered movement upon transfer to NGM growth plates. Adult animals shared a similar fate and frequently extruded their uteri and intestines through the vulval opening or anus after one or two jack-knife-like contractions (Figures 1B and 1C; Movies S2A and S2B). Typically, the median survival of srp-6(ok319) animals was ~ 1 min (Figure 1A), whereas > 90% of the srp-6(+) animals were alive after 30 min, and most were viable after immersion for ~ 24 h (not shown). Intestinal cells of srp-6(ok319) animals lost membrane integrity within ~ 60 sec of water immersion as shown by uptake of propidium iodide (Figures 1L-1Q). Consistent with the site of injury, a srp-6∷GFP transgene showed expression within intestinal cells (Figure S2).
Figure 1. srp-6 Nulls Displayed the Osl Phenotype.
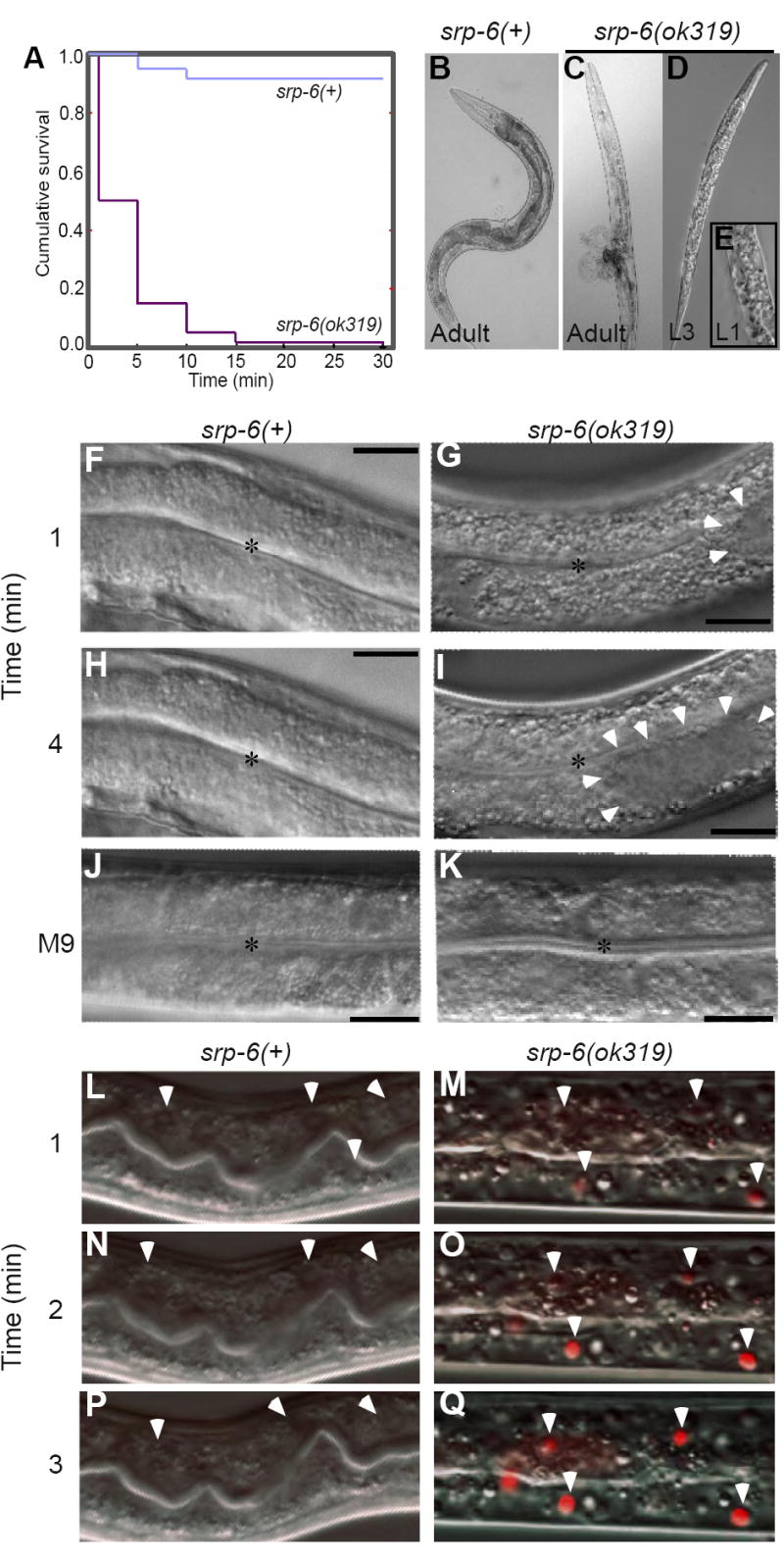
(A) Kaplan-Meier survival curves of srp-6(+) and srp-6(ok319) animals after immersion in 25 °C water (n ≈ 300 animals/group; P < 0.001, log-rank test).
(B-K) Morphology of srp-6(+) (B, F, H, J) and srp-6(ok319) (C-E, G, I, K) animals during hypotonic stress. Low resolution DIC images of adults (B, C) and larvae (D, E) after 5 min in water. The L1 was fixed in 3% formaldehyde. For higher resolution DIC imaging (F-K; bar = 50 μm), L4s were immersed in water (F-I) or M9 (J, K). Intestinal cell cytoplasm of srp-6(ok319) animals was refractile (G, I) and clear cytoplasmic vacuoles (arrowheads) expanded and coalesced (*intestinal lumen).
(L-Q) Intestinal cell plasma membrane integrity was lost in srp-6(ok319) (M, O, Q), but not in srp-6(+) (L, N, P) animals after exposure to water. Confocal microscopy was used to monitor propidium iodide uptake over time. Arrowheads indicate intestinal cell nuclei in dying animals.
Death did not occur by drowning as srp-6(ok319) animals died rapidly upon transfer to 2% agarose pads prepared with water. The phenotype of srp-6(ok319) animals in water, which was designated hypo-osmotic stress lethal (Osl) by the C. elegans nomenclature committee (http://biosci.umn.edu/CGC/Nomenclature/genes.htm), was autosomal recessive (not shown). A second srp-6 deletion mutant, srp-6(tm1994) also was Osl (not shown). Of note, animals with enhanced fibroblast growth factor signalling amass fluid in their pseudocoelomic space under isotonic conditions (Kokel et al., 1998). This Clear (Clr) phenotype was distinct morphologically from that of Osl and clr-1(e1745) mutants survived when placed in water (not shown).
Osl was not Due to Defective Morphogenesis
The secretory-excretory system, which consists of the excretory, gland, duct and pore cells, is responsible for osmoregulation in C. elegans (Nelson and Riddle, 1984). srp-6(ok319) animals expressing a clh-4∷GFP transgene showed an apparently normal excretory system (Figure S3). More importantly, srp-6(RNAi) phenocopied Osl at a time when C. elegans development was complete (Figure S4). We concluded that Osl was due to a physiological rather than an anatomic defect.
srp-6(ok319) Animals Could Undergo a Regulatory Volume Decrease
In response to hypotonic shock, cells undergo a regulatory volume decrease (RVD) to restore cell volume and osmolality (Jakab et al., 2002). RVD involves a rapid rise in intracellular Ca2+ concentrations ([Ca2+]i) followed by an efflux of inorganic ions, organic osmolytes and water. In C. elegans, changes in cellular size and [Ca2+]i can be assessed by approximating the volume of the intact organism and by measuring cytoplasmic fluorescence with the ratiometric calcium indicator, Fura-2, respectively (Lamitina and Strange, 2005). Both srp-6(ok319) and srp-6(+) animals showed a comparable increase in volume and [Ca2+]i minutes after hypotonic shock at 25 °C (Figures 2A and 2C). However, the [Ca2+]i of srp-6(ok319) animals only transiently returned to baseline and volume never decreased. We concluded that mutant animals were either RVD-defective or that RVD was normal but could not be completed due to the concomitant activation of a cell death routine. We noticed that srp-6(ok319) animals exposed to water at 10 °C were protected from death. Since this temperature does not appreciably alter water influx and cell swelling, we repeated the experiments at this temperature. Both srp-6(+) and srp-6(ok319) animals demonstrated a comparable increase and subsequent decrease in both cell volume and [Ca2+]i (Figures 2B and 2D). These findings suggested: 1) srp-6(ok319) animals could undergo an RVD, 2) cell swelling and the increased turgor pressure per se did not kill the animals and 3) hypotonic shock triggered a temperature-sensitive, SRP-6-regulated cell death routine.
Figure 2. srp-6(ok319) Animals Mounted a Regulatory Volume Decrease.
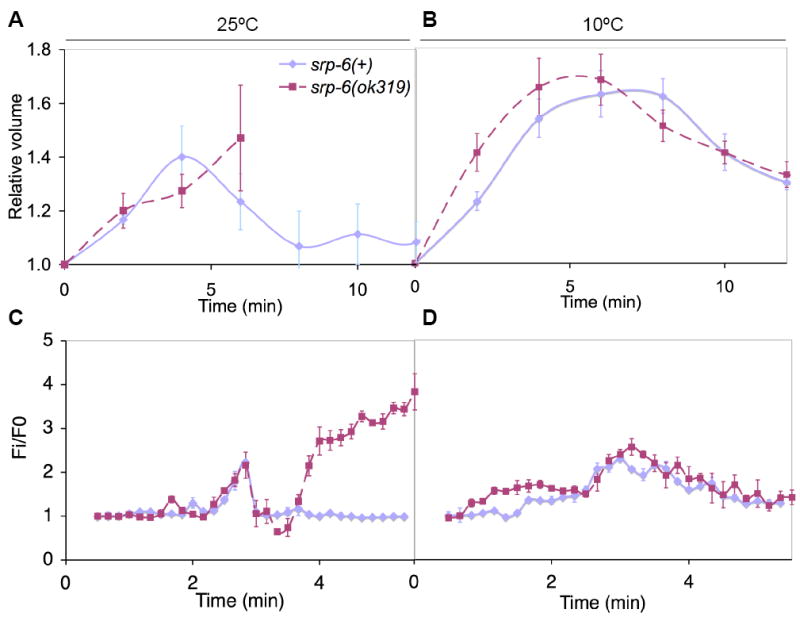
(A-D) Relative changes in cell volume (A, B) and [Ca2+]i (C, D) over time in srp-6(+) (blue) and srp-6(ok319) (red) animals exposed to water at either 25 (A, C) or 10 °C (B, D). The means (± SD) were from a representative experiment (n = 8 animals/group).
The Manner of Osl-induced Death
In C. elegans, apoptosis occurs in embryonic and larval development and typically involves the caspase, CED-3, which is regulated by pro-apoptotic (CED-4, EGL-1) and anti-apoptotic (CED-9) factors (Lettre and Hengartner, 2006). However, neither the ced-3(n717), ced-4(n1162) nor ced-91950 mutation suppressed srp-6(RNAi)-induced Osl (Figure 3A). Also, the caspase inhibitors DEVD-CHO or YVAD-CHO could not block Osl (Figure 3B).
Figure 3. Suppression of Osl.
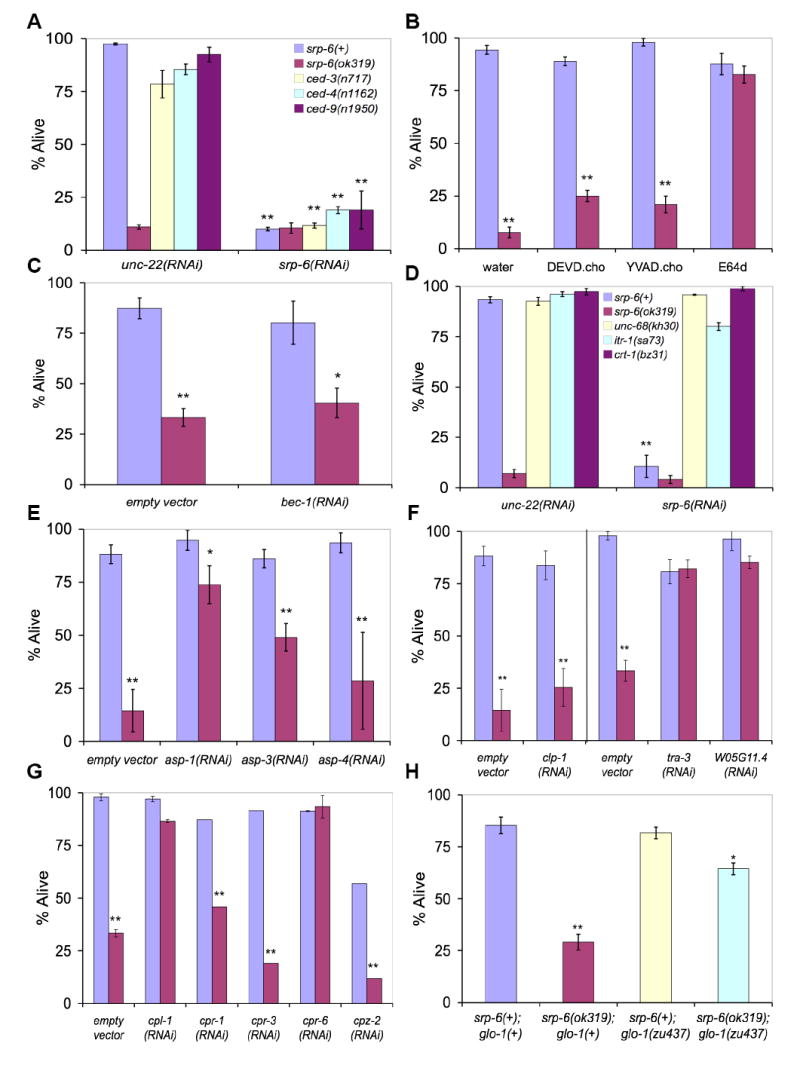
(A) Survival of ced-3(n717), ced-4(n1162) and ced-9(n1950) apoptosis mutants exposed to srp-6(RNAi) compared to control unc-22(RNAi). In all panels of this figure, mean survival (± SD) of young adults was assessed after exposure to water at 25 °C for 30 min. Asterisks indicate that the survival between groups was significantly different (n ≈ 300 animals/group, single and double asterisks indicate P < 0.05 and < 0.01, respectively).
(B) Survival of srp-6(+) (blue) compared to srp-6(ok319) (red) animals pre-treated with caspase inhibitors, DEVD-CHO or YVAD-CHO, or the papain-like cysteine peptidase inhibitor, E-64d.
(C) Survival of srp-6(+) (blue) compared to srp-6(ok319) (red) animals defective in autophagy by exposure to bec-1(RNAi) or empty vector(RNAi). Although bec-1(RNAi) did not suppress the Osl phenotype, this treatment did block normal dauer formation (not shown).
(D) Survival of unc-68(kh30), itr-1(sa73) and crt-1(bz31) calcium-handling defective mutants exposed to srp-6(RNAi) compared to unc-22(RNAi).
(E) Survival of srp-6(+) (blue) compared to srp-6(ok319) (red) animals defective in aspartic peptidase activity by exposure to asp-1(RNAi), asp-3(RNAi), asp-4(RNAi) or empty vector(RNAi). Note, survival of srp-6(ok319);empty vector (RNAi) compared to that of srp-6(ok319);asp-1(RNAi) was statistically different (P < 0.001).
(F) Survival of srp-6(+) (blue) compared to srp-6(ok319) animals (red) defective in calpain peptidase activity by exposure to clp-1(RNAi), tra-3(RNAi), W05G11.4(RNAi) or empty vector(RNAi). Note, the tra-3(RNAi) and W05G11.4(RNAi) studies were conducted with those reported in (G), but are included in this panel to facilitate comparisons between the calpain(RNAi) studies. Thus, the same empty vector controls from (G) are also included in the panel reporting the tra-3(RNAi) findings.
(G) Survival of srp-6(+) (blue) compared to srp-6(ok319) (red) animals defective in papain-like lysosomal cysteine peptidase activity by exposure to cysteine peptidase(RNAi) or empty vector(RNAi).
(H) Survival of srp-6(+) or srp-6(ok319) animals defective in gut granule (lysosomal) biogenesis. The survival of srp-6(ok319), srp-6(+);glo-1(zu437) or srp-6(ok319);glo-1(zu437) animals was compared to that of srp-6(+) animals.
In C. elegans, genes associated with the autophagic pathway are critical for normal dauer diapause (an adaptive stress response), but have not yet been linked to cell death (Melendez et al., 2003). srp-6(ok319) animals exposed to bec-1(RNAi) (orthologous to yeast APG6) showed no increase in survival (Figure 3C), but did show abnormal dauer formation (not shown).
Necrotic cell death has been described in C. elegans (Driscoll and Gerstbrein, 2003). This necrotic or degenerin pathway mimics that associated with excitotoxic injury and occurs in neurons expressing gain-of-function mutations in degenerin/epithelial Na+ channels (deg-1, mec-4, mec-10, unc-8 and unc-105), a GTP binding protein subunit (gsa-1, a Gαs) and a nicotinic acetylcholine receptor (deg-3) (Berger et al., 1998; Chalfie and Wolinsky, 1990; Yassin et al., 2001). Hyperactivation of ion channels in C. elegans leads to a rise in [Ca2+]i, and necrosis can be suppressed by mutations that block the release of Ca2+ from intracellular stores (Xu et al., 2001). We performed srp-6(RNAi) on animals lacking the ryanodine (RyR/unc-68(kh30)) or inositol-1,4,5-trisphosphate (IP3R/itr-1(sa73)) receptor. These receptor mutants and null or loss-of-function mutations of the ER Ca2+ binding protein, calreticulin (crt-1(bz31)), significantly suppressed death induced by srp-6(RNAi) (Figure 3D). Similarly, srp-6(ok319) animals treated with an intracellular Ca2+ chelator, EGTA-AM or BAPTA-AM, were protected from death (Figure S5A). srp-6(ok319) animals were not more sensitive to elevations in [Ca2+]i, as treatment with thapsigargin did not induce cell death despite a rate of rise in [Ca2+]i comparable to that observed with Osl (not shown). These data suggested that the rise in [Ca2+]i associated with a normal RVD was required, but not sufficient, to activate the Osl.
Aspartic (asp-3 and asp-4) and cysteine (calpains, clp-1 and tra-3/clp-5) peptidases are involved in the degenerin pathway (Syntichaki et al., 2002). Interestingly, asp-1(RNAi), which is expressed predominately in the lysosomes and brush border of intestinal cells (Tcherepanova et al., 2000), and to a lesser extent asp-3(RNAi), but not asp-4(RNAi), protected srp-6(ok319) animals from Osl (Figure 3E). Inhibition of calpains by tra-3/clp-5(RNAi) or W05G11.4(RNAi), but not clp-1(RNAi), also showed protection (Figure 3F). The C. elegans genome harbors at least 31 lysosomal cysteine peptidases and several are expressed in intestinal cells. RNAi with cpl-1 (catL-like) and cpr-6 (catB-like) protected srp-6(ok319) animals from death (Figure 3G). Consistent with the RNAi studies, pre-treatment of srp-6(ok319) animals with the papain-like cysteine peptidase inhibitor, E-64d, protected against Osl (Figure 3B). Although E-64d protected against death, this treatment did not block the Osl-induced rise in [Ca2+]i, suggesting that Ca2+ release occurred upstream of cysteine peptidase activation (Figure S5B). Taken together, these studies suggested that necrosis was the manner of death in Osl.
Loss of SRP-6 Peptidase Inhibitory Activity Led to Osl
Since Osl was partially dependent upon cysteine peptidases, we determined whether SRP-6 might serve as their inhibitor. Like human SERPINB3, recombinant SRP-6 inhibited papain-like lysosomal cysteine peptidases (clan CA, family C1) such as catL and catK, but not any of the serine peptidases within a broad array of purified enzymes (Table S1). Interestingly, SRP-6 also inhibited calpain-2 (clan CA, family C2). The inhibition of these enzymes was characteristic of serpin-target peptidase interactions as shown by typical stoichiometries of inhibition (SI) (Figures 4A and 4B), second-order rate constants (Figures 4C and 4D), covalent complex formation (Figures 4E and 4F) and peptidase-mediated RSL cleavages (Figure 4G).
Figure 4. SRP-6 Inhibited Papain-like Lysosomal and Calpain Cysteine Peptidases.
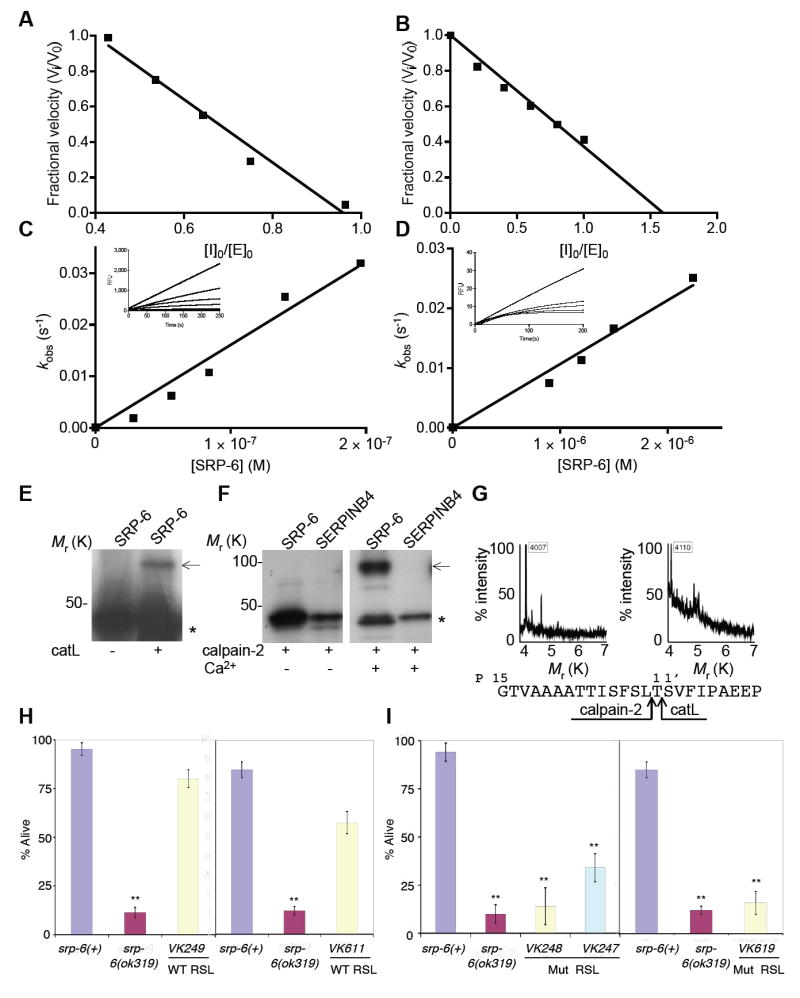
(A-B) The stoichiometry of inhibition (SI) for the interaction of SRP-6 with catL (A) or calpain-2 (B). Inhibitor:enzyme ratio of ~1:1 and ~1:1.5 completely inhibited catL and calpain-2 activity, respectively. SIs were determined from the mean of three separate experiments with representative plots shown.
(C-D) The second-order rate constant (ka) for the interaction between SRP-6 and catL (C) or calpain-2 (D). The rate of inhibition (kobs) was measured over time (inset) and plotted against the molarity of the inhibitor to give a kapp of 9 × 104 M-1 s-1 and 1.2 × 104 M-1 s-1, respectively. Correcting for the enzymes Km for the substrate, yielded a ka of 10 × 104 ± 0.1 M-1 s-1 and 2.3 × 104 ± 0.4 M-1 s-1 (mean ka ± SD from 3-4 experiments with representative plots shown), respectively.
(E-F) Complex formation between mixtures of 35S-SRP-6 and catL (E) or calpain-2 (± 5 mM Ca2+) (F). Autoradiogram after SDS-PAGE revealed native or cleaved SRP-6 (*) and a higher molecular mass complex with catL or calpain-2 (arrow). SERPINB4 does not inhibit calpain-2.
(G) The serpin reactive centers for the SRP-6-catL (left) and SRP-6-calpain-2 (right) interactions were deduced form inhibitor-enzyme complex cleavage fragments detected by MALDI-MS. The cleavage of SRP-6 occurred after the canonical P1 and P2 positions for catL and calpain-2, respectively (bottom).
(H-I) Mean survival (± SD) in water of srp-6(+) or srp-6(ok319) animals rescued with a srp-6 extra-chromosomal array. Several transgenic lines were established with each construct, but only a representative line is shown. Animals were assayed for survival as described in Figure 3A (n ≈ 300 or ≈ 75 animals/group for transgenic lines under control of the srp-6 or nhx-2 promoter, respectively). (H) VK249 (srp-6(ok319);vkEx249[srp- 6(+);Pnhx-2∷GFP]) was established using a wild-type srp-6 gene and a co-injection marker, Pnhx-2∷GFP. VK611 (srp-6(ok319);vkEx611[Pnhx-2srp-6(+);Pnhx-2∷GFP]) was similar to VK249, except that srp-6 was driven by the intestinal cell specific promoter from nhx-2. Since the survival of srp-6(ok319) animals was unaffected by the Pnhx-2∷GFP transgene (not shown), only srp-6(ok319) survival data were reported. The survival of the srp-6(ok319) animals was compared to that of srp-6(+) animals. (I) VK248 (srp-6(ok319);vkEx248[srp-6(T327R);Pnhx-2∷GFP]), VK618 (srp-6(ok319);vkEx618[Pnhx-2srp-6(T327R);Pnhx-2∷GFP]) and VK247 (srp-6(ok319);vkEx247[srp-6(L339A;T340A)]) contained a extra-chromosomal array with either a P14 (T327R) or P2-P1 (L339A;T340A) RSL mutant srp-6.
To confirm that the loss of cysteine peptidase inhibitory activity of SRP-6 was the cause of Osl, we performed transgenic rescue experiments of srp-6(ok319) animals using a srp-6 gene with either a wild-type or mutated RSL. The mutations were placed at the P14 hinge region (T337R) or the reactive center P1 and P2 residues (L339A;T340A). The classical P14 mutation blocks covalent complex formation with any target peptidase. The P2 and P1 mutations block papain-like lysosomal cysteine peptidase binding, as these enzymes prefer hydrophobic residues at the P2 position and bind cooperatively to residues at the P1 position (Schick et al., 1998). Each of these constructs was co-injected with a Pnhx-2∷GFP reporter plasmid (the nhx-2 promoter drives expression exclusively within intestinal cells (Nehrke, 2003)) into the gonads of young adult srp-6(ok319) animals. Transgenic lines with the wild-type srp-6 gene (VK249) showed nearly complete suppression of Osl (Figure 4H). In contrast, srp-6(ok319) animals harboring the srp-6 transgene with either the P14 mutation (VK248) or the P2-P1 mutations (VK247) were still Osl (Figure 4I). To determine whether intestinal cell expression was sufficient to rescue srp-6(ok319) animals, we replaced the srp-6 promoter with that from nhx-2. The nhx-2 driven wild-type srp-6, but not the P14 mutant, also rescued srp-6 nulls (VK611 and VK618, Figures 3H and 3I, respectively). We concluded that the loss of intestinal cell SRP-6 cysteine peptidase inhibitory activity caused Osl.
Lysosome-like Gut Granules Were Required for Osl
By transmission electron microscopy (TEM), the membranes of lysosomal-like gut granule membranes were disrupted several minutes after hypotonic shock (Figures 5A-5F). At this stage, most mitochondria and other cellular structures were still intact. To confirm that lysosomal instability and release of cathepsins was an early feature of Osl, confocal microscopy was used to follow the fate of gut granules labelled coincidently with the endocytic marker, tetramethylrhodamine (TMR)-dextran and the general fluorescent cysteine peptidase substrate, (Z-FR)2-R110 (Figures 5G-5R). Dual labelling confirmed that the majority of gut granules appeared to be lysosomes containing endocytosed molecules and active cysteine peptidases. The labelled granules of only live srp-6(ok319) animals began to lyse minutes after hypotonic shock (Figures 5G-5R; Movies 3A and 3B, 4A and 4B, 5A and 5B). Widefield fluorescence microscopy revealed that gut granule lysis was followed by a wave of intense cytoplasmic (Z-FR)2-R110 fluorescence, indicating the presence of diffuse cysteine peptidase activity prior to death (Figures 5S-5AD, Movies 6A and 6B). These studies showed that the disintegration of lysosomal-like gut granules and subsequent cytoplasmic cysteine peptidase activity were active components of the cell death routine and not a post-mortem event. To test this hypothesis, we generated srp-6(ok319);glo-1(zu437) double mutants. glo-1 is a Rab-like GTPase required for the biogenesis of acidic, lysosomal-like gut granules (Hermann et al., 2005). glo-1(zu437) mutants lack these lysosomes and appear to direct their contents into the intestinal lumen. srp-6(ok319);glo-1(zu437) animals exposed to hypotonic shock were protected from death (Figure 3H). Consistent with these findings, the lysosomes of srp-6(ok319);tra-3(RNAi) (Figures 5AE-5AL) and EGTA-AM treated srp-6(ok319) animals, two treatments that block Osl, remained intact after hypotonic shock (Figure S5C). Taken together, these data demonstrated that lysosomal-like gut granule instability was required for Osl. Since lysosomes did not lyse in srp-6(+) animals, we concluded that SRP-6 protected against lysosomal rupture.
Figure 5. Instability of Lysosomal Gut Granules in Osl.
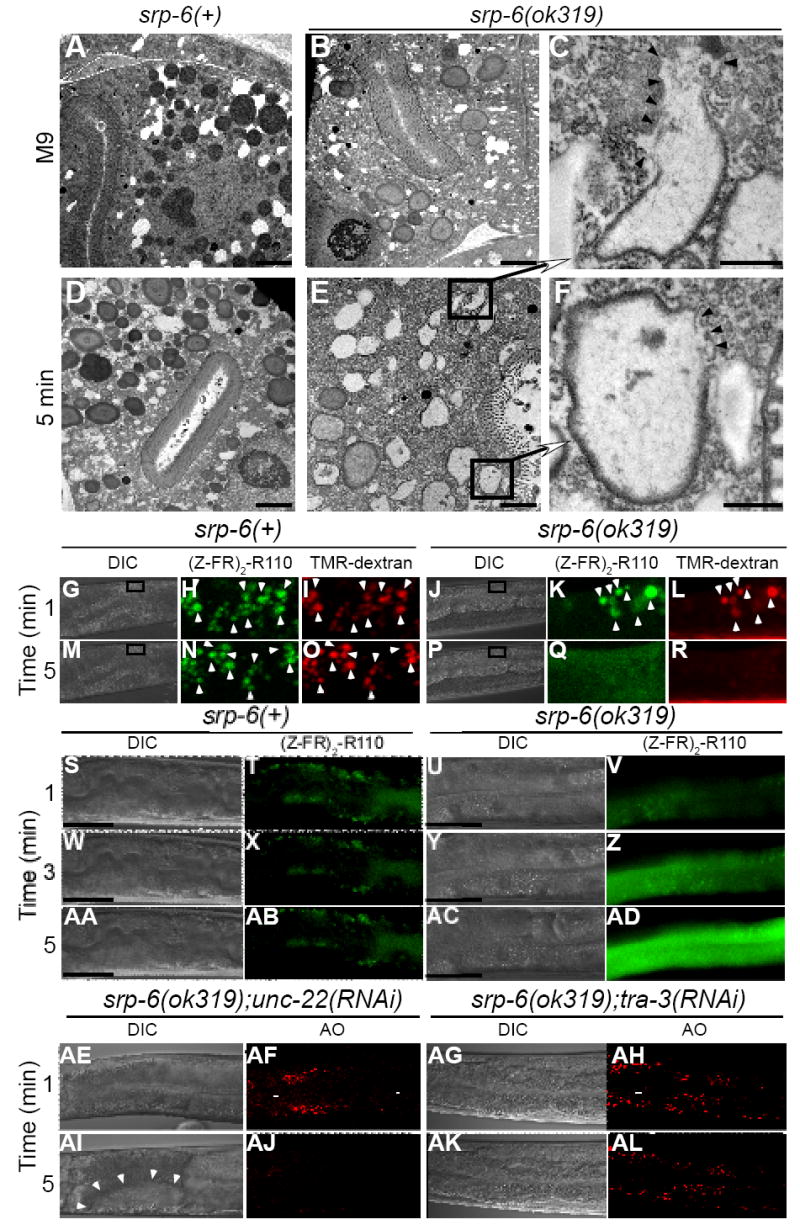
(A-F) Transmission electron microscopy of srp-6(+) (A, D) or srp-6(ok319) (B, E) animals 5 min after immersion in M9 (A, B) or water (D, E) (bar = 2 μm). Higher magnification (C, F) of lysosomal-like granules in (E) (bar = 500 nm). Arrowheads demarcate loss of granule membranes.
(G-R) Live cell confocal microscopic imaging of the fluorescent cysteine peptidase substrate, (Z-FR)2-R110 (H, K, N, Q) and the fluorescent endocytic maker, TMR-dextran (I, L, O, R) in srp-6(+) (G-I, M-O) or srp-6(ok319) (J-L, P-R) animals after immersion in water. The fluorescent images were magnified from a DIC image (boxed inset) within the intestinal cell cytoplasm (G, M, J, P). The lysosomal-like gut granules of both srp-6(ok319) and srp-6(+) animals acquired both labels (arrowheads) and were mostly coincident in the merged images (Movies 5A and 5B).
(S-AD) Live widefield DIC and fluorescence microscopy of srp-6(+) and srp-6(ok319) animals labelled with (Z-FR)2-R110 and immersed in water. Fluorescent gut lysosomes in the srp-6(+) animal remained intact (T, X, AB; bar = 50 μm). In the srp-6(ok319) animals, the disappearance of fluorescent lysosomes was accompanied by a transient wave of intense cytoplasmic fluorescence that propagated down the intestinal cell (V, Z, AD; Movies 6A and 6B).
(AE-AL) Live DIC (AE, AG, AI, AK) and confocal microscopy (AF, AH, AJ, AL) of AO-labelled srp-6(ok319);unc-22(RNAi) (AE, AF, AI, AJ) or srp-6(ok319);tra-3(RNAi) (AG, AH, AK, AL) animals immersed in water. Vacuoles are indicated by arrowheads.
To determine whether SRP-6 also protected the cell after lysosomal disruption, we compared the survival of srp-6(ok319) to srp-6(+) animals after inducing direct lysosomal injury with acridine orange (AO). Lysosomes trap this weak basic dye and lyse upon exposure to blue light (Hiruma et al., 2007). As expected, most of the gut granules from both animals lysed minutes after exposure to blue light (Figures 6A-6H). However, the srp-6(ok319) animals showed markedly increased intestinal cell vacuolization and mortality (Figure 6M). Surprisingly, when we examined the surviving srp-6(+) animals ~30 min after treatment, their lysosomal compartment was reconstituted as shown by the re-appearance of granular AO staining (Figures 6I-6L). These data showed that SRP-6 also protected the cell from injury after lysosomal rupture.
Figure 6. SRP-6 Protected Animals After Lysosomal Rupture.
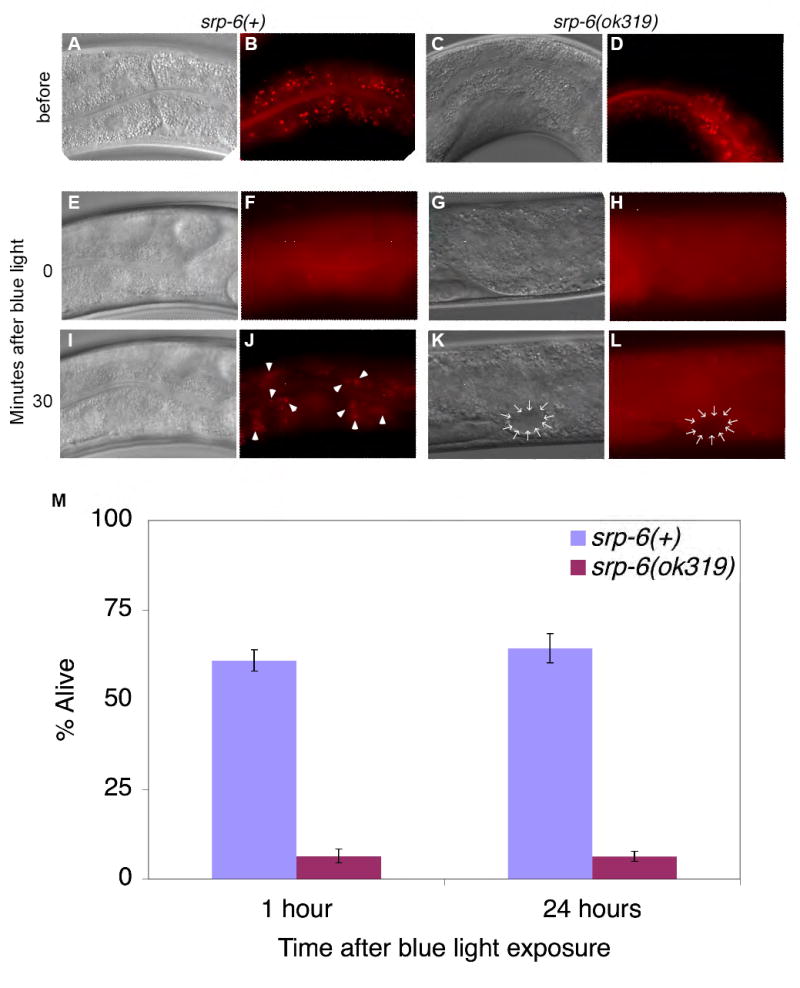
(A-L) Widefield DIC and fluorescence microscopy of a representative srp-6(+) (A, B) and srp-6(ok319) (C, D) animal labelled with the intestinal lysosomal marker, AO. After exposure to blue light, AO+ granules lysed in the srp-6(+) (F), and srp-6(ok319) (H) animals. The srp-6(ok319) animal also showed intestinal vacuolization (K, L; arrows). AO+ granules re-appeared (arrowheads) only in surviving srp-6(+)animals.
(M) Mean (± SD; n ≈ 100 animals/group) survival of srp-6(+) and srp-6(ok319) animals treated with AO and blue light.
SRP-6 Protected Against Death Induced by Other Stressors
To determine whether the protective role of SRP-6 was restricted to Osl or could be extended to other stressors, we compared the survival rate of srp-6(ok319) to srp-6(+) animals after exposure to heat-shock (Lithgow et al., 1995), hypoxia (sodium azide) (Scott et al., 2002) and hyperoxia (paraquat) (Tawe et al., 1998). In addition, ectopic expression of mec-4(d), which is normally expressed only in touch receptor neurons, can induce necrosis in several cell types including intestinal cells (Harbinder et al., 1997). To determine if SRP-6 functioned downstream in the same death pathway induced by MEC-4(d), we generated srp-6(ok319) and srp-6(+) transgenic lines using the heat-shock inducible gene, Phsp-16mec-4(d).
In response to all four stressors, the survival rate of srp-6(ok319) animals was significantly decreased compared to srp-6(+) controls (Figures 7A-7D). Similar to that observed with Osl, these stressors also induced intestinal cell vacuolization and gut granule dissolution (Figures 7E-7T), and were protected by tra-3(RNAi) and cpl-1(RNAi) (Figures S6A-S6D).
Figure 7. SRP-6 Protected Animals from Different Stressors.

(A-D) Kaplan-Meier curves comparing the survival of srp-6(+) and srp-6(ok319) animals after exposure to (A) thermal stress, (B) hypoxia, (C) hyperoxia or (D) a Phsp-16mec-4(d) transgene. (n ≈ 75 animals/group; P < 0.001, log-rank test).
(E-T) DIC (E-L) and fluorescent (M-T) images of intestinal cell vacuolization (arrowheads) and AO+ intestinal cell lysosomes (arrowheads), respectively, of srp-6(+) (E-H and M-P) and srp-6(ok319) animals (I-L and Q-T) exposed the stressor indicated above the survival curve.
(E) Hypothetical core stress response pathway regulated by SRP-6. Different stressors converge on a core stress response pathway that triggers an increase in [Ca2+]i and modulation of at least one other stress-transducing factor (STF). Cytosolic Ca2+ and STF activate calpains, which associate with lysosomal membranes and enhance the lysosomal response to stress by facilitating, for example, autophagy. Calpains also increase lysosomal membrane permeability allowing for the small leakage of cysteine peptidases into the surrounding cytosol. Cytosolic cysteine peptidases also could provide an adaptive function by enhancing, for example, cytoskeletal rearrangements. However, in the absence of SRP-6, excessive calpain activity leads to massive lysosomal rupture, overwhelming release of unregulated lysosomal cysteine peptidases and necrotic cell death.
DISCUSSION
Previously, we showed that several serpinsIC, like SRP-6, neutralized lysosomal cysteine peptidases (C1 family) such as cathepsins K, L, S and V (Silverman et al., 2004). Based on the Osl phenotype, we can now ascribe biological relevance to this activity, as the pro-survival function of SRP-6 in vivo was dependent upon its ability to neutralize cysteine peptidases. Since at least 5 of the 13 human serpinsIC inhibit cysteine peptidases, this survival function is probably conserved in higher vertebrates as well. Indeed, tumor cells induced to over- or under-express SERPINB3, the prototypical human cysteine peptidase inhibitor, show increased or decreased survival, respectively (reviewed in (Silverman et al., 2004)).
SRP-6 inhibition of a calpain extended the activity of serpinsIC to include a different member of the papain clan (C2 family). Calpastatins are the only other intracellular proteins known to inhibit calpains (Wendt et al., 2004). However, calpastatins are present in the genomes of higher organisms (vertebrates) and are absent from those of simpler metazoans, such as C. elegans and D. melanogaster (Goll et al., 2003). Thus, serpinsIC may serve as the only calpain inhibitors in simpler organisms. It is not known if calpastatins subsumed this activity in vertebrates or evolved a different function, such as shielding the enzyme itself from proteolytic attack (Melloni et al., 2006). Since calpastatins, unlike serpins, are reversible inhibitors, the retention of both types of inhibitory functions in higher organisms would add additional means of protecting against misdirected calpain activity (e.g., muscle and neuronal degeneration, cataract formation) (Zatz and Starling, 2005).
srp-6 nulls were also significantly more susceptible to the lethal effects of thermal stress, oxygen free radicals, hypoxia and channel hyperactivity. Although these stimuli initiated injury by disparate means, such as protein aggregation (Hirsch et al., 2006), reactive oxygen species (Tawe et al., 1998), energy failure (Scott et al., 2002) or Ca2+ and Na+ influx (Xu et al., 2001), respectively; they showed a remarkably similar death phenotype in srp-6(ok319) animals. These findings suggested that the proximal signalling events triggered by these different noxious stimuli converged upon an adaptive peptidase-driven core stress response pathway, which in the absence of SRP-6 regulatory activity, triggered massive lysosomal lysis and necrotic cell death rather than acclimation to insult (Figure 7U). Thus, peptidase-inhibitor imbalance played a determinant role in shifting the outcome from adaptation and survival to cellular injury and death. The adaptive function of this peptidase pathway was unknown but could, for example, help restore normal cellular function by 1) degrading damaged or unfolded intracellular proteins, 2) activating counter-regulatory ion channels or receptors, 3) enhancing autophagy or 4) facilitating cytoskeleton reorganization (Jakab et al., 2002). Taken together, peptidases associated with necrotic cell death pathways, like those with apoptotic pathways (Kuranaga and Miura, 2007), are likely to subserve normal physiological processes, such as the adaptation to stress, and are not solely reserved for cellular execution.
Cytoplasmic swelling and vacuolization, an obligatory rise in [Ca2+]I and lysosomal instability suggested that the Osl and degenerin pathways were similar (Chalfie and Wolinsky, 1990). Although Osl occurred more rapidly and employed a slightly different complement of cysteine and aspartic peptidases, different death-inducing stimuli and tissue specific factors could account for these variations. The ability of SRP-6 to protect against the toxicity induced by ectopic MEC-4(d) expression also suggested that the peptidase-driven death routines triggered by Osl and MEC-4(d) were highly homologous. Of note, SRP-6 was unlikely to protect against endogenous MEC-4(d), as their expression patterns are non-overlapping (intestinal cell and touch receptor neurons, respectively).
In excitotoxic injury, calcium activated calpains appear to associate with lysosomes and facilitate the release of acid hydrolases (Yamashima et al., 2003). However, the linearity of this Ca2+-calpain-cathepsin pathway has been difficult to substantiate, in vivo. Since, the rise in [Ca2+]i occurred minutes after exposure to hypotonic shock and was not blocked by the cysteine peptidase inhibitor, E-64d, this event was likely to be proximal in the signalling pathway. This conclusion was also consistent with studies showing that the rise in [Ca2+]i was required but not sufficient to induce necrosis. Thus, the rise in [Ca2+]i had to be followed or accompanied by at least one other signalling element to activate the downstream proteolytic events. We have designated this element, stress-transducing factor(s) (STF). The function of STF was unknown, but could, for example, involve phosphorylation events that facilitated the activation and targeting of calpains to lysosomal membranes (Yamashima et al., 1996).
Although calpains and lysosomal cysteine peptidases functioned downstream of increases in [Ca2+]i, it was not known whether these peptidases worked in parallel or series. If these peptidases worked in parallel pathways, then death required “two peptidase hits”, as down-regulation of either peptidase by RNAi was sufficient to block death. However, the parallel pathway theory was inconsistent with studies showing that srp-6(ok319) animals exposed to calpain(RNAi) prior to hypotonic shock survived with their lysosomes intact, suggesting that calpains enhanced lysosomal lysis and therefore acted proximal to the release of lysosomal cysteine peptidases. Moreover, as calpain(RNAi) could not block AO and blue light-induced lysosomal lysis and the release of lysosomal cysteine peptidases (not shown), these latter enzymes, which were sufficient in themselves to kill srp-6(ok319) animals, could act distally to calpains. Taken together, these studies were more consistent with these peptidases working in series, with calpains operating upstream (pre-lysosomal) and lysosomal cysteine peptidases operating downstream (post-lysosomal) in a core stress response pathway capable of triggering necrotic cell death. Since srp-6(ok319) animals showed increased calpain-dependent lysosomal lysis after exposure to at least 5 different noxious stimuli, and decreased lysosomal cysteine peptidase-dependent survival after AO-induced lysosomal rupture, we concluded that SRP-6 was critically positioned at the nexus of this stress response pathway by regulating both the induction and sequelae of excessive lysosomal permeability. This conclusion was consistent with the ability of SRP-6 to neutralize calpains and lysosomal cysteine peptidases.
Since Christian de Duve described lysosomes over 50 years ago, there has been little consensus regarding their function as either “…suicide bags, pre-packaged destruction awaiting a programmed instruction to strike…” or “…a more benign, if less exciting role in the cell’s economy, namely as a waste-disposal system…” (Lloyd and Forster, 1986). Using real-time imaging, the disintegration of lysosomes was not a consequence of post-mortem autolysis, but occurred in srp-6(ok319) animals prior to their death. The absence of cell death in srp-6(ok319) animals devoid of acidic gut granules due to a mutation in glo-1 confirmed unequivocally that these organelles were required for Osl-induced necrotic cell death. However, as shown by the AO studies, massive lysosomal rupture was neither irreversible nor lethal, providing that sufficient anti-peptidase activity resided within the cell. Thus, fulminant necrosis, such as Osl, can no longer be considered as an accidental, passive or irreversible death routine, but one in which different upstream signals converge upon a regulable peptidase pathway that integrates the lysosomal compartment in determining cell fate. If the serpinsIC as a class serve as the antidote to this type of cell death routine, strategies designed to up-regulate their activity might be used to protect against the massive cellular necrosis induced by hemispheric thromboembolic stoke and transmural myocardial infarction in adults and necrotizing enterocolitis in preterm infants. Alternatively, if serpinsIC are pro-survival factors, these findings begin to explain why increased expression of certain family members are poor prognostic indicators in different types of human malignancies (Bolger et al., 1997), and provides a rationale for designing therapies to disrupt serpinIC activities within tumor cells.
EXPERIMENTAL PROCEDURES
C. elegans strains
Standard procedures for C. elegans strain maintenance, crosses and other genetic manipulations were used (Brenner, 1974). All strains were grown at 20-25 °C (unless otherwise stated) on standard NGM plates seeded with E. coli OP50 as the food source. N2 Bristol was the wild-type strain. The mutant strains srp-6(ok319), crt-1(bz31), unc-68(kh30), itr-1(sa73), ced-1(e1735), ced-2(e1752), ced-3(n717), ced-4(n1162), ced-9(n1950), glo-1(zu437) and srp-6(ok319);glo-1(zu437) were generated in the laboratories of the authors or were provided by the Caenorhabditis Genetics Center (http://www.cbs.umn.edu/CGC/). srp-6(ok319) transgenic animals and plasmid constructions are described in Supplemental Experimental Procedures.
Hypotonic Shock and Cellular Stress Assays
For survival curves, staged animals were collected, washed with M9 and placed on an unseeded NGM plate for 30 min to allow for the expulsion of gut contents. For hypotonic shock assays, animals were placed into a 60 mm plastic Petri dish containing deionized, double distilled water (hereafter referred to as water) at 25 °C. Animals were examined every 5 min and those that became rigid, did not respond to touch using a platinum wire, ceased pharyngeal pumping, did not recover when transferred back to NGM plates or had extruded their viscera through the vulva were scored as dead. Animals from the initial hypotonic shock experiments never regained mobility and DIC imaging at ~24h showed extensive post-mortem autolysis. For thermal stress experiments, animals were incubated at 37 °C for 1 h on NGM plates seeded with OP50. To ensure that heat-treated animals were not mistakenly scored as dead due to decreased movement, nematodes were recovered by washing in 25 °C M9 solution before transferring to NGM plates (Lithgow et al., 1995). Animals were scored for death every few hours as described above. For hypoxia experiments, animals were incubated at 25 °C for 1 h in M9 solution containing 0.1 M sodium azide (Fisher Scientific) (Scott et al., 2002). For hyperoxia experiments, animals were incubated at 25 °C for 2 h in M9 containing 8 mM paraquat (Sigma) (Tawe et al., 1998). To ensure that azide- or paraquat-treated animals were not mistakenly scored as dead due to drug-induced paralysis, animals were recovered by washing five times with M9 at 25 °C before transferring to NGM plates. Animals were scored for death every few hours as described above. For Phsp-16mec-4(d) experiments, transgenic L1s and L2s were heat shocked at 30 °C overnight on NGM plates seeded with OP50. As MEC-4(d) induces paralysis, we followed animals individually over time and counted only those with extensive autolysis as dead. Kaplan-Meier survival curves were plotted for all groups.
For certain assays, water survival studies were conducted over a finite time period. Animals were washed off the plates with M9 media, counted and transferred to a Büchner funnel fitted with a 10 μm TCTP Isopore membrane filter. Animals were washed 3 times with 10 ml of either M9 (control) or water by applying light vacuum pressure at 25 mm Hg. The animals, supported by the filter, were transferred to a Petri dish containing either M9 or water at a concentration of 100 worms/ml. After 30-60 min, SYTOX Green (Molecular Probes) was added to the solution at a final concentration of 5 μM. After 5 min, the animals were visualized under UV fluorescence using a Leica MFLZIII dissecting microscope with an excitation filter of 492 nm and an emission filter of 530 nm. Fluorescent worms that were rigid and unresponsive to touch were counted as dead. The percentage alive = (number of alive worms)/(total number of worms) × 100. Alternatively, animals on NGM plates were transferred directly onto hypotonic plates (2% agarose made with water). After 30 min, animals were recovered with M9 media and examined using SYTOX Green as described above.
Direct lysosomal rupture was induced in live animals by labelling with 1 mM acridine orange (AO; Molecular Probes) followed by irradiation with blue light (Hiruma et al., 2007). To follow the course of lysosomal rupture in individual animals, blue light was delivered via a mercury HBO 103 W lamp with an excitation filter of 450-490 nm. Lysosomal lysis, as shown by the dispersion of AO red fluorescence, was monitored using excitation and emission wavelengths of 572 nm and 630 nm, respectively. For bulk survival analyses, populations of worms were exposed to 425-475 nm blue light at an intensity at 450 W/m2 for 5 min using a BilliblanketPlus Phototherapy system (Ohmeda Medical). Fluorescent microscopy confirmed that >90% of the labelled gut granules lysed in 100% of the animals. Worms were allowed to recover for 1 h and scored for viability at several time points over a ~24 h period.
Statistical Analysis
Kaplan-Meier survival curves were plotted using GBSTAT and analyzed using the log-rank test. For survival analysis at individual time points, the mean % alive ± SD was derived from at least 3 experiments. Differences in the distributions (alive vs. dead) between groups were assessed statistically by chi square analysis (one degree of freedom) and by pooling the raw data from triplicate experiments (≈ 100 animals/group/experiment, or ≈ 300 total animals/group). P-values < 0.05 were considered significant with single and double asterisks indicating a P < 0.05 and < 0.01, respectively.
Imaging
The imaging of [Ca2+]i with Fura-2 AM and Fluo-4 AM, lysosome-like gut granules with 10,000 MW dextran-TMR and membrane permeability with propidium iodide, along with confocal and transmission electron microscopy methods are described in Supplemental Experimental Procedures.
RNAi
All RNAi was induced by feeding and is described in Supplemental Experimental Procedures.
Volumetric Analysis
The degree of animal swelling was based on approximating the volume of a cylinder and is described in Supplemental Experimental Procedures.
Biochemical Studies
The source of enzymes, inhibitors and substrates as well as the kinetic, stoichiometry of inhibition, complex formation and RSL cleavage assays are described in Supplemental Experimental Procedures.
Supplementary Material
Supplemental Data Supplemental data include one table, six figures, twelve movies and Supplementary Experimental Procedures and References can be found with this article online at http://www.cell.com/cgi/content/full
Acknowledgments
We thank Lewis Jacobson, Michael Hengartner and Erik Jorgensen for showing us the way of the worm, David H. Hall for assistance in C. elegans anatomy and James Moore, Glenn Papworth and Katie Baty for calcium and confocal imaging. This work was supported by the NIH (CA87007, CA86007 and HG001843), the Twenty-Five Club of Magee-Womens Hospital and the Mario Lemieux Foundation. This work is dedicated to the memory Stanley J. Korsmeyer, mentor, friend and inspiration.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Berger AJ, Hart AC, Kaplan JM. G alphas-induced neurodegeneration in Caenorhabditis elegans. J Neurosci. 1998;18:2871–2880. doi: 10.1523/JNEUROSCI.18-08-02871.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger BS, Dabbas M, Lopes A, Monaghan JM. Prognostic value of preoperative squamous cell carcinoma antigen level in patients surgically treated for cervical carcinoma. Gynecol Oncol. 1997;65:309–313. doi: 10.1006/gyno.1997.4619. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfie M, Wolinsky E. The identification and suppression of inherited neurodegeneration in Caenorhabditis elegans. Nature. 1990;345:410–416. doi: 10.1038/345410a0. [DOI] [PubMed] [Google Scholar]
- Church FC, Pike RN, Tollefsen DM, Buckle AM, Ciaccia AV, Olson ST. Regulation of Hemostasis by Heparin-binding Serpins. In: Silverman GA, Lomas DA, editors. Molecular and Cellular Aspects of the Serpinopathies and Disorders of Serpin Activity. Singapore: World Scientific Publishing Co. Pte. Ltd.; 2007. pp. 509–554. [Google Scholar]
- Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- de Duve C. Lysosomes revisited. Eur J Biochem. 1983;137:391–397. doi: 10.1111/j.1432-1033.1983.tb07841.x. [DOI] [PubMed] [Google Scholar]
- Dougherty KM, Pearson JM, Yang AY, Westrick RJ, Baker MS, Ginsburg D. The plasminogen activator inhibitor-2 gene is not required for normal murine development or survival. Proc Natl Acad Sci U S A. 1999;96:686–691. doi: 10.1073/pnas.96.2.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll M, Gerstbrein B. Dying for a cause: invertebrate genetics takes on human neurodegeneration. Nat Rev Genet. 2003;4:181–194. doi: 10.1038/nrg1018. [DOI] [PubMed] [Google Scholar]
- Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiological reviews. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci. 2007;32:37–43. doi: 10.1016/j.tibs.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Harbinder S, Tavernarakis N, Herndon LA, Kinnell M, Xu SQ, Fire A, Driscoll M. Genetically targeted cell disruption in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1997;94:13128–13133. doi: 10.1073/pnas.94.24.13128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GJ, Schroeder LK, Hieb CA, Kershner AM, Rabbitts BM, Fonarev P, Grant BD, Priess JR. Genetic analysis of lysosomal trafficking in Caenorhabditis elegans. Mol Biol Cell. 2005;16:3273–3288. doi: 10.1091/mbc.E05-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch C, Gauss R, Sommer T. Coping with stress: cellular relaxation techniques. Trends Cell Biol. 2006;16:657–663. doi: 10.1016/j.tcb.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Hiruma H, Katakura T, Takenami T, Igawa S, Kanoh M, Fujimura T, Kawakami T. Vesicle disruption, plasma membrane bleb formation, and acute cell death caused by illumination with blue light in acridine orange-loaded malignant melanoma cells. Journal of photochemistry and photobiology. 2007;86:1–8. doi: 10.1016/j.jphotobiol.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Jakab M, Furst J, Gschwentner M, Botta G, Garavaglia ML, Bazzini C, Rodighiero S, Meyer G, Eichmueller S, Woll E, et al. Mechanisms sensing and modulating signals arising from cell swelling. Cell Physiol Biochem. 2002;12:235–258. doi: 10.1159/000067895. [DOI] [PubMed] [Google Scholar]
- Kokel M, Borland CZ, DeLong L, Horvitz HR, Stern MJ. clr-1 encodes a receptor tyrosine phosphatase that negatively regulates an FGF receptor signaling pathway in Caenorhabditis elegans. Genes Dev. 1998;12:1425–1437. doi: 10.1101/gad.12.10.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuranaga E, Miura M. Nonapoptotic functions of caspases: caspases as regulatory molecules for immunity and cell-fate determination. Trends Cell Biol. 2007;17:135–144. doi: 10.1016/j.tcb.2007.01.001. [DOI] [PubMed] [Google Scholar]
- Lamitina ST, Strange K. Transcriptional targets of DAF-16 insulin signaling pathway protect C. elegans from extreme hypertonic stress. Am J Physiol Cell Physiol. 2005;288:C467–C474. doi: 10.1152/ajpcell.00451.2004. [DOI] [PubMed] [Google Scholar]
- Law RH, Zhang Q, McGowan S, Buckle AM, Silverman GA, Wong W, Rosado CJ, Langendorf CG, Pike RN, Bird PI, et al. An overview of the serpin superfamily. Genome Biol. 2006;7:216–226. doi: 10.1186/gb-2006-7-5-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettre G, Hengartner MO. Developmental apoptosis in C. elegans: a complex CEDnario. Nat Rev Mol Cell Biol. 2006;7:97–108. doi: 10.1038/nrm1836. [DOI] [PubMed] [Google Scholar]
- Lithgow GJ, White TM, Melov S, Johnson TE. Thermotolerance and extended life-span conferred by single-gene mutations and induced by thermal stress. Proc Natl Acad Sci U S A. 1995;92:7540–7544. doi: 10.1073/pnas.92.16.7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd JB, Forster S. The lysosome membrane. Trends Biochem Sci. 1986;11:365–368. [Google Scholar]
- Luke CJ, Pak SC, Askew DJ, Askew YS, Smith JE, Silverman GA. Selective conservation of the RSL-encoding, proteinase inhibitory-type, clade l serpins in Caenorhabditis species. Front Biosci. 2006;11:581–594. doi: 10.2741/1820. [DOI] [PubMed] [Google Scholar]
- Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003;301:1387–1391. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- Melloni E, Averna M, Stifanese R, De Tullio R, Defranchi E, Salamino F, Pontremoli S. Association of calpastatin with inactive calpain: a novel mechanism to control the activation of the protease? J Biol Chem. 2006;281:24945–24954. doi: 10.1074/jbc.M601449200. [DOI] [PubMed] [Google Scholar]
- Nehrke K. A reduction in intestinal cell pHi due to loss of the Caenorhabditis elegans Na+/H+ exchanger NHX-2 increases life span. J Biol Chem. 2003;278:44657–44566. doi: 10.1074/jbc.M307351200. [DOI] [PubMed] [Google Scholar]
- Nelson FK, Riddle DL. Functional study of the Caenorhabditis elegans secretory-excretory system using laser microsurgery. J Exp Zool. 1984;231:45–56. doi: 10.1002/jez.1402310107. [DOI] [PubMed] [Google Scholar]
- Scarff KL, Ung KS, Nandurkar H, Crack PJ, Bird CH, Bird PI. Targeted Disruption of SPI3/Serpinb6 Does Not Result in Developmental or Growth Defects, Leukocyte Dysfunction, or Susceptibility to Stroke. Mol Cell Biol. 2004;24:4075–4082. doi: 10.1128/MCB.24.9.4075-4082.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schick C, Bromme D, Bartuski AJ, Uemura Y, Schechter NM, Silverman GA. The reactive site loop of the serpin SCCA1 is essential for cysteine proteinase inhibition. Proc Natl Acad Sci USA. 1998;95:13465–13470. doi: 10.1073/pnas.95.23.13465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott BA, Avidan MS, Crowder CM. Regulation of hypoxic death in C. elegans by the insulin/IGF receptor homolog DAF-2. Science. 2002;296:2388–2391. doi: 10.1126/science.1072302. [DOI] [PubMed] [Google Scholar]
- Silverman GA, Bird PI, Carrell RW, Coughlin PB, Gettins PG, Irving JI, Lomas DA, Luke CJ, Moyer RW, Pemberton PA, et al. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins: Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J Biol Chem. 2001;276:33293–33296. doi: 10.1074/jbc.R100016200. [DOI] [PubMed] [Google Scholar]
- Silverman GA, Whisstock JC, Askew DJ, Pak SC, Luke C, Cataltepe S, Irving JA, Bird PI. Human clade B serpins (ov-serpins) belong to a cohort of evolutionarily-dispersed intracellular proteinase inhibitor clades that protect cells from promiscuous proteolysis. Cell Mol Life Sci. 2004;61:301–325. doi: 10.1007/s00018-003-3240-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syntichaki P, Xu K, Driscoll M, Tavernarakis N. Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature. 2002;419:939–944. doi: 10.1038/nature01108. [DOI] [PubMed] [Google Scholar]
- Tawe WN, Eschbach ML, Walter RD, Henkle-Duhrsen K. Identification of stress-responsive genes in Caenorhabditis elegans using RT-PCR differential display. Nucleic Acids Res. 1998;26:1621–1627. doi: 10.1093/nar/26.7.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcherepanova I, Bhattacharyya L, Rubin CS, Freedman JH. Aspartic proteases from the nematode Caenorhabditis elegans. Structural organization and developmental and cell-specific expression of asp-1. J Biol Chem. 2000;275:26359–26369. doi: 10.1074/jbc.M000956200. [DOI] [PubMed] [Google Scholar]
- Wendt A, Thompson VF, Goll DE. Interaction of calpastatin with calpain: a review. Biol Chem. 2004;385:465–472. doi: 10.1515/BC.2004.054. [DOI] [PubMed] [Google Scholar]
- Xu K, Tavernarakis N, Driscoll M. Necrotic cell death in C. elegans requires the function of calreticulin and regulators of Ca(2+) release from the endoplasmic reticulum. Neuron. 2001;31:957–971. doi: 10.1016/s0896-6273(01)00432-9. [DOI] [PubMed] [Google Scholar]
- Yamashima T, Saido TC, Takita M, Miyazawa A, Yamano J, Miyakawa A, Nishijyo H, Yamashita J, Kawashima S, Ono T, et al. Transient brain ischaemia provokes Ca2+, PIP2 and calpain responses prior to delayed neuronal death in monkeys. The European journal of neuroscience. 1996;8:1932–1944. doi: 10.1111/j.1460-9568.1996.tb01337.x. [DOI] [PubMed] [Google Scholar]
- Yamashima T, Tonchev AB, Tsukada T, Saido TC, Imajoh-Ohmi S, Momoi T, Kominami E. Sustained calpain activation associated with lysosomal rupture executes necrosis of the postischemic CA1 neurons in primates. Hippocampus. 2003;13:791–800. doi: 10.1002/hipo.10127. [DOI] [PubMed] [Google Scholar]
- Yassin L, Gillo B, Kahan T, Halevi S, Eshel M, Treinin M. Characterization of the deg-3/des-2 receptor: a nicotinic acetylcholine receptor that mutates to cause neuronal degeneration. Mol Cell Neurosci. 2001;17:589–599. doi: 10.1006/mcne.2000.0944. [DOI] [PubMed] [Google Scholar]
- Zatz M, Starling A. Calpains and disease. N Engl J Med. 2005;352:2413–2423. doi: 10.1056/NEJMra043361. [DOI] [PubMed] [Google Scholar]
- Zhang M, Park SM, Wang Y, Shah R, Liu N, Murmann AE, Wang CR, Peter ME, Ashton-Rickardt PG. Serine protease inhibitor 6 protects cytotoxic T cells from self-inflicted injury by ensuring the integrity of cytotoxic granules. Immunity. 2006;24:451–461. doi: 10.1016/j.immuni.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Zong WX, Thompson CB. Necrotic death as a cell fate. Genes Dev. 2006;20:1–15. doi: 10.1101/gad.1376506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data Supplemental data include one table, six figures, twelve movies and Supplementary Experimental Procedures and References can be found with this article online at http://www.cell.com/cgi/content/full