

Abstract
Chronic nicotine exposure, in smokers or in experimental rodents administered nicotine, produces elevated levels of nicotinic acetylcholine receptors in several brain regions. However, there are few data on up-regulation of receptors in specific neuronal subtypes. We tested whether functional up-regulation of nicotinic responses occurs in cultured GABAergic neurons of the ventral midbrain. Fura-2 measurements of nicotinic responses were made on ventral midbrain neurons from knock-in mice heterozygous for the α4-M2 domain Leu9'Ala mutation, which confers nicotine hypersensitivity. Chronic nicotine exposure at a concentration (10 nM for 3 days) that activates only the hypersensitive α4*(Leu 9' Ala) receptors, but not wild-type receptors, resulted in significant potentiation of ACh (100 μM)-elicited responses. Experiments were also performed on midbrain neuronal cultures heterozygous for the α4*(Leu9'Ala) mutation as well as for a GFP protein fused to a GABA transporter that reliably reveals GABAergic neurons. In cultures chronically treated with 10 nM nicotine, there was significantly increased α4* nicotinic-induced Ca2+ influx elicited by low concentration of ACh (3 μM). Furthermore, chronic exposure to the competitive antagonist dihydro-β-erythroidine, but not to the noncompetitive antagonist mecamylamine, induced up-regulation of ACh elicited nicotinic responses. These results suggest that occupation of α4* nicotinic receptor binding site(s), at the interface between two subunits, is sufficient to promote assembly and/or up-regulation of functional receptors in GABAergic neurons. Up-regulation in neurons is both “cell-autonomous”, occurring at the cell itself, and “receptor autonomous”, occurring at the receptor itself, and may be a thermodynamic necessity of ligand-protein interactions.
Keywords: nicotinic receptor, up-regulation, GABAergic neurons, ventral midbrain, fura-2, neuronal cultures
Introduction
Nicotine is the most widely abused drug, afflicting over one billion people worldwide. The primary targets of nicotine are neuronal nicotinic acetylcholine receptors (nAChRs), of which there are 12 different subunits that combine in a variety of stoichiometries to form pentameric ligand-gated cation channels [1]. Most α4 subunit containing (α4*) receptors also contain the β2 subunit (β2*)[2], although other subunits are probably present in specific neuronal types. This so-called α4β2* nAChR receptor family comprises the most prevalent high-sensitivity nAChRs in the brain[3]; that is, nicotine at the sub-micromolar concentrations found in tobacco smoke binds to, and activates, α4β2 receptors more than other subtypes.
Activation of α4β2* nicotinic receptor subunits is necessary and sufficient for tolerance, sensitization and reward behavior [4-6]. Nearly a quarter-century ago, pioneering studies in vivo using radio-ligand binding showed that chronic nicotine administered to rodents results in elevated levels of nicotinic receptors [7, 8]. Up-regulation of α4β2* receptors with chronic nicotine also occurs in vitro in transfected clonal cell lines [9-13] and in primary neuronal cultures [14]. Furthermore, the mechanism of receptor up-regulation is apparently due to posttranscriptional mechanisms since nAChR mRNA expression levels are unaltered with chronic nicotine [12, 13,15]. Although the mechanism of receptor up-regulation is unclear, several models have been proposed. Chronic nicotine might increase receptor assembly or decrease receptor degradation [9, 14, 16]; act as a pharmacological chaperone to facilitate assembly of subunits [10]; facilitate subunit protein folding into the correct conformation that would allow for subunit assembly [17]; decrease turnover rate of the receptor [18]; induce a conformational switch into a more easily activated high affinity receptors [19, 20]; or facilitate maturation of nicotinic receptors in the endoplasmic reticulum of the secretory pathway [21].
We previously found that chronic nicotine can functionally up-regulate nicotinic responses in cultured ventral midbrain neurons [14]. In a subsequent publication using cultured midbrain neurons from the α4(Leu9'Ala) mice, we showed that nicotine at a low enough concentration to interact selectively with α4* receptors was sufficient to up-regulate nicotinic responses [5]. In the present study, we examined the influence of chronic nicotine in α4(Leu 9' Ala) midbrain neuronal cultures in only one subtype of neurons--GABAergic neurons.
Methods
Calcium imaging in cultured ventral midbrain neurons
Changes in [Ca2+]i were measured in ventral midbrain neuronal cultures using the ratiometric fura-2 method [5, 14, 22]. Ventral midbrain tissue (including the SN and VTA) obtained from embryonic day 14 embryos was digested with 1 mg/ml papain, and cells were mechanically separated by gentle pipetting. Cells were plated onto 35 mm dishes with glass coverslip bottoms in Neurobasal culture medium containing 2% B27 supplement, 0.5 mm Glutamax, and 5% horse serum, all obtained from Invitrogen (Carlsbad, CA). Cell cultures were kept in a temperature-, humidity-, and CO2/O2-controlled incubator. Measurements were made at 28 to 35 days in culture. For chronic drug application, (-)-nicotine hydrogen tartrate (10 nM), dihydro-β-erythroidine hydrobromide (DHβE, 10 μM), mecamylamine hydrochloride (MEC, 10 μM), or control solution (cell culture media) were incubated in the culture dishes for three days with replacement of fresh drug solution at each day of incubation. The chronic drug or control solution was removed ∼ 1 hr before the imaging experiments. Dishes were rinsed thoroughly twice with extracellular solution and during this 1 hr period, cells were loaded with 1.25 μm fura-2 AM (Invitrogen) in a 0.01% pluronic acid solution for 30 min at room temperature (22 ± 2°C) and in the dark. During experiments, cultures were continually perfused with an extracellular solution (in mm): 150 NaCl, 10 HEPES, 10 glucose, 4 KCl, 2 CaCl2, and 2 MgCl2. Various channel blockers to suppress nonspecific responses were included in the extracellular solution (in μm): 0.5 TTX(voltage-gated Na+ channels), 0.5 atropine (muscarinic receptors), 10 CNQX (AMPA and kainate receptors), 20 bicuculline (GABAA receptors), 50 AP-5 (NMDA receptors) and 0.01 methyllycaconitine (MLA, α7 receptors). Ascorbic acid at 1 mm was also added to the extracellular solution to protect neurons from photodamage. Imaging was performed with an inverted fluorescence microscope (IX71, Olympus, Melville, NY) using a 40× oil-immersed objective (UApo/340, 1.35 numerical aperture). To generate ratiometric estimates of [Ca2+]i, pairs of images were obtained with alternate excitation wavelengths of 340/380 nm and emission wavelength of 510 nm. A U-tube connected to a pipette, placed ∼0.5 mm away from the targeted cell population, was used to deliver nicotine. Nicotinic receptors desensitize at agonist concentrations too low to cause appreciable activation; therefore, between applications, negative pressure was maintained on the U-tube to prevent nicotine from leaking out of the pipette. Nicotine was applied onto cultured cells when solution flow in the U-tube was momentarily (2 s) reversed by stopping outflow with a computer-controlled pinch valve. Images were captured with a 16 bit CCD camera (Cascade 650, Photometrics, Tucson, AZ) and analyzed with Slidebook 4.1 imaging software (Intelligent Imaging Innovations, Denver, CO).
Spectral Confocal Imaging
GAT1-GFP fluorescence images were collected from an inverted confocal microscope equipped with spectrally resolved multiple detector systems (C1si, Nikon, Tokyo, Japan). Each pixel of the X-Y image has complete spectral emission data, comprising the lambda stack [23-25]. Spectral images were taken, with excitation at 440 nm by a modulated diode laser at 20% power, over 512 × 512 pixels at 12-bit intensity resolution using a 60X plan apochromat water (1.2 NA) objective. The pinhole was set to a nominal diameter of 61.3 μm. Lambda stacks of images were acquired at wavelengths between 479 and 634 nm, at 5 nm steps.
In all cases, detector gains and laser attenuators were adjusted to lie within the linear range of the detector.
Statistical Analysis
All values are reported as mean ± S.E.M and the number of experiments was also reported. Significant differences (at p < 0.05) were determined between two groups using a t-test for continuous data meeting parametric assumptions of normality and equal variance. Otherwise the Mann-Whitney rank sum test was used for nonparametric data. For comparisons between three groups, significant difference (at p < 0.05) were determined using a one-way analysis of variance for continuous data meeting parametric assumptions of normality and equal variance. Otherwise the Kruskal-Wallis one-way analysis of variance on ranks was performed with Dunn's method of pairwise post-hoc analysis. Degrees of freedom (df) were reported for results of statistical analyses.
Results
Interaction of chronic nicotine with α4* receptors in cultured ventral midbrain neurons is sufficient for up-regulation of nicotinic responses
We examined whether selective interaction of chronic nicotine with α4* nAChRs in cultured ventral midbrain neurons is sufficient to induce up-regulation of functional nicotinic Ca2+ flux through nicotinic receptors using fura-2 measurements. We used cultured neurons from brain tissue of knock-in mice heterozygous for the α4 (Leu 9'Ala) mutation [5, 22]. The Leu9'Ala mutation is near the gate (second transmembrane region) of the channel and renders the receptor 40-50-fold more sensitive to ACh or nicotine [5, 22]. Appreciable nAChR signals appear in midbrain cultures after a period of several weeks. Therefore, imaging was performed on neurons 4-5 weeks in culture.
Either saline (control) or a low chronic dose of nicotine (10 nM for 3 days) was applied to the dishes of the cultured ventral midbrain neurons. Nicotine at 10 nM does not activate wild-type α4β2 nicotinic receptors, does activate (< 7% maximal response) α4(L9'A) β2 nAChRs, and falls within a “window” that does not completely desensitize receptors [22]. Nicotine was washed from the dish of cultured neurons for at least 1 hr. When we tested nicotinic responses elicited by 2 sec application of 100 μM Ach (activates 60% and 100% of wildtype and hypersensitive receptors, respectively), we found a significant up-regulation (66 ± 7%) of peak fura-2 measured calcium fluxes in ventral midbrain neurons from chronic nicotine (0.21 ± 0.02) as compared to chronic saline (0.13 ± 0.01; t-test, p < 0.001, df = 689; Figure 1A).
Figure 1. Fura-2 recordings of cultured ventral midbrain neurons after chronic exposure to nicotine.

Fura-2 imaging was performed on ventral midbrain cultures from embryos heterozygous for the hypersensitive α4(Leu 9'Ala) mutation.
A, Time course of fura-2-recorded Ca2+ fluxes, showing that midbrain neurons previously exposed to chronic nicotine (10 nM for 3 d) had greater ACh (100 μM for 2 sec) induced nicotinic responses than control neurons.
B, Since voltage-gated calcium channel blockers could not be used because they also target nicotinic receptors[22], in control experiments fura-2 recordings in cultured neurons were conducted during membrane depolarization with 50 mM KCl (2 sec). There was no difference in the KCl-elicited Ca2+ influx between control and chronic nicotine treated neurons, indicating that functional voltage-gated calcium channel responses are not up-regulated. Therefore the functional up-regulation in A is due directly to nicotinic receptor activity. Inset shows a plot of fura-2 responses over various KCl concentrations, showing that the responses do not saturate at 50 mM KCl. Fura-2 measurements were taken in the presence of ion channel and receptor blockers: atropine, AP-5, CNQX, bicuculline, TTX, and MLA.
These results support the idea that selective interaction of chronic nicotine with the α4*(Leu 9' Ala) receptors is sufficient for up-regulation of nicotinic responses in ventral midbrain neurons, and confirmed previous experiments on cultures from knock-in mice with hypersensitive α4*-Leu 9'Ala receptors [5].
One potential confounding issue may be that the up-regulated fura-2 responses could reflect calcium influx through up-regulated voltage-gated calcium channels, which activate subsequent to membrane depolarization following activation of nicotinic receptors. Various channel blockers to suppress nonspecific responses were included in the extracellular solution: TTX (voltage-gated Na+ channels), atropine (muscarinic receptors), CNQX (AMPA and kainate receptors), bicuculline (GABAA receptors), AP-5 (NMDA receptors) and methyllycaconitine (MLA, α7 receptors). In previous studies of whole-cell patch-clamp recordings of α4β2 nAChRs, toxins and pharmacological inhibitors of a variety of voltage-gated calcium channels directly blocked α4β2 nAChRs with similar or higher affinity [22], vitiating their use in these experiments. Therefore, we activated voltage-gated Ca2+ channels by depolarizing the cultured neurons in the presence of the various channel blockers in the extracellular solution; we found no significant difference in fura-2 measured Ca2+ influx in neurons chronically treated with nicotine (10 nM for 3 days) vs saline treated controls (p = 0.5, t-test, df = 253). Thus, we ruled out that the up-regulated nicotinic responses with chronic nicotine was due to secondary activation of voltage-gated calcium channels.
Thus in ventral midbrain neurons chronically treated with nicotine the functional up-regulation of fura-2 measurements was, in fact, due to functional up-regulation of Ca2+ flux through up-regulated nicotinic receptors rather than indirectly due to voltage-gated calcium channels in ventral midbrain neurons.
Interaction of chronic nicotine with α4* receptors in cultured GABAergic neurons is sufficient for up-regulation of α4* responses
To begin exploring the mechanism of functional up-regulation in GABAergic neurons of the ventral midbrain, we further tested whether ventral midbrain GABAergic neurons, in particular, responded to chronic nicotine with up-regulated functional α4* receptors in a cell culture system using fura-2 measurements. Previous measurements with this system showed that chronic incubation with 1 μM nicotine functionally up-regulated nicotine responses with pharmacology suggestive of α4β2* receptors. Also, in cultured neurons transfected with fluorescent α4β2* receptors, chronic incubation with 1 μM nicotine numerically up-regulated receptors as measured by fluorescence of the subunits [14].
In the present study, we tested whether ventral midbrain GABAergic neurons, in particular, responded to chronic nicotine with up-regulated functional α4* receptors. To distinguish living GABAergic mouse neurons in culture, we chose a knock-in strain containing a GFP moiety in the GAT1 GABA transporter, because (1) rigorous electrophysiological tests on this strain showed that the set of fluorescent neurons equals the set of GABAergic neurons; (2) the GAT1-GFP strain is maintained on a mixed Sv129-C57BL6 background similar to the α4-YFP strain; and (3) the GAT1-GFP strain has normal electrophysiological, behavioral, and reproductive characteristics[26]. We bred GAT1-GFP mice with wild-type mice. In pilot experiments on the midbrain cultures from embryonic mice, we found that the cultures contained >95% GABAergic neurons. These observations that nearly all the neurons are GABAergic, combined with previous experiments showing up-regulation of functional α4β2 receptors transfected into neurons [14], provided proof that chronic nicotine up-regulates functional α4* receptors in nearly pure cultures of ventral midbrain GABAergic neurons. Contact with additional neuronal types, such as their typical pre-synaptic input cells, is not necessary for nicotine-induced up-regulation in cultured GABAergic neurons.
The availability of the α4* hypersensitive strain allowed for extending these results to test two additional points: whether activation of α4* receptors (without concurrent activation of other nAChRs) on GABAergic neurons is sufficient for functional up-regulation of these α4* receptors; and whether the previous pharmacological evidence for α4 responses could be supplanted by more direct isolation of α4* responses. The α4-L9'A strain has been well characterized electrophysiologically, neurochemically, and behaviorally; it has only a modestly decreased number of α4β2* receptors[5, 22]. We bred homozygous GAT1-GFP mice with homozygous α4-L9'A mice (from a colony that is also maintained on a mixed Sv129/C57BL6 background), producing embryonic mice heterozygous for both the hypersensitive α4L9'A knock-in allele [5] and the GAT1-GFP knock-in allele[26]. We exposed the embryonic cultures to a low concentration of nicotine (10 nM) for 3 days, so that only the hypersensitive α4* receptors were chronically activated[5]. Then, by measuring Ca2+ flux in fluorescent neurons containing hypersensitive α4L9'A* nAChRs, we were confident that a low ACh dose (3 μM for 2 s) activated primarily the α4* nAChRs on GABAergic neurons. We found that chronic incubation in nicotine for 3 d resulted in a 30 ± 9% and 82 ± 15% increase in peak and area of ACh-induced Ca2+ response (p < 0.002 and p < 0.001, t-test, df = 343, respectively) as compared to control (Figure).
Because the experiments allowed us both to activate chronically, and then to test, only α4 responses in a well-characterized neuronal culture from ventral midbrain, we conclude that α4* receptor activating concentrations of nicotine in GABAergic neurons are sufficient for functional up-regulation of these α4* receptors.
Chronic competitive antagonism with DHβE but not MEC up-regulates α4* responses
Because nicotinic antagonists can also up-regulate α4* nicotinic responses in HEK293 cell lines [9, 19], we also examined the effects of chronic exposure of two antagonists on the nicotinic responses in cultured ventral midbrain neurons from knock-in mice with hypersensitive Leu9'Ala α4* receptors. Fura-2 responses elicited by 2 sec application of 100 μM ACh were significantly larger (34 ± 4%) in neurons treated for 3 days) with the nicotinic receptor competitive antagonist dihydro-β-erythroidine (DHβE), 10 μM (0.12 ± 0.01 vs 0.09 ± 0.01 in DHβE vs control, respectively, p = 0.037, t-test, df = 92)(Figure 3A). However, there was no significant difference in the fura-2 measured nicotinic responses in neurons incubated with the noncompetitive antagonist mecamylamine (10 μM, 3 days) (0.09 ± 0.01 vs 0.09 ± 0.01 in mecamylamine vs control, respectively, p = 0.6, t-test, df = 99).
Figure 3. Fura-2 recordings of cultured ventral midbrain neurons after chronic exposure to nicotinic antagonists.

Fura-2 imaging was performed on ventral midbrain cultures from embryos heterozygous for the hypersensitive α4 (Leu 9'Ala) mutation.
A, Time course of fura-2-recorded Ca2+ fluxes showing that cultured midbrain neurons in chronic DHβE (10 μM for 3 d), had greater ACh (100 μM for 2 sec) induced responses than control neurons.
B, Time course of fura-2-recorded Ca2+ fluxes showing that midbrain neuronal cultures in chronic noncompetitive antagonist, mecamylamine (10 μM for 3 d), had no significant difference in ACh (100 μM for 2 sec) induced responses as compared to control neurons. C, Bar graph showing that the area of the ACh induced fura-2 responses in DHβE treated neurons was significantly augmented over control but not for mecamylamine (MEC) treated neurons. Fura-2 measurements were taken in the presence of ion channel and receptor blockers: atropine, AP-5, CNQX, bicuculline, TTX, and MLA.
Previous studies in cell lines [9-11] showed that chronic administration of the competitive antagonist DHβE functionally up-regulates α4* nicotinic responses, with no effect of the non-competitive antagonist mecamylamine. The results from this study extend this result to GABAergic ventral midbrain neurons.
Discussion
Up-regulation is “cell autonomous” and “receptor autonomous”
In the present study we have extended the results of previous studies examining up-regulation of α4* nAChRs in ventral midbrain neurons from wild-type mice using nicotinic receptor pharmacology [14] and from the gain-of-function α4(Leu9'Ala)* knock-in mice [5]. We present data showing that selective activation of α4* nAChRs is sufficient for α4* receptor up-regulation in almost pure cultures of GABAergic ventral midbrain neurons. We term this result “cell autonomy”, by analogy with the classical genetic concept of a protein whose mutation produces a phenotype in the cell that expresses the protein, without the need for interactions with other cells.
Cultured midbrain neurons have yielded extensive information about α4* receptor up-regulation. In previous studies on cultured midbrain neurons from wild-type mice, chronic nicotine up-regulated functional responses with pharmacology appropriate to α4* receptors[14]. Also, transfected fluorescent α4* subunits were up-regulated at the protein level[14]. Experiments with cultures from α4-hypersensitive mice then showed that up-regulation occurred in chronic nicotine concentrations (10 nM) that activated only α4* receptors[5]. In the present experiments, we add the two facts that functional up-regulation occurs in homotypic GABAergic cultures lacking their typical presynaptic partners and that functional up-regulation occurs when tested with ACh concentrations that activate only the α4 receptors. It can thus be concluded that activation of α4* receptors on GABAergic neurons is sufficient for numerical up-regulation of functional α4* receptors on these same GABAergic neurons. We note that because desensitization occurs with a high probability following channel opening, we also allow the possibility that up-regulation arises more strongly from desensitization than from activation. We term this result “receptor autonomy”, because activation of no other nicotinic receptors appears necessary within the cell-autonomous process that leads to up-regulation.
The mechanism of up-regulation of nicotinic receptors remains unsettled. “Cell autonomy” and “Receptor autonomy” are certainly consistent with the data from clonal cell lines transfected with nAChRs, that nicotine acts as a “pharmacological chaperone” or “maturational enhancer” for α4β2* receptors [10, 21, 27]. Nicotinic receptor up-regulation could also arise from mechanisms that regulate receptor turnover[10], from proteins that govern assembly and trafficking[9, 20, 21, 28-31], or from additional factors that regulate desensitization[32]. Another potential mechanism of nAChR up-regulation may be due to signal transduction mechanisms initiated by calcium influx through the receptors. One strong counterargument to the Ca influx hypothesis is that the competitive antagonist DHβE also induces functional up-regulation (Figure 3). Interestingly, the noncompetitive antagonist, mecamylamine, did not produce any functional up-regulation (Figure 3), consistent with a previous study performed in HEK293 cells [9]; this point is discussed in the next section. Furthermore, human α4 harboring a Ser 247 Phe mutation (at the M2 6' position) renders the channel nonfunctional in HEK tsA201 cells and yet chronic nicotine induces robust up-regulation of the receptor in this clonal cell line [10]. This point also argues against a calcium signaling mechanism of nicotinic receptor up-regulation.
In the future, it will be vital to examine changes in specific nAChR subunit expression in specific neuronal types in vivo. Such information will allow us to better understand how altered nicotinic receptor expression in specific neuronal circuits will alter their functional output and ultimately lead us to a better understanding how chronic nicotine use develops into an addiction.
Thermodynamic aspects of the pharmacological chaperone concept at α4β2 receptors
Recent papers provide incisive discussions about the mechanism of up-regulation[9, 13, 19, 21, 27, 33, 34]. That up-regulation is both “cell-autonomous” and “receptor-autonomous”, proceeds post-translationally [12, 13, 15, 18, 19], occurs before insertion in the plasma membrane [9, 21, 27], and does not involve an obvious Ca signaling pathway (present data) allows one to consider relatively straightforward concepts in up-regulation. Even in biochemistry, thermodynamics rules the fate of molecular complexes, if kinetic barriers permit. It is therefore useful to ask whether, and to what extent, up-regulation is determined by the energetics of ligand binding and by chemical potentials that are proportional to log(concentration). However, we do not distinguish between entropy and enthalpy when referring to the thermodynamics of nicotine-receptor interactions [35-38]. In the context of drug-receptor binding, thermodynamics provides that the drug-receptor interaction eventually reaches its most stable state (again, kinetic barriers permitting) (Figure 4). First, we note that a drug's overall affinity for a receptor includes contributions from the possibly complex conformational states that occur only (or predominantly) if the drug is bound. In other words, if a drug-bound receptor isomerizes to form an open channel, then channel opening usually “locks” the agonist onto the receptor[39]; and the overall dissociation constant is lowered by the presence of all the open states (Figure 4B). Indeed, if desensitization follows binding and is at least as likely as opening, then desensitization also significantly further locks the agonist onto the receptor and decreases the overall dissociation constant of the drug-receptor complex (Figure 4B).
Figure 4. Thermodynamics of nicotinic receptor up-regulation.
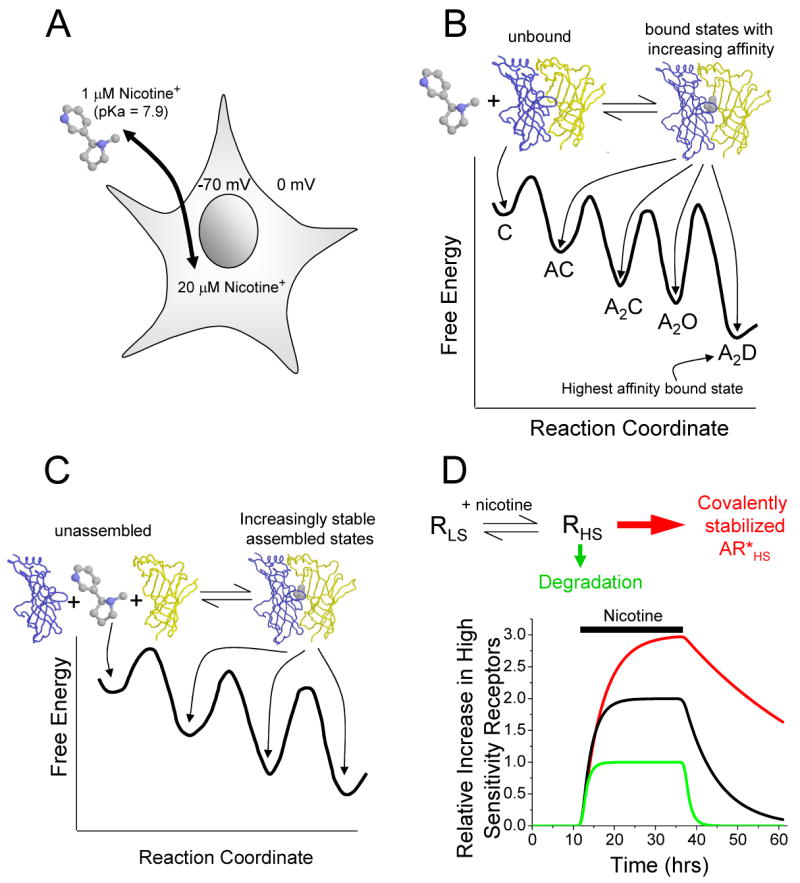
A, Since nicotine is cell-permeant in its deprotonated form and a weak base (pKa = 7.9), it accumulates within cells according to the membrane potential—roughly 20-fold at -70 mV. Thus, intracellular nicotine can interact with nicotinic receptor subunits in compartments where they are being assembled and transported. The concentration of nicotine accumulated inside the neurons is 100 to 1000 fold greater than the dissociation constant for the receptors and therefore can bind to a majority of the intracellular pool of receptors.
B, When nicotine binds to the interface of nicotinic receptor subunits (modified from the AChBP structure from Celie et al [55]), the bound complex has a lower total free energy than the unbound state, and the complex eventually finds the bound state of highest affinity (lowest total free energy) [56]. C, closed; A, agonist; O, open; and D, desensitized.
C, Nicotine helps stabilizes a full pentamer in the assembled state by binding to the interfaces of the nicotinic receptor subunits.
D, The concentration of nicotine found in the bloodstream of smokers selectively up-regulates the high sensitivity (HS) over the low sensitivity (LS) subpopulation of nicotinic receptors since the EC50s of the low and high subpopulations are ∼1 μM and ∼100 μM, respectively. This situation, which summarizes the concepts of panels A, B, and C, is denoted by the black line. In addition, the assembled receptor may be diverted away from the degradative pathway (green line). Additional interactions with other proteins or covalent modifications may result in further “covalent stabilization” (red line), resulting in additional nicotinic receptor up-regulation. Since receptor synthesis and assembly occur over hours, we expect that the turnover rate should be at least as long.
If a drug is present at a concentration comparable to, or higher than, the overall dissociation constant for the high-affinity interaction, then the presence of the drug leads to a significant fraction of high-affinity complexes; if, on the other hand, the drug is present at a concentration lower than the overall binding constant, the equilibrium is shifted only slightly. We now add a kinetic argument: in most straightforward kinetic schemes, the stable states are likely to form, in the presence of ligand, somewhat faster but on the same time scales as they disappear when ligand is removed. If formation of the stable states requires minutes to hours, we expect the stabilization to last at least as long [12, 13]. This stabilization of high-affinity or high-sensitivity states, which outlasts the presence of the ligand, is a form of up-regulation that can be analyzed without reference to detailed cell biological mechanisms. Indeed, much of up-regulation may simply be stabilization. According to this reasoning, it is not surprising that nicotine, when present at the sub-micromolar concentrations in the brain of smokers, most strongly up-regulates α4β2* receptors; for α4β2* receptors have higher overall affinity than most other nAChRs.
That nicotine binds at some subunit interfaces of a pentamer provides another level of stabilization [16, 19, 40]. Because nicotine binds to both α and β subunits simultaneously, it stabilizes the ternary complex, α–nicotine-β. Nicotine therefore helps to stabilize a full pentamer in an assembled state[14] (Figure 4C).
It is now well accepted that α4β2 receptors exist in a mixed population whose EC50 values differ by 50- to 100-fold [14, 19, 41, 42] (∼ 1 μM and ∼ 100 μM). The reasoning given in the previous paragraph implies that micromolar nicotine concentrations preferentially stabilize, and therefore preferentially up-regulate the high-affinity but not the low-affinity form of the pentamer. The physical difference between the states is not yet clear. It could be an isomerization[19, 20]. Alternatively, several papers suggest that the two forms differ in subunit composition: the high- and low-sensitivity forms would consist of the α42β23 and α43β22 pentamers, respectively [43-45]. The physical basis of the distinction is indeed important because the nature of the difference may determine the barriers to interconversion; for instance, if the subunit composition gives rise to the difference, interconversion is unlikely to happen after the endoplasmic reticulum; and if stoichiometry is involved, then available subunit ratios also influence the outcome[18]. However, thermodynamic arguments about stabilization by nicotine do not require one to know this difference. Nicotine at concentrations ∼ 1 μM must stabilize the high-sensitivity form 50 - 100 times more than the low-affinity form, corresponding to (RT/F)ln(100) = 2.6 kcal/mol. The additional stabilization provided by desensitization might change these values; there are inconsistent observations about the relative desensitization properties of the two forms.
In this view, up-regulation by stabilization of functional, high-sensitivity nicotinic receptors by nicotine is a thermodynamic necessity. The process is probably enhanced by the fact that nicotine is cell-permeant and can therefore interact with receptors when they are being synthesized and transported [9, 21], not simply after stable insertion into the cell membrane. Furthermore, because nicotine, like most alkaloids, is a weak base, it accumulates within cells according to the membrane potential—roughly 20-fold at -70 mV [46] (Figure 4A). The endoplasmic reticulum probably has little transmembrane potential, allowing the nicotine concentration that interacts with nascent receptors to reach values roughly equal to that of the cytoplasm. It is not surprising that nicotine exhibits an EC50 of < 100 nM for up-regulation in clonal cell lines transfected with α4 and β2 subunits[27]. Nicotinic receptors are also up-regulated by impermeant agonists including simple tetramethylammonium and carbachol, showing that surface interactions alone may provide a sufficient thermodynamic driving force; however full up-regulation by impermeant agonists does require rather high concentrations (0.1 – 1 mM) [21, 33, 47].
According to these considerations, one expects any drug to up-regulate nicotinic receptors by stabilizing them if (a) it binds at a subunit interface, (b) has an EC50 or overall dissociation constant in the micromolar range, and (c) permeates cell membranes. The competitive antagonist DHβE is therefore not a surprising up-regulator in the present and previous experiments [9, 19, 21]. One thermodynamic distinction between agonist and antagonist binding to neuronal nicotinic receptors is that agonist binding is both enthalpy and entropy driven, while antagonist binding is completely entropy driven [35].
Channel blockers typically bind on the axis of the channel pore and therefore touch all subunits; if such binding stabilizes the assembled state of the receptor, it too might up-regulate. “Foot-in-the-door” open-channel blockers like QX-222 at muscle nicotinic receptors strongly stabilize agonist binding[48]; therefore the most dramatic effects of channel blockers might be to enhance agonist-induced up-regulation when the blocker is applied simultaneously with agonist. However there are few examples of such well-defined actions for neuronal AChRs. Neither hexamethonium [49] nor chlorisondamine [50] are foot-in-the door blockers at high-sensitivity α4β2 receptors; it is not surprising that the latter has no effect on nicotine-induced up-regulation in animals[51]. Also unsurprisingly, the channel blockers hexamethonium, mecamylamine, or decamethonium by themselves do not produce appreciable up-regulation [9, 33].
Biochemical specificity sometimes exceeds that afforded by free energy differences alone. Proofreading processes in DNA replication and in protein synthesis reduce the error rate far below that expected from free energy differences. “Pharmacological chaperones” might accomplish an analogous task by allowing integral membrane proteins to escape retention and degradation by quality control systems in endoplasmic reticulum [52]. Typically these proofreading mechanisms use high-energy phosphate bonds to lock in a structure even after the thermodynamic driving force has changed to disfavor the structure. Perhaps another class of proteins exists to modulate the purely thermodynamic specificity of Cys-loop receptor assembly. If so, one expects to find that such a protein binds at subunit interfaces. Considering the structural similarity between snake venom toxins (which bind at the interface) and the lynx / SLURP family[53, 54], the lynx / SLURP proteins would be good candidates.
In conclusion, thermodynamic arguments, modified by kinetics and compartmentalization, can explain much of the phenomenology associated with nicotine-induced up-regulation of α4β2 receptors. However, several apparently contradictory results, particularly with other drugs, remain unexplained by thermodynamics alone and require more detailed mechanistic explanations.
Figure 2. Fura-2 recordings of cultured GABAergic ventral midbrain neurons after chronic exposure to nicotine.
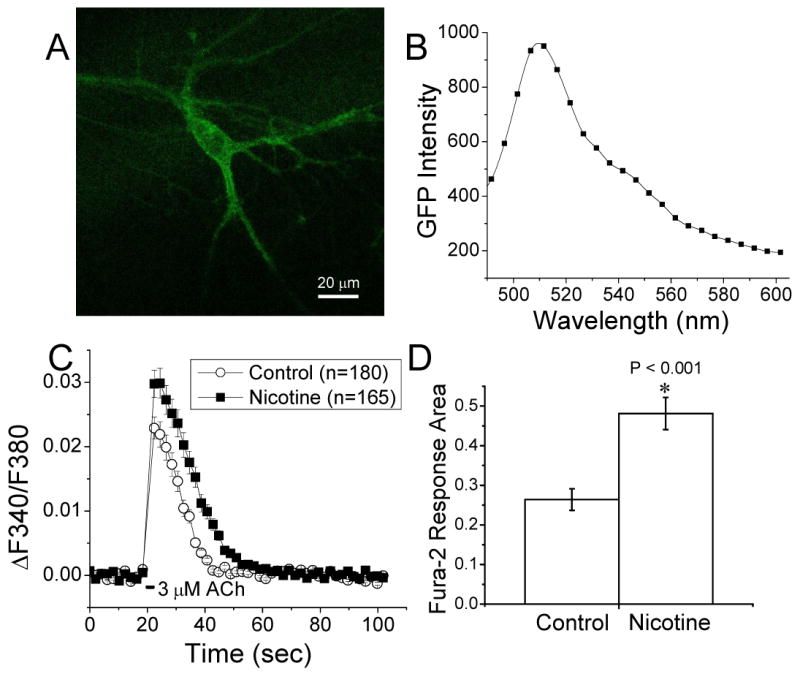
Fura-2 imaging was performed on ventral midbrain cultures from embryos heterozygous for the hypersensitive α4 (Leu 9'Ala) mutation and the GAT1-GFP mutation [26], which allowed us to visually identify GABAergic neurons.
A, A z-projection of spectrally unmixed confocal images of a cultured α4 (Leu 9'Ala) Het neuron expressing GAT1-GFP.
B, A spectrum of the neuron showing characteristic GFP emission with peak emission at ∼510 nm.
C, Time course of fura-2-recorded Ca2+ fluxes showing that GAT1-GFP+ neurons in chronic nicotine cultures (10 nM for 3 d) had greater ACh (3 μM for 2 sec) induced responses than control neurons.
D, Bar graph showing that the area of the ACh induced fura-2 responses in nicotine treated neurons was also significantly augmented over control. Fura-2 measurements were taken in the presence of ion channel and receptor blockers: atropine, AP-5, CNQX, bicuculline, TTX, and MLA.
Acknowledgments
We like to thank Sheri McKinney and Qi Huang for excellent cell cultures. This research was supported by the NIH (grants DA-17279, NS-11756), by the California Tobacco-Related Disease Research Project (grant 12RT-0245), by Philip Morris USA/International at Caltech, by the Elizabeth Ross Foundation and NARSAD (postdoctoral fellowships to R. N.), and by the Plum Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tapper AR, Nashmi R, Lester H. Neuronal Nicotinic Acetylcholine Receptors and Nicotine Dependence. In: Pollock JD, editor. The Cell Biology of Addiction. Cold Spring Harbor, NY 11724: Cold Spring Harbor Laboratory Press; 2005. [Google Scholar]
- 2.Marks MJ, Whiteaker P, Collins AC. Deletion of the α7, β2, or β4 nicotinic receptor subunit genes identifies highly expressed subtypes with relatively low affinity for [3H]epibatidine. Mol Pharmacol. 2006;70:947–59. doi: 10.1124/mol.106.025338. [DOI] [PubMed] [Google Scholar]
- 3.Whiting PJ, Liu R, Morley BJ, Lindstrom JM. Structurally different neuronal nicotinic acetylcholine receptor-subtypes purified and characterized using monoclonal-antibodies. J Neurosci. 1987;7:4005–16. doi: 10.1523/JNEUROSCI.07-12-04005.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Picciotto MR, Zoli M, Rimondini R, Lena C, Marubio LM, Pich EM, et al. Acetylcholine receptors containing the β2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–7. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- 5.Tapper A, McKinney S, Nashmi R, Schwarz J, Deshpande P, Labarca C, et al. Nicotine action on α4* receptors: sufficient for reward, tolerance and sensitization. Science. 2004;306:1029–32. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- 6.Maskos U, Molles BE, Pons S, Besson M, Guiard BP, Guilloux JP, et al. Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature. 2005;436:103–7. doi: 10.1038/nature03694. [DOI] [PubMed] [Google Scholar]
- 7.Marks MJ, Stitzel JA, Collins AC. Time course study of the effects of chronic nicotine infusion on drug response and brain receptors. J Pharmacol Exp Ther. 1985;235:619–28. [PubMed] [Google Scholar]
- 8.Schwartz RD, Kellar KJ. Nicotinic cholinergic receptor binding sites in the brain: regulation in vivo. Science. 1983;220:214–6. doi: 10.1126/science.6828889. [DOI] [PubMed] [Google Scholar]
- 9.Darsow T, Booker TK, Pina-Crespo JC, Heinemann SF. Exocytic trafficking is required for nicotine-induced up-regulation of α4β2 nicotinic acetylcholine receptors. J Biol Chem. 2005;280:18311–20. doi: 10.1074/jbc.M501157200. [DOI] [PubMed] [Google Scholar]
- 10.Kuryatov A, Luo J, Cooper J, Lindstrom J. Nicotine acts as a pharmacological chaperone to up-regulate human α4β2 acetylcholine receptors. Mol Pharmacol. 2005;68:1839–51. doi: 10.1124/mol.105.012419. [DOI] [PubMed] [Google Scholar]
- 11.Gopalakrishnan M, Molinari EJ, Sullivan JP. Regulation of human α4β2 neuronal nicotinic acetylcholine receptors by cholinergic channel ligands and second messenger pathways. Mol Pharmacol. 1997;52:524–34. [PubMed] [Google Scholar]
- 12.Ke L, Eisenhour CM, Bencherif M, Lukas RJ. Effects of chronic nicotine treatment on expression of diverse nicotinic acetylcholine receptor subtypes. I. Dose- and time-dependent effects of nicotine treatment. J Pharmacol Exp Ther. 1998;286:825–40. [PubMed] [Google Scholar]
- 13.Bencherif M, Fowler K, Lukas RJ, Lippiello PM. Mechanisms of up-regulation of neuronal nicotinic acetylcholine receptors in clonal cell lines and primary cultures of fetal rat brain. J Pharmacol Exp Ther. 1995;275:987–94. [PubMed] [Google Scholar]
- 14.Nashmi R, Dickinson ME, McKinney S, Jareb M, Labarca C, Fraser SE, et al. Assembly of α4β2 nicotinic acetylcholine receptors assessed with functional fluorescently labeled subunits: effects of localization, trafficking, and nicotine-induced upregulation in clonal mammalian cells and in cultured midbrain neurons. J Neurosci. 2003;23:11554–67. doi: 10.1523/JNEUROSCI.23-37-11554.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF, et al. Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J Neurosci. 1992;12:2765–84. doi: 10.1523/JNEUROSCI.12-07-02765.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang F, Nelson ME, Kuryatov A, Olale F, Cooper J, Keyser K, et al. Chronic nicotine treatment up-regulates human α3β2 but not α3β4 acetylcholine receptors stably transfected in human embryonic kidney cells. J Biol Chem. 1998;273:28721–32. doi: 10.1074/jbc.273.44.28721. [DOI] [PubMed] [Google Scholar]
- 17.Harkness PC, Millar NS. Changes in conformation and subcellular distribution of α4β2 nicotinic acetylcholine receptors revealed by chronic nicotine treatment and expression of subunit chimeras. J Neurosci. 2002;22:10172–81. doi: 10.1523/JNEUROSCI.22-23-10172.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peng X, Gerzanich V, Anand R, Whiting PJ, Lindstrom J. Nicotine-induced increase in neuronal nicotinic receptors results from a decrease in the rate of receptor turnover. Mol Pharmacol. 1994;46:523–30. [PubMed] [Google Scholar]
- 19.Buisson B, Bertrand D. Chronic exposure to nicotine upregulates the human α4β2 nicotinic acetylcholine receptor function. J Neurosci. 2001;21:1819–29. doi: 10.1523/JNEUROSCI.21-06-01819.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vallejo YF, Buisson B, Bertrand D, Green WN. Chronic nicotine exposure upregulates nicotinic receptors by a novel mechanism. J Neurosci. 2005;25:5563–72. doi: 10.1523/JNEUROSCI.5240-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sallette J, Pons S, Devillers-Thiery A, Soudant M, Prado de Carvalho L, Changeux JP, et al. Nicotine upregulates its own receptors through enhanced intracellular maturation. Neuron. 2005;46:595–607. doi: 10.1016/j.neuron.2005.03.029. [DOI] [PubMed] [Google Scholar]
- 22.Fonck C, Cohen BN, Nashmi R, Whiteaker P, Wagenaar D, Rodrigues-Pinguet N, et al. Novel seizure phenotype and sleep disruptions in knock-in mice with hypersensitive α4 nicotinic receptors. J Neurosci. 2005;25:11396–113411. doi: 10.1523/JNEUROSCI.3597-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lansford R, Bearman G, Fraser SE. Resolution of multiple green fluorescent protein color variants and dyes using two-photon microscopy and imaging spectroscopy. J Biomed Opt. 2001;6:311–8. doi: 10.1117/1.1383780. [DOI] [PubMed] [Google Scholar]
- 24.Dickinson ME, Bearman G, Tilie S, Lansford R, Fraser SE. Multi-spectral imaging and linear unmixing add a whole new dimension to laser scanning fluorescence microscopy. Biotechniques. 2001;31(1272):4–6. doi: 10.2144/01316bt01. 8. [DOI] [PubMed] [Google Scholar]
- 25.Nashmi R, Fraser SE, Lester H, Dickinson ME. FRET measurements using multispectral imaging. In: P A, D RN, editors. Molecular Imaging: FRET Microscopy and Spectroscopy. New York: Oxford University Press; 2005. pp. 180–92. [Google Scholar]
- 26.Chiu CS, Jensen K, Sokolova I, Wang D, Li M, Deshpande P, et al. Number, density, and surface/cytoplasmic distribution of GABA transporters at presynaptic structures of knock-in mice carrying GABA transporter subtype 1-green fluorescent protein fusions. J Neurosci. 2002;22:10251–66. doi: 10.1523/JNEUROSCI.22-23-10251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tumkosit P, Kuryatov A, Luo J, Lindstrom J. Beta3 subunits promote expression and nicotine-induced up-regulation of human nicotinic alpha6* nicotinic acetylcholine receptors expressed in transfected cell lines. Mol Pharmacol. 2006;70:1358–68. doi: 10.1124/mol.106.027326. [DOI] [PubMed] [Google Scholar]
- 28.Xu J, Zhu Y, Heinemann SF. Identification of sequence motifs that target neuronal nicotinic receptors to dendrites and axons. J Neurosci. 2006;26:9780–93. doi: 10.1523/JNEUROSCI.0840-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Z, Tearle AW, Nai Q, Berg DK. Rapid activity-driven SNARE-dependent trafficking of nicotinic receptors on somatic spines. J Neurosci. 2005;25:1159–68. doi: 10.1523/JNEUROSCI.3953-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keller SH, Lindstrom J, Ellisman M, Taylor P. Adjacent basic amino acid residues recognized by the COP I complex and ubiquitination govern endoplasmic reticulum to cell surface trafficking of the nicotinic acetylcholine receptor α-subunit. J Biol Chem. 2001;276:18384–91. doi: 10.1074/jbc.M100691200. [DOI] [PubMed] [Google Scholar]
- 31.Marchand S, Devillers-Thiery A, Pons S, Changeux JP, Cartaud J. Rapsyn escorts the nicotinic acetylcholine receptor along the exocytic pathway via association with lipid rafts. J Neurosci. 2002;22:8891–901. doi: 10.1523/JNEUROSCI.22-20-08891.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33:905–19. doi: 10.1016/s0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 33.Whiteaker P, Sharples CG, Wonnacott S. Agonist-induced up-regulation of α4β2 nicotinic acetylcholine receptors in M10 cells: pharmacological and spatial definition. Mol Pharmacol. 1998;53:950–62. [PubMed] [Google Scholar]
- 34.Chimienti F, Hogg RC, Plantard L, Lehmann C, Brakch N, Fischer J, et al. Identification of SLURP-1 as an epidermal neuromodulator explains the clinical phenotype of Mal de Meleda. Hum Mol Genet. 2003 doi: 10.1093/hmg/ddg320. [DOI] [PubMed] [Google Scholar]
- 35.Borea PA, Varani K, Gessi S, Gilli P, Gilli G. Binding thermodynamics at the human neuronal nicotine receptor. Biochem Pharmacol. 1998;55:1189–97. doi: 10.1016/s0006-2952(97)00578-9. [DOI] [PubMed] [Google Scholar]
- 36.Borea PA, Dalpiaz A, Gessi S, Gilli G. Thermodynamics of 5-HT3 receptor binding discriminates agonistic from antagonistic behaviour. Eur J Pharmacol. 1996;298:329–34. doi: 10.1016/0014-2999(95)00813-6. [DOI] [PubMed] [Google Scholar]
- 37.Tairi AP, Hovius R, Pick H, Blasey H, Bernard A, Surprenant A, et al. Ligand binding to the serotonin 5HT3 receptor studied with a novel fluorescent ligand. Biochemistry. 1998;37:15850–64. doi: 10.1021/bi981812z. [DOI] [PubMed] [Google Scholar]
- 38.Maksay G. Thermodynamics of gamma-aminobutyric acid type A receptor binding differentiate agonists from antagonists. Mol Pharmacol. 1994;46:386–90. [PubMed] [Google Scholar]
- 39.Chang Y, Weiss DS. Channel opening locks agonist onto the GABAC receptor. Nat Neurosci. 1999;2:219–25. doi: 10.1038/6313. [DOI] [PubMed] [Google Scholar]
- 40.Sallette J, Bohler S, Benoit P, Soudant M, Pons S, Le Novere N, et al. An extracellular protein microdomain controls up-regulation of neuronal nicotinic acetylcholine receptors by nicotine. J Biol Chem. 2004;279:18767–75. doi: 10.1074/jbc.M308260200. [DOI] [PubMed] [Google Scholar]
- 41.Buisson B, Gopalakrishnan M, Arneric SP, Sullivan JP, Bertrand D. Human α4β2 neuronal nicotinic acetylcholine receptor in HEK 293 cells: A patch-clamp study. J Neurosci. 1996;16:7880–91. doi: 10.1523/JNEUROSCI.16-24-07880.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marks MJ, Whiteaker P, Calcaterra J, Stitzel JA, Bullock AE, Grady SR, et al. Two pharmacologically distinct components of nicotinic receptor-mediated rubidium efflux in mouse brain require the β2 subunit. J Pharmacol Exp Ther. 1999;289:1090–103. [PubMed] [Google Scholar]
- 43.Zwart R, Vijverberg HP. Four pharmacologically distinct subtypes of α4β2 nicotinic acetylcholine receptor expressed in Xenopus laevis oocytes. Mol Pharmacol. 1998;54:1124–31. [PubMed] [Google Scholar]
- 44.Zhou Y, Nelson ME, Kuryatov A, Choi C, Cooper J, Lindstrom J. Human α4β2 acetylcholine receptors formed from linked subunits. J Neurosci. 2003;23:9004–15. doi: 10.1523/JNEUROSCI.23-27-09004.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nelson ME, Kuryatov A, Choi CH, Zhou Y, Lindstrom J. Alternate stoichiometries of α4β2 nicotinic acetylcholine receptors. Mol Pharmacol. 2003;63:332–41. doi: 10.1124/mol.63.2.332. [DOI] [PubMed] [Google Scholar]
- 46.Jia L, Flotildes K, Li M, Cohen BN. Nicotine trapping causes the persistent desensitization of α4β2 nicotinic receptors expressed in oocytes. J Neurochem. 2003;84:753–66. doi: 10.1046/j.1471-4159.2003.01578.x. [DOI] [PubMed] [Google Scholar]
- 47.Peng X, Gerzanich V, Anand R, Wang F, Lindstrom J. Chronic nicotine treatment up-regulates α3 and α7 acetylcholine receptor subtypes expressed by the human neuroblastoma cell line SH-SY5Y. Mol Pharmacol. 1997;51:776–84. doi: 10.1124/mol.51.5.776. [DOI] [PubMed] [Google Scholar]
- 48.Lester HA. The permeation pathway of neurotransmitter-gated ion channels. Ann Rev Biophys Biomol Struc. 1992;21:267–92. doi: 10.1146/annurev.bb.21.060192.001411. [DOI] [PubMed] [Google Scholar]
- 49.Bourinet E, Fournier F, Lory P, Charnet P, Nargeot J. Protein-kinase-c regulation of cardiac calcium channels expressed in Xenopus oocytes. Pflugers Arch Eur J Physiol. 1992;421:247–55. doi: 10.1007/BF00374834. [DOI] [PubMed] [Google Scholar]
- 50.Briggs CA, Gubbins EJ, Marks MJ, Putman CB, Thimmapaya R, Meyer MD, et al. Untranslated region-dependent exclusive expression of high-sensitivity subforms of α4β2 and α3β2 nicotinic acetylcholine receptors. Mol Pharmacol. 2006;70:227–40. doi: 10.1124/mol.105.020198. [DOI] [PubMed] [Google Scholar]
- 51.el-Bizri H, Clarke PB. Regulation of nicotinic receptors in rat brain following quasi-irreversible nicotinic blockade by chlorisondamine and chronic treatment with nicotine. Br J Pharmacol. 1994;113:917–25. doi: 10.1111/j.1476-5381.1994.tb17080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Conn PM, Janovick JA, Brothers SP, Knollman PE. ‘Effective inefficiency’: cellular control of protein trafficking as a mechanism of post-translational regulation. J Endocrinol. 2006;190:13–6. doi: 10.1677/joe.1.06771. [DOI] [PubMed] [Google Scholar]
- 53.Miwa JM, Ibanez-Tallon I, Crabtree GW, Sanchez R, Sali A, Role LW, et al. lynx1, an endogenous toxin-like modulator of nicotinic acetylcholine receptors in the mammalian CNS. Neuron. 1999;23:105–14. doi: 10.1016/s0896-6273(00)80757-6. [DOI] [PubMed] [Google Scholar]
- 54.Miwa JM, Stevens TR, King SL, Caldarone BJ, Ibanez-Tallon I, Xiao C, et al. The Prototoxin lynx1 Acts on Nicotinic Acetylcholine Receptors to Balance Neuronal Activity and Survival In Vivo. Neuron. 2006;51:587–600. doi: 10.1016/j.neuron.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 55.Celie PH, van Rossum-Fikkert SE, van Dijk WJ, Brejc K, Smit AB, Sixma TK. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron. 2004;41:907–14. doi: 10.1016/s0896-6273(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 56.Auerbach A, Akk G. Desensitization of mouse nicotinic acetylcholine receptor channels. A two-gate mechanism. J Gen Physiol. 1998;112:181–97. doi: 10.1085/jgp.112.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]