

Abstract
Strains of the yeast Saccharomyces cerevisiae differ in their sensitivities to tobacco osmotin, an antifungal protein of the PR-5 family. However, cells sensitive to tobacco osmotin showed resistance to osmotin-like proteins purified from the plant Atriplex nummularia, indicating a strict specificity between the antifungal protein and its target cell. A member of a gene family encoding stress proteins induced by heat and nitrogen limitation, collectively called Pir proteins, was isolated among the genes that conveyed resistance to tobacco osmotin to a susceptible strain. We show that overexpression of Pir proteins increased resistance to osmotin, whereas simultaneous deletion of all PIR genes in a tolerant strain resulted in sensitivity. Pir proteins have been immunolocalized to the cell wall. The enzymatic digestion of the cell wall of sensitive and resistant cells rendered spheroplasts equally susceptible to the cytotoxic action of tobacco osmotin but not to other osmotin-like proteins, indicating that the cell membrane interacts specifically with osmotin and facilitates its action. Our results demonstrate that fungal cell wall proteins are determinants of resistance to antifungal PR-5 proteins.
Plants defend themselves from pathogenic fungi by a variety of means, including the production of several proteins with antifungal properties. Although some of these have been shown to have specific enzymatic activity, i.e., chitinases (PR-3) and β-(1,3)-glucanases (PR-2), many have no known catalytic function and their mechanisms of action are not understood. For example, the bases for activity against fungi of thionins, defensins, PR-1, PR-4, and PR-5 proteins are not known (1). Osmotin is a member of the PR-5 family that was originally identified as the predominant protein that accumulated in tobacco cells as a function of osmotic adaptation (2). Subsequently, osmotin and other osmotin-like proteins were shown to have antifungal activity in vitro against a broad range of fungi, including several plant pathogens (1). Leaves of transgenic potato plants expressing tobacco osmotin exhibited partial resistance to Phytophtora infestans (3), demonstrating that PR-5 proteins can be used as a source of resistance to fungal infection. The fungal growth inhibition by osmotin and zeamatin, a maize PR-5 protein, correlated with plasma membrane permeabilization and dissipation of the membrane potential (4, 5), suggesting a physical interaction between PR-5 proteins and the plasma membrane of sensitive fungi, but the precise mechanism of cytotoxicity remains unknown.
Many of the PR proteins, including osmotin, exhibit clear specificity of their toxicity against fungi, indicating that there must be determinants of sensitivity and resistance in fungal cells (1, 5). Even the most studied plant antifungal proteins, chitinases and β-(1,3)-glucanases, which act as cell wall degrading enzymes, are not uniformly active against all fungi that contain substrates for these enzymes as important cell wall components. This differential activity is not understood and specific genetic factors that condition sensitivity or resistance to antifungal enzymes have not been identified. Knowledge of the bases for this selectivity would be very helpful in determining strategies to overcome the resistance of important pathogens. The resistance of fungi to these toxic proteins could be the result of the nature of interacting targets present on the cell wall or plasma membrane of fungi as was shown for killer toxins of yeast (6, 7). If these targets could be identified, structural modifications to the antifungal proteins might be engineered to improve their specific toxicity against insensitive fungi.
To study the bases for the specificity and the mechanism of toxicity of PR-5 antifungal proteins we began a search for a biological system that would allow the genetic identification of determinants governing resistance and sensitivity to PR-5 proteins. We report here the existence of genetic variants of Saccharomyces cerevisiae with increased sensitivity to tobacco PR-5. We have isolated a gene family encoding integral cell wall proteins whose overexpression resulted in resistance to osmotin, a tobacco PR-5 protein. Null mutations in the structural genes of these cell wall proteins increased the sensitivity to osmotin of tolerant cells.
MATERIALS AND METHODS
Strains and Media.
Strains GRF167 and BWG7a have been described elsewhere (8, 9). The set of strains containing combined deletions of the PIR genes was kindly provided by A. Toh-e (University of Tokyo) and has been described in detail (10). Their relevant genotypes are DT8–1D (PIR1 PIR2 PIR3), DT8–1B (pir1::LEU2), DT8–1A (pir2::HIS3), DT8–1C (pir1::LEU2 pir2::HIS3), YAT1540 (pir2::HIS3 pir3::URA3), and YAT1588 (pir1::LEU2 pir2::HIS3 pir3::URA3). The ORE1/PIR2 locus was deleted in BWG7a and GRF167 by the one-step gene disruption method (11) after replacement of the internal 0.7-kb PstI fragment of ORE1 by the URA3 gene. Precise gene disruption was confirmed by Southern blotting. Standard procedures for yeast culture, sporulation, tetrad dissection, and growth media preparation were followed (11, 12). Lyticase (Sigma, L8137) was used for cell wall and ascus digestion. Osmotin was purified from salt-adapted tobacco cell suspension cultures (Nicotiana tabacum L. var. Wisconsin 38) to apparent homogeneity as described (2). PR-5 proteins A8 and A9 were purified similarly from cell suspension cultures of the plant Atriplex nummularia. The potency of different batches of purified osmotin was determined from the minimum amount of protein that prevented the growth of BWG7a cells. Typical lethal doses of purified osmotin ranged between 10 and 20 μg/ml.
Measurement of Sensitivity to Osmotin.
Tolerance to tobacco osmotin was determined in 0.5-ml liquid cultures containing various concentrations of purified osmotin. Cultures were started at an OD600nm of 0.01–0.05 from diluted overnight cultures, incubated at 30°C with shaking for 16–20 hr, and the OD600nm reached was determined with appropriate dilutions. The amount of osmotin that reduced growth by 50% is denoted as IC50. Osmotin-induced cell death was measured by incubating cells or spheroplasts at a density of ≈6 × 107 cells/ml with various concentrations of purified osmotin for 1 hr at 30°C. After dilution in water and plating, the number of viable counts was determined. When spheroplasts were used, 0.8 M sorbitol was included in all solutions. The amount of osmotin that reduced viable counts by 50% under these experimental conditions is referred to as LD50.
Gene Library and Cloning.
Total DNA from strain GRF167 was partially digested with Sau3A1, size fractionated by agarose gel electrophoresis, and inserted into YEp24 (13). More than 14,000 clones in Escherichia coli (XL1-Blue, Stratagene) were obtained with an average insert size of 5.6 kb. Plasmid DNA of pooled E. coli clones was used to transform BWG7a cells (14). Osmotin-resistant clones were obtained by plating on yeast extract/peptone/dextrose (YPD) containing 50 μg/ml of purified osmotin. For subsequent use, the PIR2 gene was subcloned as a 1.5-kb BamHI–SalI fragment in YEp24. The coding regions of PIR homologues from strains GRF167 and BWG7a were amplified by PCR using gene-specific synthetic oligonucleotides annealing to the start and stop codon regions. The identity of individual PIR clones was determined by restriction mapping and partial nucleotide sequencing. Amplified DNA fragments were cloned in plasmid p414GPD (15) for constitutive expression.
Labeling of Secreted Proteins and Northern Blot Analysis.
Secretory proteins were labeled as described by Russo et al. (16), precipitated by trichloroacetic acid, resolved on a SDS/7.5–15% PAGE gradient gel and visualized by x-ray autoradiography. For Northern blotting, total RNA was extracted (17), electrophoresed on agarose-formaldehyde gel, transferred to nitrocellulose membrane, and probed with 32P-labeled DNA fragments corresponding to the coding regions of the PIR genes under standard high-stringency conditions (13).
Immunoelectron Microscopy.
For antibody production, the open reading frame of PIR3 from strain BWG7a was translationally fused to TrxA using the plasmid pET-32a (Novagen). The construct was transformed into E. coli strain AD494(DE3) and synthesis of the recombinant protein was induced with 1 mM isopropyl β-d-thiogalactoside for 20 hr at 18°C. The Pir3::TrxA fusion protein was purified using S-tag agarose beads (Novagen) and SDS/PAGE. Approximately 200 μg of protein was injected into a hen and two booster injections of 100 μg each were given at 2-week intervals. Polyclonal antibodies were purified by polyethylene glycol fractionation (18, 19). Pir3 polyclonal antibody recognizes Pir1 and Pir2 proteins since they are 75 and 79.5% identical to Pir3, respectively. For immunocytochemistry, yeast cells were fixed and embedded as described by Mulholland et al. (20). Thin sections were blocked with 1% gelatin in TTBS (tris buffered saline/0.05% Tween 20, pH 7.6), and incubated with anti-Pir3 polyclonal antibodies diluted 1:5,000 in TTBS/1% rabbit normal serum/1% BSA. Preimmune antibodies were substituted for anti-Pir3 as a control. Rabbit anti-chicken IgG conjugated to 10 nm gold particles diluted 1:50 in TTBS/1% BSA was used as secondary antibody. All observations were made on a EM200 transmission electron microscope (Philips Electronic Instruments, Mahwah, NJ).
RESULTS
Differential Sensitivity to Osmotin Among Yeast Strains.
With the aim to use the yeast S. cerevisiae as a model to identify determinants of resistance/sensitivity to antifungal proteins, several yeast strains were surveyed for their sensitivity to tobacco osmotin, an antifungal protein of the PR-5 family. Most laboratory strains that were tested had varying degrees of resistance to osmotin, but strain BWG7a displayed a uniquely high sensitivity to tobacco osmotin. Addition of as little as 10 μg/ml (≈0.4 μM) of osmotin to the medium prevented the growth of BWG7a cells (IC50 ≈3 μg/ml), whereas a saturating concentration of osmotin (240 μg/ml) only partially inhibited the growth of the highly tolerant strain GRF167 (IC50 ≈200 μg/ml) (data not shown). Treatment of BWG7a cells with tobacco osmotin for various lengths of time, followed by dilution and plating, showed that the cytotoxic effect of osmotin in sensitive yeast cells was irreversible, as demonstrated by the steep decrease in viable counts after 1 hr in the presence of osmotin (Fig. 1). The sensitivity of BWG7a cells is specific to osmotin purified from tobacco cells because the homologous osmotin-like proteins A9 (Fig. 1) and A8 (not shown) purified from A. nummularia cells had little or no effect on BWG7a cells up to a concentration of 100 μg/ml, the maximum concentration tested. However, the same batches of proteins were active against other fungal species tested, such as Verticillium dahliae and Trichoderma longibrachiatum (data not shown). It has been proposed that the antifungal activity of osmotin and zeamatin, a PR-5 protein from maize, is the result of fungal membrane permeabilization due to interactions between the plasma membrane of sensitive fungi and putative pore-forming domains in these proteins (4, 5, 21). Osmotins isolated from tobacco and A. nummularia also differed greatly in their toxicity toward spheroplast (Fig. 1), indicating that osmotin-like proteins interact specifically with sensitive yeast cells and do not insert freely in lipid bilayers.
Figure 1.

Differential sensitivity of yeast strains to the cytotoxic effect of PR-5 proteins. (Upper) Cells of strains GRF167 (○, □) and BWG7a (▪, •) were incubated in YPD medium with (□, ▪) and without (○, •) 50 μg/ml of purified tobacco osmotin for the time lengths indicated. Subsequently, cells were diluted and plated, and the number of viable counts was determined after incubation at 30°C for 2 days. (Lower) Approximately 107 cells per ml of walled cells (□, ▪) and spheroplasts (○, •) of strain BWG7a were incubated at 30°C for 1 hr in YPD or 0.8 M sorbitol, respectively, containing the indicated concentrations of tobacco osmotin (▪, •) and the osmotin-like protein A9 (○, □) purified from cultured cell suspensions of A. nummularia. Viable counts were determined as indicated above and are shown normalized to the value without added proteins.
Isolation of Genes Conferring Resistance to Osmotin.
To characterize the genetic basis for sensitivity/tolerance to tobacco osmotin, strains BWG7a and GRF167 were crossed. Neither parental phenotype was dominant because the resulting diploid displayed an intermediate degree of sensitivity to osmotin (IC50 ≈45 μg/ml). Haploid segregants from six tetrads demonstrated varying degrees of resistance to osmotin, suggesting that more than one gene was contributing to osmotin resistance. To identify these genetic determinants of resistance to osmotin, a genomic library of the highly resistant GRF167 strain was made in the multicopy shuttle vector YEp24 and used to transform BWG7a cells. Resistant clones were isolated by their ability to grow on YPD medium containing 50 μg/ml of osmotin. Plasmids harbored by 35 osmotin-resistant clones were isolated and analyzed by restriction mapping. Based on their restriction patterns, the plasmid inserts corresponded to three different loci that were named ORE1, ORE2, and ORE3 (for osmotin resistance). Although transformation with either ORE gene conferred significant osmotin tolerance to BWG7a cells (Fig. 2), the resistance of these transformants was still lower than that of GRF167 (data not shown). This partial complementation of sensitivity by each individual ORE gene is consistent with the genetic segregation data, which suggested that more than one gene was responsible for the high osmotin resistance of GRF167. Because 28 of the 35 resistant clones corresponded to ORE1, this gene was selected for further study. The proteins encoded by ORE2 and ORE3 are not structurally related to ORE1 and will be described elsewhere.
Figure 2.
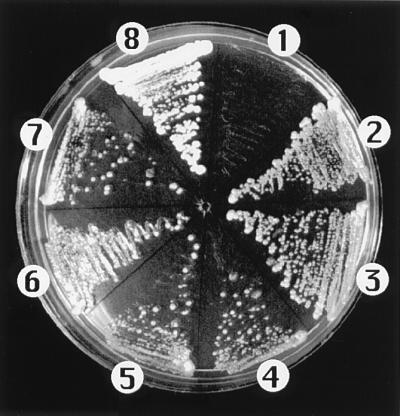
Resistance to tobacco osmotin conferred by genes ORE1, ORE2, and ORE3. Cells of BWG7a transformed with the empty vector YEp24 (1), and transformants of BWG7a (2–7) expressing ORE1 (2 and 3), ORE2 (4 and 5), and ORE3 (6 and 7), and GRF167 (8) were streaked on YPD medium containing 50 μg/ml of tobacco osmotin. Only control BWG7a cells failed to grow.
Boundaries of the ORE1 gene within the genomic inserts were determined by restriction fragment subcloning and retransformation for osmotin resistance. Sequence determination of ORE1 and comparison to the GenBank/EMBL nucleic acid databases demonstrated its identity to the PIR2/HSP150 gene. Therefore, ORE1 will be referred to as PIR2 hereafter. The highly similar Pir2 and Hsp150 proteins contain a 19-amino acid long stretch repeated 10 times and are predicted to be structural proteins (10, 16). Hsp150 expression is induced by heat stress and nitrogen starvation and most of the newly synthesized protein is secreted to the medium (16, 22). Conceivably, the osmotin sensitivity of BWG7a might arise from defective synthesis or secretion of the endogenous Hsp150/Pir2 protein, a defect suppressed by the ectopic expression of PIR2 from GRF167. This, however, does not seem to be the case, since BWG7a cells secrete a protein of about 150 kDa (the expected size for glycosylated Pir2/Hsp150), whose abundance is greatly induced by heat shock, and that is absent in an isogenic strain where PIR2 has been disrupted (Fig. 3). At least part of the heat-shock induction of Pir2 seems transcriptional, as shown by the higher level of PIR2 mRNA found both in sensitive and resistant cells (Fig. 3 and ref. 22). PIR2 mRNA accumulation is also induced by osmotin in the sensitive strain but not in the tolerant one (Fig. 3). This differential accumulation could be due to cellular stress related to the cytotoxic effect of osmotin on BWG7a cells rather than being a protective response against osmotin, since heat-shock induction of Pir2 prior to osmotin treatment did not prevent osmotin-induced cell death (results not shown).
Figure 3.

The synthesis of the Pir2/Hsp150 protein is induced by heat shock and osmotin in sensitive yeast cells. (Upper) Newly synthesized proteins in BWG7a cells (lanes 1 and 2) and an isogenic Δpir2 derivative (lane 3) were labeled with [35S]methionine at either 23°C (lane 1) or 37°C (lanes 2 and 3) for 1 hr. Proteins from the growth media were resolved by SDS/PAGE and visualized by autoradiography. Arrow indicates the band corresponding to Pir2/Hsp150. Molecular sizes in kDa are at right. (Lower) Five micrograms of total RNA from strains BWG7a and GRF167 incubated for 30 min at 23°C or 37°C without osmotin, or at 23°C in the presence of 10 μg/ml or 50 μg/ml of tobacco osmotin, were subjected to Northern blot analysis using PIR2 as probe.
Functional Equivalence of Pir Isoforms.
Disruption of PIR2 in GRF167 and BWG7a did not change substantially their resistance/sensitivity to osmotin (data not shown). However, at least two other PIR homologues (PIR1 and PIR3) have been identified in S. cerevisiae-encoding proteins that share 84.4% and 79.5% identity with the amino acid sequence of Pir2 (10). Only combined deletions of the PIR genes rendered heat-shock sensitive cells (10). Consistent with the multigenic nature of the Pir proteins, we found that resistant cells became sensitive to osmotin only after deletion of all three PIR genes, whereas single and double mutations did not increase sensitivity significantly (Fig. 4). These results suggest that Pir isoforms are functionally redundant and confer tolerance to osmotin in naturally resistant yeast strains. However, only the PIR2 gene was isolated during our genetic screen for determinants of osmotin resistance. To investigate differences in gene activity between PIR genes, the amount of gene expression of the chromosomal copies of the PIR genes was estimated from the steady levels of the corresponding mRNAs. As illustrated in Fig. 5, PIR2 mRNA is clearly the most abundant transcript among PIR messages. PIR1 mRNA is noticeably less abundant than that of PIR2, and PIR3 mRNA is barely detectable under similar hybridization conditions and identical exposure times. As shown before (Fig. 3), the expression of PIR2 is further enhanced by heat treatment, whereas that of PIR1 and PIR3 is not responsive to heat, as determined from longer exposure times.
Figure 4.

Pir proteins are functionally redundant. The osmotin tolerance of congenic strains containing various combinations of PIR deletions (see Materials and Methods) was compared with that of the parental wild-type strain DT8–1D and BWG7a in liquid cultures. Values are the average of two independent experiments and are normalized to the OD600nm of control cultures without osmotin.
Figure 5.

Relative abundance of mRNAs synthesized from genes PIR1, PIR2, and PIR3. Five micrograms of total RNA purified from BWG7a (lanes 1 and 2) and GRF167 (lanes 3 and 4) cells growing at 30°C (lanes 1 and 3) or transferred to 37°C for 60 min (lanes 2 and 4) were hybridized to DNA probes corresponding to the open reading frames of the PIR1, PIR2, and PIR3 genes. Lane 5 shows RNA extracted from YAT1588 cells transformed with plasmid p414GPD carrying the indicated PIR gene.
To further investigate functional differences between the Pir isoforms regarding resistance to osmotin, the open reading frames of PIR1, PIR2, and PIR3 from BWG7a and GRF167 were amplified by PCR and cloned into the vector p414GPD to attain similar levels of gene expression of all six Pir isoforms. Regardless of their origin, the production of any Pir protein conferred similar resistance to osmotin when expressed at equivalent levels in the sensitive strains BWG7a and YAT1588, the latter bearing a triple deletion of the PIR genes (data not shown). Therefore, the isolation of PIR2 but not PIR1 or PIR3 during the screening for tolerance was probably due to the higher level of gene expression driven by the native PIR2 promoter and not to functional differences between Pir isoforms. Moreover, these results demonstrate that Pir isoforms isolated from tolerant and sensitive strains are equally able to increase resistance to osmotin.
Pir Proteins Are Cell Wall Determinants for Osmotin Resistance.
Although Hsp150/Pir2 is secreted in S. cerevisiae, some Hsp150/Pir2 is still cell bound and its homologue in Schizosaccharomyces pombe remains associated with the cell (16). Moreover, the peptide sequence of the Pir proteins is composed of tandem repeats, a feature consistent with a structural role. Therefore, we investigated the cellular localization of the Pir proteins using polyclonal antibodies made against nonglycosylated recombinant Pir3 protein. The results of immunolocalization experiments, depicted in Fig. 6, indicate that Pir proteins are targeted to the cell wall. The antibodies detected Pir proteins in the cell wall of wild-type cells BWG7a and DT8–1D (both PIR1 PIR2 PIR3) but not in the null mutant YAT1588 (Δpir1 Δpir2 Δpir3). The densities of gold particles per μm2 were 132 ± 20 in BWG7a, 170 ± 20 in DT8–1D, and only 18 ± 7 in YAT1588 (n = 10 cells) after subtracting the background counts obtained with preimmune antibodies. To prove the cross-reactivity of Pir1 and Pir2 proteins with anti-Pir3 antibodies and to localize each Pir isoform individually, YAT1588 cells expressing PIR1, PIR2, or PIR3 genes from BWG7a in plasmid p414GPD were also subjected to immunocytochemical analysis. All three Pir isoforms were detected at the cell wall (results not shown). The localization of Pir proteins strongly suggests that these proteins are integral components of the cell wall of S. cerevisiae.
Figure 6.

Immunoelectron localization of Pir proteins to the cell wall. Thin sections of cells of strains BWG7a (A and B), DT8–1D (C and D), and YAT1588 (E and F) were incubated with anti-Pir antibodies (A, C, and E) or preimmune antibodies (B, D, and F). Secondary antibodies were conjugated to 10 nm gold particles, which appear as small black dots at the site of positive reaction. The average density of gold particles per μm2 in the samples depicted in A through F were, respectively, 227 ± 20, 95 ± 3, 288 ± 29, 118 ± 11, 112 ± 7, and 94 ± 12 (n = 10 cells). (Bar = 0.2 μm.)
Consistent with the cell wall localization of the Pir proteins, the resistance to osmotin mediated by PIR2 required an intact cell surface. The cytotoxic effect of osmotin was assayed using cells and spheroplasts of BWG7a expressing different amounts of Pir2. The result demonstrated that PIR2 conveyed osmotin resistance only when an intact cell wall was in place because spheroplasting of cells overexpressing Pir2 restored sensitivity to osmotin (Fig. 7). Similarly, whereas DT8–1D cells (PIR1 PIR2 PIR3) were more tolerant than congenic YAT1588 cells (Δpir1 Δpir2 Δpir3) (see Fig. 4), the spheroplasts of both strains were equally sensitive to osmotin. The LD50 for osmotin was 20.8 ± 2.8 μg/ml for spheroplasts of strain DT8–1D and 16.5 ± 1.5 μg/ml for those of YAT1588 (n = 2). These values are very similar to the LD50 determined for the spheroplasts from BWG7a (12.5 ± 3.6 μg/ml, see Fig. 6). Spheroplasts of other strains naturally resistant to osmotin were also sensitive to tobacco osmotin (results not shown).
Figure 7.

PIR2 is a cell wall determinant of resistance to osmotin. The tolerance to tobacco osmotin of cells (○, □) and spheroplasts (•, ▪) of strain BWG7a transformed (□, ▪) or not (○, •) with PIR2 in YEp24 was determined by incubation in either YPD medium (for cells) or 0.8 M sorbitol (for spheroplasts) containing increasing concentrations of osmotin as indicated. After 1 hr at 30°C, cells and spheroplasts were diluted and plated. Illustrated is the percentage of viable counts relative to the controls without osmotin. Values are the average of at least two measurements.
DISCUSSION
We have shown that tobacco osmotin, an antifungal protein of the PR-5 family, is active against certain strains of the yeast S. cerevisiae. Sensitive yeast cells are rapidly killed by osmotin. Within the first hour following osmotin addition, the viability of a sensitive culture dropped to about 1% of the starting cell count (Fig. 1). Our results show that differential resistance among yeast strains may be explained, at least in part, by changes in the architecture of the cell wall. First, the Pir proteins have been localized to the cell wall (Fig. 6), and when the expression of all Pir isoforms was abolished by appropriate gene deletions, the sensitivity to osmotin increased (Fig. 4). Second, overexpression of Pir2 increased resistance to osmotin but only when the cell wall was present, since spheroplasting of transformants expressing Pir2 restored sensitivity to osmotin (Fig. 7). The high sensitivity of BWG7a to tobacco osmotin cannot be explained by a malfunction of its complement of Pir proteins because (i) all Pir isoforms from BWG7a are competent to increase osmotin tolerance when overproduced, (ii) the expression of the PIR genes in BWG7a is indistinguishable from that found in the highly tolerant GRF167 strain (Fig. 5), (iii) Pir2 is exported efficiently to the extracellular medium upon heat stress (Fig. 3), and (iv) similar immunolabeling was found in the cell wall of BWG7a and the tolerant strain DT8–1D using Pir-specific antibodies (Fig. 6). Likely, the cell wall-localized Pir proteins can affect the ability of osmotin to reach and permeabilize the plasma membrane, the apparent target for osmotin action (1, 4, 21). Two models are possible that account for our results. First, BWG7a cells may harbor a defect in the cell wall architecture, yet to be identified but distinct from the Pir proteins themselves, which makes the cell wall more permeable to osmotin. Alternatively, osmotin-resistant cells may better retain the Pir proteins in the cell wall instead of releasing them to the medium. The overexpression of Pir proteins may compensate for such possible defects and reduce the accessibility of osmotin to the plasma membrane. In a second model, a structural component of the wall in sensitive cells may be recognized by osmotin, thereby facilitating its translocation across the cell wall toward the plasma membrane. The overexpression of the Pir proteins could mask or alter the structure of the hypothetical cell wall receptor. It is noteworthy that BWG7a cells are very sensitive to tobacco osmotin but resistant to the osmotin-like proteins A8 and A9 purified from A. nummularia, indicating that BWG7a is not generally sensitive to PR-5 proteins and that there is a specific interaction between BWG7a cells and tobacco osmotin. Cell wall receptors have been identified for other antifungal proteins that are active against S. cerevisiae. For example, the killer toxin K1, a pore-forming protein, uses the cell wall β-(1–6)-d-glucan cross-linked with glycoproteins as a cell-surface receptor (7), and the KT28 toxin binds to mannoprotein receptors on the yeast cell surface (6). Assessing the nature of the hypothetical receptor on the cell wall of BWG7a will be facilitated by the analysis of BWG7a mutants with altered cell wall and increased resistance to tobacco osmotin (unpublished results) in the same way that kre mutants have lead to identification of the cell surface receptor of the K1 killer toxin (7).
It has been proposed that the antifungal activity of osmotin and zeamatin, a PR-5 protein from maize, is the result of fungal membrane permeabilization due to interactions between the plasma membrane of sensitive fungi and putative pore-forming domains in these proteins (4, 5, 21). Our results indicate that the plasma membrane of resistant fungal cells may also be targeted by tobacco osmotin because spheroplasts derived from resistant yeast cells are as susceptible to the cytotoxic effect of osmotin as those from sensitive cells. However, yeast spheroplasts are resistant to other osmotin-like proteins (Fig. 1), demonstrating a strict specificity between the antifungal protein and its target cell. These results rule out the trivial possibility that osmotin inserts nonspecifically in the membrane lipid bilayer and strongly suggests the existence of a plasma membrane-based receptor for osmotin or a facilitator of its activity. This scenario bears similarities with the proposed model of K1 binding to yeast cells. The experimental evidence indicates a two-step mechanism corresponding to two classes of binding sites for K1, a low-affinity and high-velocity binding to the cell wall receptor and a high-affinity and low-velocity binding component reflecting the energy-dependent binding to a secondary plasma membrane receptor and subsequent formation of an ion channel (23, 24).
Based on the evidence provided here, we suggest that the cell wall of osmotin-resistant yeast strains may act as a physical barrier for osmotin, presumably masking binding sites in the plasma membrane that mediate osmotin action, whereas the cell wall of sensitive strains might be a facilitator for osmotin binding and translocation to the plasma membrane. This model of strain-specific recognition by osmotin is consistent with the observation that osmotin and osmotin-like proteins of the PR-5 family of antifungal proteins vary in their effectiveness against specific fungal pathogens (1, 5). We also propose that the yeast S. cerevisiae can be used as a powerful genetic tool to identify molecular determinants of resistance and sensitivity to plant antifungal proteins and, eventually, should facilitate the understanding of the mechanism of action of the PR-5 class of antifungal proteins.
Acknowledgments
We thank Dr. A. Toh-e for strains with pir deletions, J. Clithero for DNA sequencing, Dr. J. Mulholland for assistance with immunoelectron microscopy, D. Sherman for gold-conjugated secondary antibodies, and the Electron Microscopy Center (Purdue University) for the use of the transmission electron microscope. This work was supported by grants from Pioneer Hi-Bred International, The Consortium for Plant Biotechnology Inc., and U.S. Department of Agriculture Grant 94-37100-0754.
ABBREVIATION
- YPD
yeast extract/peptone/dextrose
References
- 1.Yun D J, Bressan R A, Hasegawa P M. In: Plant Breeding Reviews. Janick J, editor. Vol. 14. New York: Wiley; 1997. pp. 39–88. [Google Scholar]
- 2.Singh K A, Bracker C A, Hasegawa P M, Handa A K, Buckel S, Hermodson M A, Pfankoch E, Regnier F E, Bressan R A. Plant Physiol. 1987;85:529–536. doi: 10.1104/pp.85.2.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu D, Raghothama P M, Hasegawa P M, Bressan R A. Proc Natl Acad Sci USA. 1994;91:1888–1892. doi: 10.1073/pnas.91.5.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts W K, Selitrennikoff C P. J Gen Microbiol. 1990;136:1771–1778. [Google Scholar]
- 5.Abad L R, Paino D’Urzo M, Liu D, Narasimhan M L, Reuveni M, Zhu J K, Niu X, Singh N K, Hasegawa P M, Bressan R A. Plant Sci. 1996;118:11–23. [Google Scholar]
- 6.Schmitt M, Radler F. Antimicrob Agents Chemother. 1990;34:1615–1618. doi: 10.1128/aac.34.8.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bussey H. Mol Microbiol. 1991;5:2339–2343. doi: 10.1111/j.1365-2958.1991.tb02079.x. [DOI] [PubMed] [Google Scholar]
- 8.Mendoza I, Rubio F, Rodriguez-Navarro A, Pardo J M. J Biol Chem. 1994;269:8792–8796. [PubMed] [Google Scholar]
- 9.Becker M D, Guarente L. Methods Enzymol. 1991;194:182–187. doi: 10.1016/0076-6879(91)94015-5. [DOI] [PubMed] [Google Scholar]
- 10.Toh-e A, Yasunaga S, Nisogi H, Tanaka K, Oguchi T, Matsui Y. Yeast. 1993;9:481–494. doi: 10.1002/yea.320090504. [DOI] [PubMed] [Google Scholar]
- 11.Rothstein R. In: DNA Cloning. Glover D M, editor. Vol. 2. Oxford: IRL; 1985. pp. 45–66. [Google Scholar]
- 12.Sherman F. Methods Enzymol. 1991;194:12–17. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- 13.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 14.Elble R. BioTechniques. 1992;13:18–20. [PubMed] [Google Scholar]
- 15.Mumberg D, Muller R, Funk M. Gene. 1995;156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- 16.Russo P, Kalkkinen N, Sereneva H, Paakkola J, Makarow M. Proc Natl Acad Sci USA. 1992;89:3671–3675. doi: 10.1073/pnas.89.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carlson M, Botstein D. Cell. 1982;28:145–154. doi: 10.1016/0092-8674(82)90384-1. [DOI] [PubMed] [Google Scholar]
- 18.Song C S, Yu J H, Bai D H, Hester P Y, Kim K H. J Immunol. 1985;135:3354–3359. [PubMed] [Google Scholar]
- 19.Zhu J K, Bressan R A, Hasegawa P M. Proc Natl Acad Sci USA. 1993;90:8557–8561. doi: 10.1073/pnas.90.18.8557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mulholland J, Preuss D, Moon A, Wong A, Drubin D, Botstein D. J Cell Biol. 1994;125:381–391. doi: 10.1083/jcb.125.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woloshuk C P, Meulenhoff J S, Sela-Buurlage M, Elzen P J M, Cornelissen B J C. Plant Cell. 1991;3:619–628. doi: 10.1105/tpc.3.6.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Russo P, Simonen M, Uimari A, Teesalu T, Makarow M. Mol Gen Genet. 1993;239:273–280. doi: 10.1007/BF00281628. [DOI] [PubMed] [Google Scholar]
- 23.Kurzweilová H, Sigler K. Arch Microbiol. 1994;162:211–214. doi: 10.1007/BF00314477. [DOI] [PubMed] [Google Scholar]
- 24.Schmitt M J, Compain P. Arch Microbiol. 1995;164:435–443. doi: 10.1007/BF02529742. [DOI] [PubMed] [Google Scholar]