

Abstract
Endothelial nitric oxide synthase (eNOS) catalyzes the conversion of L-arginine to L-citrulline and nitric oxide (NO), an important modulator of vascular function. eNOS is regulated post-translationally through phosphorylation/dephosphorylation at a number of specific phosphorylation sites including Ser-116 in the bovine eNOS sequence. Whether phosphorylation of eNOS at Ser-116 in endothelial cells is stimulatory or inhibitory has not previously been definitively determined. In this study we show that mimicking phosphorylation of eNOS at Ser-116 by Asp mutation reduces basal NO release from endothelial cells. Preventing phosphorylation at this site by Ala mutation increases the amount of NO release from endothelial cells in response to agonist stimulation. In addition, mimicking phosphorylation of Ser-116 increases eNOS association with caveolin-1 and reduces the vascular reactivity of intact aortic rings. eNOS phosphorylation at Ser-116, therefore, appears to contribute to negative modulation of eNOS activity and hence to regulation of vascular tone.
Keywords: nitric oxide, eNOS, phosphorylation, protein-protein interactions
1. Introduction
The vascular homeostatic enzyme, endothelial nitric oxide synthase (eNOS) catalyzes the conversion of L-arginine to L-citrulline and nitric oxide (NO). NO, thus produced in the endothelium has a key role in regulation of vascular tone (Ignarro, 1989), platelet aggregation (Radomski et al, 1987), platelet and leukocyte adhesion to the endothelium (Kubes et al, 1991;Radomski et al, 1987), vascular smooth muscle cell proliferation (Garg and Hassid, 1989), endothelial cell apoptosis (Hoffmann et al, 2001), angiogenesis (Papapetropoulos et al, 1997), and vascular leakage during acute inflammation (Cirino et al, 2003). eNOS functions as a homodimer with each of the two subunits consisting of an N-terminal oxygenase domain, and a C-terminal reductase domain, bridged by a Ca2+-calmodulin (CaM)-binding domain. The N-terminal oxygenase domain contains a heme moiety and binding sites for L-arginine, zinc, and the cofactor, tetrahydrobiopterin (BH4). The C-terminal reductase domain contains binding sites for FAD, FMN, and NADPH (Alderton et al, 2001;Venema et al, 1996).
eNOS is subject to tight regulation at the transcriptional, post-transcriptional (mRNA stability), and post-translational levels (Fleming and Busse, 2003). Regulation of eNOS by protein-protein interactions and phosphorylation at serine (Ser), threonine (Thr), and tyrosine (Tyr) are two of the most important post-translational regulatory mechanisms. To date, six specific sites of phosphorylation have been identified in bovine eNOS at Tyr-83, Ser-116, Thr-497, Ser-617, Ser-635, and Ser-1179 (equivalent to Tyr-81, Ser-114, Thr-495, Ser-615, Ser-633, and Ser-1177 in the human sequence) (Fulton et al, 2005;Venema, 2002). Tyr-83, Ser-635, and Ser-1179 phosphorylation are associated with eNOS activation and increases in NO release. Thr-497 phosphorylation negatively regulates eNOS activity with dephosphorylation of this site being associated with increased eNOS activity (Fleming et al, 2001;Harris et al, 2001;Michell et al, 2001). Controversy exists about whether phosphorylation of Ser-116 is stimulatory or inhibitory. Phosphorylation of Ser-116 in endothelial cells in response to increased fluid shear stress, a stimulus known to substantially increase eNOS activity, was first demonstrated by mass spectroscopy (Gallis et al, 1999). Likewise, it has been shown (Drew et al, 2004) that high-density lipoprotein and apolipoprotein AI increase eNOS activity in endothelial cells by a mechanism involving Ser-116 phosphorylation. These studies therefore support the conclusion that Ser-116 phosphorylation is stimulatory. On the other hand, other studies have shown that the eNOS-activating agonist, vascular endothelial growth factor (VEGF) induces Ser-116 dephosphorylation in endothelial cells and that, when COS-7 cells are transfected with wild-type and mutant, nonphosphorylatable (116A) forms of eNOS, and then stimulated with the Ca2+ ionophore, A23187 that eNOS activity of the mutant is increased rather than decreased (Kou et al, 2002). Further evidence that Ser-116 phosphorylation may be inhibitory rather than stimulatory has also been provided based on studies with plasmid-transfected COS-7 cells (Bauer et al, 2003). An additional study (Boo et al, 2002) reported a finding of no effect of fluid shear stress, VEGF, or 8-bromo cAMP on the phosphorylation status of Ser-116 in endothelial cells. Conflicting conclusions have therefore been drawn from these various studies. One potential explanation for these different conclusions is that determinations of the functional consequences of eNOS Ser-116 phosphorylation have been limited to investigations in COS-7 cells that are phenotypically very different from endothelial cells in that they have a different complement of signaling kinases and phosphatases and lack some of the eNOS-interacting proteins that have an important influence on eNOS activity. In the present study therefore we have investigated the functional consequences of Ser-116 phosphorylation of eNOS in endothelial cells rather than in COS-7 cells. We have also defined for the first time a potential mechanism by which this phosphorylation event may affect eNOS activity and, more importantly, have demonstrated a role for Ser-116 dephosphorylation in agonist-induced vascular relaxation.
2. Materials and Methods
2.1. Materials
Anti-eNOS antibody, anti-caveolin-1 antibody, and anti-Hsp90 antibody were obtained from BD Transduction Laboratories (San Diego, CA, USA). Anti-phospho-Ser-116 eNOS antibody was obtained from Upstate Inc. (Charlottesville, VA, USA). Anti-soluble guanylate cyclase antibodies were purchased from Cayman Chemical (Ann Arbor, MI, USA). VEGF was obtained from R&D Systems, Inc. (Minneapolis, MN, USA). Thapsigargin was obtained from Sigma Chemical (St Louis, MO, USA). E. coli BJ5183 cells were purchased from Stratagene (La Jolla, CA, USA). Mouse IgG Trueblot™ horseradish peroxidase-conjugated secondary antibody was obtained from eBioscience (San Diego, CA, USA).
2.2. Cell culture
Primary cultures of bovine aortic endothelial cells (BAECs) were obtained from VEC Technologies Inc. (Rensselaer, NY, USA). Primary cultures were subcultured 1 :4 at 3-5 day intervals and used for experiments during passage two to passage six. Cultures were maintained in M199 medium (GIBCO™ Invitrogen Corporation, Grand Island, NY, USA) supplemented with 10% fetal bovine serum, 5% iron-supplemented calf serum, 0.6 μg/ml thymidine, 2.2 mg/ml sodium bicarbonate, 500 IU/ml penicillin, and 500 IU/ml streptomycin. Human embryonic kidney 293 (HEK293) cells were obtained from Stratagene. HEK293 and COS-7 cells were maintained in Dulbecco’s modification of eagle’s medium (DMEM) with 4.5 g/L glucose and L-glutamine (Mediatech Inc., Herndon, VA, USA) supplemented with 10% fetal bovine serum, 500 IU/ml penicillin, and 500 μg/ml streptomycin.
2.3. Mutagenesis
Asp or Ala mutants of eNOS phosphorylation sites were generated using the QuikChange® Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer’s instructions. This procedure is based on PCR amplication using primers of approximately 40 nucleotides in length in which the 116D, 497D, 617D, 635D, 1179D, and 635D/1179D mutations were created by replacing the Ser-116, Thr-497, Ser-617, Ser-635, Ser-1179, and Ser-635/Ser-1179 codons with Asp codons. A 116A mutation was created by replacing the Ser-116 codon with an Ala codon. Nucleotide sequence changes were as follows: 116D, TCC→GAC; 497D, ACC→GAC; 617D, AGC→GAC; 635D, TCC→GAC; 1179D, AGC→GAC; 116A, TCC→GCC. The sequences of all mutated constructs were confirmed by the Molecular Biology Core Facility of the Medical College of Georgia.
2.4. Construction and purification of recombinant eNOS adenoviruses
Adenoviruses expressing full-length wild-type, Asp, and Ala mutants of eNOS were generated by the AdEasy system (He et al, 1998). Briefly, bovine full-length, Asp, or Ala mutant eNOS cDNAs were all subcloned into the Hind III and Xba I sites of the multiple cloning site of the pAdTrack-CMV shuttle vector. The correct DNA sequence was verified by sequencing in the Molecular Biology Core Facility of the Medical College of Georgia. The pAd-Track-CMV-eNOS construct was linearized with Pme I and subsequently cotransformed into E. coli BJ5183 cells with an adenoviral backbone plasmid, pAdEasy-1. Recombinants were selected by kanamycin resistance and verified by restriction enzyme digestion. The confirmed recombinant plasmid was then transfected into the adenoviral packaging HEK 293 cell line. Viral production was monitored over 7-10 days by visualization of green fluorescent protein expression. After 7-10 days, virus was harvested and purified by banding on a cesium chloride gradient. The purified virus was then dialyzed and stored at -80° C.
2.5. Expression of eNOS adenoviruses in endothelial cells
After serum-starvation for 2 h, BAECs were infected with various MOIs of adenoviruses for 24 h to determine the titer that gave maximal expression of eNOS without significant cell death. Cell viability was assessed by trypan blue exclusion. In subsequent experiments using gene transfer of eNOS, confluent BAECs in 100 mm dishes were infected for 24 h at an optimal MOI.
2.6. Measurement of NO release
NO release from BAECs was measured by the bioassay procedure first described by Ishii et al. (Ishii et al, 1991) and used in many of our previous studies (Harris et al, 2001;Harris et al, 2006;Li et al, 2005) with the modifications described in the present study. Confluent BAECs were infected as described for adenovirus expression. COS-7 cells were infected with optimal amount of both α and β subunits of soluble guanylyl cyclase (sGC) adenoviruses overnight at 37°C and then used as reporter cells. Immunoblotting of these cells with anti-sGCα and anit-sGCβ antibodies show that they express higher levels of expression of both sGC subunits than are expressed in the rat aortic smooth muscle cells that we have used as reporter cells in previous studies. After 10 min incubation of infected BAECs or eNOS knockdown BAECs with Locke’s buffer containing 20 units/ml superoxide dismutase and 0.3 mM 3-isobutyl-1-methylxanthine in the presence or absence of VEGF, the BAEC bathing medium was transferred to confluent COS-7 reporter cells which were incubated for 3 min and then extracted in 0.1 M HCl. cGMP concentrations in the lysates were quantitied using an enzyme immunoassay kit (Cayman Chemical Co., Ann Arbor, MI, USA) according to the manufacturer’s instructions.
2.7. Immunoprecipitation and immunoblotting
Immunoprecipitation and immunoblotting of BAECs was performed as previously described (Li et al, 2005). Briefly, after adenovirus infection of BAECs, BAECs were lysed in ice-cold lysis buffer containing 50 mM Tris-HCl, pH 7.4, 100 mM NaF, 15 mM Na4P2O7, 1 mM Na3VO4, 1% Triton X-100, 1 mM PMSF, 10 μg/ml pepstatin A, and 5 μg/ml aprotinin. Lysates were centrifuged at 10,000× g to remove insoluble material and were then precleared by adding 50 μl of protein A/G agarose (Santa Cruz Biotechnology, Santa Cruz, CA, USA) followed by incubation for 2 h at 4°C with rocking. The agarose beads were then pelleted by centrifugation at 1,000× g. Precleared lysates were immunoblotted with anti-eNOS antibody or were used for immunoprecipitation experiments. For immunoprecipitation anti-eNOS antibody (10 μl) was added to the precleared lysate and samples were incubated overnight at 4°C with rocking. Protein A/G agarose was then added and samples were incubated for an additional 3 h at 4°C. Beads were subsequently washed two times with ice-cold lysis buffer. Immunoprecipitated proteins were eluted from the beads by boiling for 5 min in SDS sample buffer. The supernatants were subjected to SDS polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and immunoblotted with anti-caveolin-1 (1: 1000 dilution), or anti-Hsp90 (1:1000 dilution) antibodies. Bound antibody was visualized using Super Signal West Pico Chemiluminescent Substrate (Pierce Chemical, Rockford, IL, USA) following exposure of the blot to peroxidase-conjugated secondary antibody. The membranes were stripped and reprobed with anti-eNOS (1:1000 dilution) antibody to show equal amounts of eNOS immunoprecipitation in all samples.
2.8. eNOS knockdown cells
Retroviral knockdown BAECs were obtained as described previously (Zhang et al, 2006). Briefly, retroviruses were generated encoding siRNA 3122. BAEC were seeded at a density of 1.3×106 cells/100mm dish and infected with approximately 105 cfu/ml of active retrovirus. 24h later, cells were selected for viral uptake using the antibiotic, puromycin (0.8 μg/ml) for 7-10 days. To obtain eNOS constructs impervious to knockdown, several silent mutations which do not change the amino acid sequence, were generated in wild-type (WT), and 116A eNOS using the following primers and the mutagenesis kit. Sense: 5′-GCACGATATCGAAAGCAAAGGGCTGCAGCCCGCCCC CATG-3′; Antisense: 5′-CATGGGGGCGGGCTGCAGCCCTTTGCTTTCGATATCGT GC-3′. These primers introduced a total of 4 nucleotide substitutions in the DNA coding for bovine eNOS residues 1032-1037 with changing the amino acid sequence (HDIESK). WT or mutant eNOS was subcloned into Hind III and Xba I sites of the pADTrack-CMV shuttle vector and subjected to the same procedures as described above for construction and purification of recombinant eNOS adenoviruses not having silent mutations.
2.9. Adenoviral gene transfer into the endothelium of mouse aortae and isometric studies
Congenic eNOS -/- knockout mice backbred at least 10 generations to C5BL/6J mice (Huang et al, 1995;Shesely et al, 1996) (8 weeks old) were purchased from Jackson Laboratory. Mice were anesthetized with ketamine/xylazine and thoracic aortae were then injected with WT or 116D adenoviruses (Scotland et al, 2002). The virus-filled vessels were incubated in serum-free DMEM medium with 4.5 g/L glucose overnight. For isometric force recording, the same protocols and equipment (Multi-myograph with PowerLab software) were used as we have described previously (Harris et al, 2003). Aortae were cut into 3 mm ring segments and mounted on stainless-steel holders in organ baths containing physiological saline (130 mM NaCl, 4.7 mM KCl, 1.18 mM KH2PO4, 1.17 mM MgSO4·7H2O, 1.6 mM CaCl2·2H2O, 14.9 mM NaHCO3, 5.5 mM dextrose, and 0.03 mM Na2EDTA) aerated with 95% O2/5% CO2. Preparations were allowed to equilibrate for 60 min under constant passive force of (~1 mN) before the experiment began. After vascular ring preparation, a dose-response curve to phenylephrine (10-9 to 10-5 M) was constructed to determine the EC50 of the contractile response. After phenylephrine had been washed out, vessel segments were contracted with an EC50 concentration of phenylephrine. When the contraction reached a plateau phase, an accumulated dose response to ACh (10-9 to 10-4 M) were then conducted.
2.10. Statistics
All data are representative of three separate experiments and reported as mean ± S.E. Overall differences between groups were analyzed using one-way ANOVA and tested with Tukey-Kramer Multiple-Comparison Test for determining differences between the means when more than two groups were compared. An independent t-test was used when only two groups were compared. In all tests, significance was accepted at P < 0.05.
3. Results
3.1. Effects of Asp mutation of eNOS on basal NO release in endothelial cells
The effects of site-specific phosphorylation on enzyme activity can often be mimicked by Ser or Thr to Asp mutation. Therefore, in order to determine the effects of mimicking phosphorylation of Ser-116 on basal NO release from endothelial cells and to compare these effects to those of mimicking phosphorylation of the other Ser/Thr sites, we prepared and purified recombinant adenoviruses for expression of WT, 116D, 497D, 617D, 635D, 1179D, and 635D/1179D forms of eNOS in BAECs. Cells were transduced with these various adenoviruses and with a negative control (β-galactosidase) adenovirus. Viral titers were adjusted to obtain equal levels of expression of the various forms of eNOS. Relative levels of eNOS expression were assessed by immunoblotting of cell lysates with anti-eNOS antibody. As shown in Fig. 1B, adenoviral-mediated gene transfer with the eNOS adenoviruses significantly increased the levels of eNOS expression compared to the negative control. Furthermore, levels of overexpression of each of the different forms of eNOS were equivalent. Relative amounts of NO release under each of the various conditions were also determined using a cGMP reporter cell assay. As shown in Fig. 1A, overexpression of WT eNOS in BAECs increased NO production by at least 4-fold. As also illustrated in Fig. 1A, mimicking phosphorylation at the Ser-116 and Thr-497 sites reduced basal NO release compared to WT eNOS overexpression. The 497D result confirmed what had been concluded previously that phosphorylation of this site is inhibitory (Bauer et al, 2003;Michell et al, 2002). The 116D result is important because it confirms that phosphorylation of this site in endothelial cells is inhibitory rather than stimulatory. Mimicking phosphorylation of Ser-617, Ser-635, and Ser-1179, on the other hand, increased basal NO release compared to WT eNOS overexpression as did mimicking phosphorylation of both Ser-635 and Ser-1179 at the same time.
Fig. 1. Effects of Asp mutation of eNOS on basal NO release in endothelial cells.
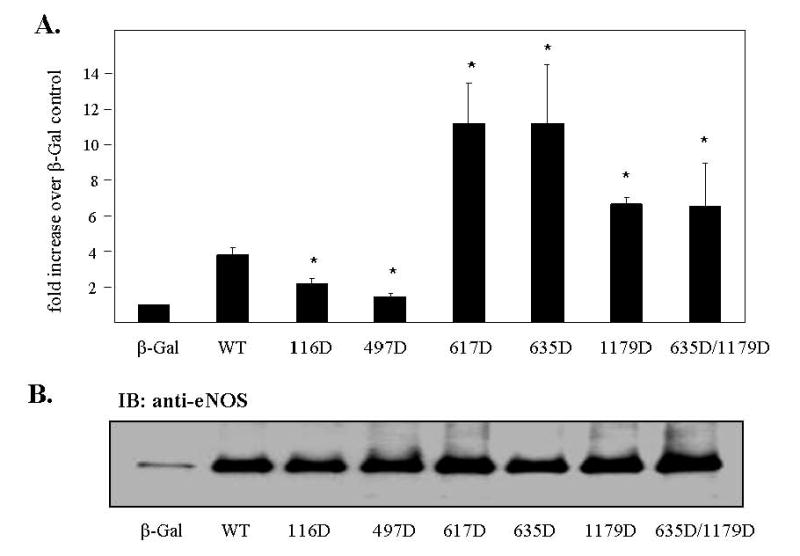
BAECs were infected for 24 h with adenoviruses expressing either β-galactosidase (β-Gal), wild-type (WT) eNOS, or Asp mutants of eNOS. COS-7 cells were infected for 24 h with both sGCα and sGCβ adenoviruses. BAECs were then incubated with blank Locke’s buffer for 10 min. The conditioned medium was transferred to COS-7 cells and incubated for 3 min. (A) Reporter cells were lysed in 0.1 M HCl. NO release was quantitated by cGMP concentrations according to the manufacturer’s instructions (means ± S.E., n=3, *P < 0.05 vs. WT). (B) BAECs were lysed and the cell lysates were immunoblotted with anti-eNOS antibody to confirm equal levels of overexpression of all the various forms of eNOS (n=3).
3.2. Effects of Asp mutation of eNOS on eNOS association with caveolin-1 in endothelial cells
In order to obtain additional support for the conclusion that Ser-116 phosphorylation has an inhibitory effect on eNOS activity, we also overexpressed WT and 116D forms of eNOS in COS-7 cells followed by measurement of NO release. However, in contrast to the experiments carried out in BAECs, when WT and 116D forms of eNOS were equivalently overexpressed in COS-7 cells, no significant differences in NO release were detected (data not shown) suggesting that mimicking Ser-116 phosphorylation per se is not sufficient to inhibit eNOS activity. We therefore investigated the possibility that eNOS phosphorylation at Ser-116 might have an indirect effect on eNOS activity by altering eNOS protein-protein interactions with inhibitory proteins such as caveolin-1 (Ju et al, 1997) or stimulatory proteins such as Hsp90 (Garcia-Cardena et al, 1998). BAECs were infected with adenoviruses expressing either WT eNOS, or 116D, 497D, 617D, 635D, or 1179D mutants of eNOS. Lysates were prepared and equal quantities of lysate proteins from each condition were immunoprecipitated with anti-eNOS antibody. Immunoprecipitates were then immunoblotted with anti-caveolin-1 antibody. As shown in Fig. 2A, mimicking phosphorylation of Ser-116 increases binding of eNOS to the eNOS-inhibitory protein, caveolin-1 (3.9 ± 1.0 fold increase over WT eNOS, mean ± S.E., n=3). Mimicking phosphorylation of Thr-497, Ser-617, Ser-635, and Ser-1179, however, had no effect on eNOS-caveolin-1 association. The membranes were stripped and reprobed with anti-eNOS antibody, showing that there were equal amounts of WT or mutant eNOS immunoprecipitated from the infected endothelial cells (Fig. 2B). This provides a potential explanation for the mechanism by which phosphorylation of this site decreases eNOS activity and decreases endothelial NO release. When similar experiments were performed to determine whether any of the Asp mutations had an effect on eNOS association with Hsp90, results were negative, suggesting that phosphorylation does not alter the Hsp90-eNOS interaction.
Fig. 2. Effects of Asp mutation of eNOS on eNOS association with caveolin-1.
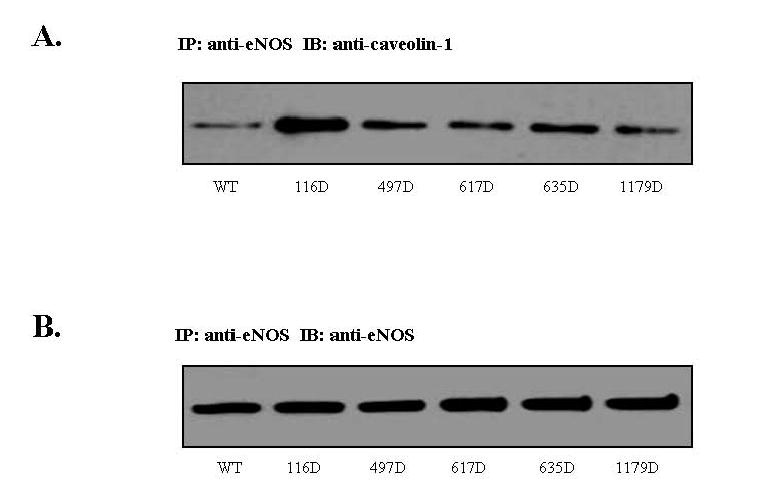
BAECs were infected for 24 h with adenoviruses expressing either WT eNOS, or Asp mutants of eNOS. (A) Lysates were prepared and equal quantities of lysate proteins from each condition were immunoprecipitated with anti-eNOS antibody. Immunoprecipitates were then immunoblotted with anti-caveolin-1 antibody (n=3). (B) The membranes were stripped and reprobed with anti-eNOS antibody (n=3).
3.3. Effects of Ala mutation of eNOS at Ser-116 on agonist-stimulated NO release in eNOS knockdown endothelial cells
As already indicated, adenoviral-mediated overexpression of a phospho-mimetic 116D form of eNOS in BAECs significantly reduced the amount of NO released under basal conditions compared to that released from BAECs in which WT eNOS was overexpressed at equivalent levels. We next sought to obtain evidence that agonist-activation of eNOS in BAECs depends in part on dephosphorylation of Ser-116. We therefore first tested whether we could detect VEGF-induced Ser-116 dephosphorylation in our BAEC cell culture model. BAECs were either treated or not treated with VEGF (20 ng/ml) for 10 min. Cell lysates were prepared and were immunoblotted with a phospho-specific antibody that recognizes eNOS only when it is phosphorylated at Ser-116. These experiments showed that VEGF promoted a significant dephosphorylation at Ser-116 within 10 min (data not shown).
In order to obtain further evidence for a role of Ser-116 dephosphorylation in agonist stimulation of eNOS activation in BAECs, we prepared an adenovirus that expresses a Ser-116 to Ala dephospho-mimetic form of eNOS that is unable to be endogenously phosphorylated or dephosphorylated at this site. Viral titers were adjusted so that infection of BAECs with WT and 116A forms of eNOS gave equal levels of eNOS overexpression. BAECs were then infected with negative control (β-galactosidase), WT eNOS, and 116A adenoviruses and subsequently either not treated or treated for 10 min with VEGF (20 ng/ml) or the intracellular Ca2+ elevator, thapsigargin (TG) (1 μM). VEGF and TG induced almost identical increases in NO release from BAECs regardless of whether cells were infected with the β-galactosidase, WT eNOS, or 116A adenoviruses (data not shown). Thus it appears that these stimuli induce the same increase in NO release from BAECs whether or not eNOS is overexpressed. Apparently, factors other than the level of eNOS expression play a dominant role in limiting the amount of NO release from BAECs in response to agonist stimulation.
As an alternative to overexpression of WT and 116A forms of eNOS in BAECs that already have high levels of expression of endogenous eNOS, we moved to a different model system in which the two forms of eNOS were reconstituted in eNOS “knockdown” BAECs. In this model, endogenous eNOS expression in BAECs is reduced by about 90% by stable transfection by a retrovirus that encodes an eNOS interfering RNA (RNAi). These cells were infected with adenovirus constructs for WT and 116A forms of eNOS that contained silent mutations that changed the nucleotide sequence of the adenovirally produced mRNAs in such a way as to make them impervious to RNAi knockdown without changing the amino acid sequences of the proteins. Cells were then stimulated with VEGF (20 ng/ml) for 10 min and relative levels of NO release were quantitied by cGMP reporter cell assay. As shown in Fig. 3A, VEGF stimulated significant increases in NO release from WT-infected knockdown BAECs. Basal and VEGF-stimulated NO production was significantly increased in cells infected with the 116A nonphosphorylatable eNOS mutant suggesting that dephosphorylation of Ser-116 contributes to the eNOS activation process. Immunoblotting of cell lysates with anti-eNOS antibody showed that expression levels of WT eNOS and 116A were equal (Fig. 3B).
Fig. 3. Effects of Ala mutation of eNOS on NO release in eNOS knockdown endothelial cells.
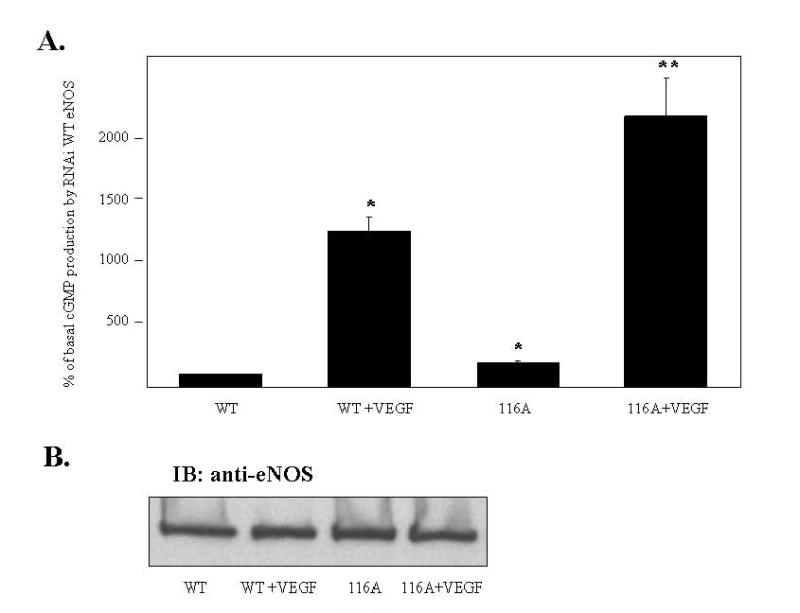
eNOS knockdown BAECs were infected for 24 h with RNAi adenoviruses expressing either WT eNOS or a 116A mutant of eNOS. COS-7 cells were infected for 24 h with both sGCα and sGCβ adenoviruses. The BAECs were then changed to serum-free media and incubated for 40 min. The BAECs were incubated with blank Locke’s buffer or VEGF (20 ng/ml) for 10 min. The conditioned medium was transferred to COS-7 cells and incubated for 3 min. (A) Reporter cells were lysed in 0.1 M HCl. NO release was quantitated by cGMP concentrations according to the manufacturer’s instructions (means ± S.E., n=3, *P < 0.05 vs. WT, **P < 0.05 vs. WT + VEGF) (B) BAECs were lysed and the cell lysates were immunoblotted with anti-eNOS antibody to confirm equal levels of expression of all the various forms of eNOS (n=3).
3.4. Effects of Asp mutation of eNOS at Ser-116 on ACh-induced vasorelaxation of eNOS-reconstituted aortic rings from eNOS knockout mice
Finally, we investigated the role of Ser-116 dephosphorylation of eNOS in agonist-induced relaxation of intact blood vessel segments. eNOS knockout mice (8 weeks old) were anesthetized and thoracic aortae were then injected with WT or 116D adenoviruses and incubated at 37°C overnight. Aortic rings were prepared and the dose-response to phenylephrine was determined and found to be similar for WT eNOS- and 116D eNOS-transduced rings (EC50 = 10-4 M, Emax = 10-2 M). Vessel segments were then contracted with an EC50 concentration of phenylephrine. When the contraction reached a plateau, an accumulated dose response to acetylcholine (Ach) was then conducted. As shown in Fig. 4, viral infection of eNOS knockout mice ring segments with WT eNOS restored the dose-response relaxation to ACh. In addition, upon viral infection of eNOS knockout mice ring segments with the phospho-mimetic 116D eNOS, the log EC50 of ACh-induced relaxation (-6.3 ± 0.3) was significantly decreased from that of WT eNOS adenovirus infection (-6.9 ± 0.1).
Fig. 4. Effects of Asp mutation of Ser-116 of eNOS on ACh-induced vasorelaxation in aortic rings from eNOS knockout mice.
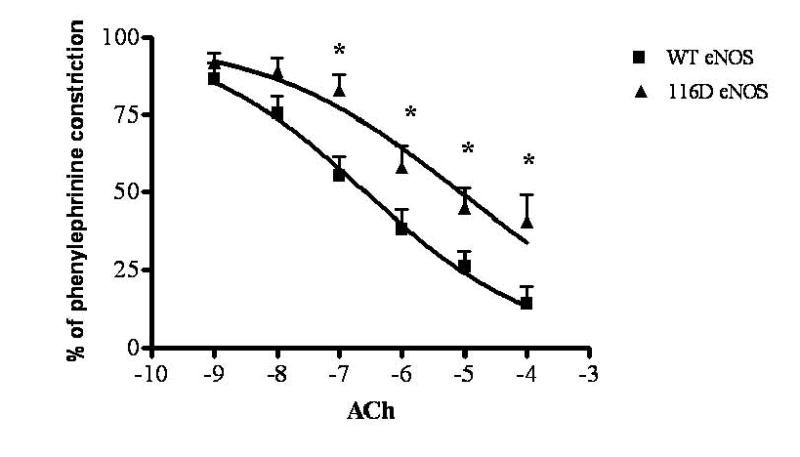
eNOS knockout mice were anesthetized with ketamine/xylazine and thoracic aortae were then injected with WT or 116D adenoviruses and incubated in DMEM serum-free media at 37°C overnight. Vessel segments were contracted with an EC50 concentration of phenylephrine. When the contraction reached a plateau, an accumulated dose response to ACh was conducted. The results shown are representative of three separate experiments (means ± S.E., n=3, * P < 0.05 vs. WT).
4. Discussion
Previous studies in cultured endothelial cells have concluded that eNOS phosphorylation at Ser-116 may be responsible in part for enzyme activation in response to fluid shear stress (Gallis et al, 1999) and high-density lipoprotein and apolipoprotein AI (Drew et al, 2004). Other studies have reported that agonist activation of eNOS in endothelial cells involves either no change in the phosphorylation status of eNOS at Ser-116 (Boo et al, 2002) or involves a dephosphorylation of eNOS at this site (Kou et al, 2002). Mutagenesis studies in COS-7 cells also suggest that phosphorylation of eNOS at Ser-116 inhibits rather than stimulates eNOS activity (Bauer et al, 2003). In the present study, by expression of phosphomimetic 116D and nonphosphorylatable 116A forms of eNOS we have confirmed for the first time in endothelial cells that phosphorylation of eNOS at Ser-116 is inhibitory rather than stimulatory and that VEGF-induced activation of the enzyme involves a dephosphorylation event rather than a phosphorylation event. In addition, by equal overexpression of phosphomimetic 617D, 635D, and 1179D forms of eNOS we have shown that mimicking phosphorylation of these three sites increases basal NO release compared to WT eNOS overexpression. This is interesting because, first of all, the purified 617D enzyme does not have greater maximal activity than purified WT eNOS in vitro (Michell et al, 2002). Secondly, because Ser-1179 is generally believed to be the most important of the eNOS phosphorylation sites it is interesting that mimicking phosphorylation of Ser-617 and Ser-635 increased activity to an even greater extent than mimicking phosphorylation of Ser-1179. Further, it is interesting that mimicking phosphorylation of both Ser-635 and Ser-1179 at the same time did not increase activity more than mimicking phosphorylation of only one of the two sites. Therefore, these two phosphorylation events do not appear to have an additive or synergistic effect on NO release in this model system.
Our results also show that eNOS phosphorylation at Ser-116 does not have a direct effect on catalytic activity of the enzyme but rather that inhibition may occur through increased association of eNOS with the plasmalemmal caveolae structural protein, caveolin-1. Plasmalemmal caveolae are small membrane invaginations that are particularly abundant in endothelial cells. Caveolin-1 is an integral membrane protein that forms a hairpin loop within caveolae membranes and interacts with eNOS through both N- and C-terminal cytoplasmic domains to inhibit eNOS activity (Ju et al, 1997). A negative control β-galactosidase adenovirus, a WT eNOS adenovirus, or a nonphosphorylatable 116A eNOS adenovirus were each expressed in regular BAECs followed by agonist stimulation of the cells. This resulted in almost identical agonist-stimulated increases in NO release from the cells regardless of which virus was used for the transduction. This suggests that maximal NO release is achieved by activation of endogenous eNOS and that this cannot be increased through simple eNOS overexpression. Another possibility is that a factor other than the level of eNOS expression (for example, levels of the eNOS cofactor, tetrahydrobiopterin) may be more important than levels of eNOS expression in limiting agonist- stimulated NO production in endothelial cells. This particular limitation, however, has been overcome in this study through the use of eNOS knockdown endothelial cells. When these cells were transduced to an equal extent by the WT eNOS and 116A eNOS adenoviruses, their was a clear increase in both basal and agonist-induced NO release (as measured by a cGMP reporter cell assay) for 116A eNOS-transduced cells as compared to WT eNOS-transduced cells indicating that dephosphorylation of Ser-116 contributes to eNOS activation in endothelial cells. cGMP reporter cell assays provide reliable and accurate measurements of NO release from cells in culture. However, because cultured cells may not perfectly mirror events that occur in intact blood vessels, we have complemented the cultured cell studies with intact vessel bioassays. These studies clearly show that mimicking phosphorylation of eNOS at Ser-116 also reduces the reactivity of intact aortic rings. We therefore conclude that eNOS phosphorylation at Ser-116 contributes to negative modulation of eNOS activity in endothelial cells and in blood vessels and thereby contributes to regulation of vascular tone.
Acknowledgments
This work was supported by grants from the National Institutes of Health (R. C. Venema) and the American Heart Association (R. C. Venema).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer PM, Fulton D, Boo YC, Sorescu GP, Kemp BE, Jo H, Sessa WC. Compensatory phosphorylation and protein-protein interactions revealed by loss of function and gain of function mutants of multiple serine phosphorylation sites in endothelial nitric-oxide synthase. J Biol Chem. 2003;278:14841–14849. doi: 10.1074/jbc.M211926200. [DOI] [PubMed] [Google Scholar]
- Boo YC, Hwang J, Sykes M, Michell BJ, Kemp BE, Lum H, Jo H. Shear stress stimulates phosphorylation of eNOS at Ser(635) by a protein kinase A-dependent mechanism. Am J Physiol Heart Circ Physiol. 2002;283:H1819–H1828. doi: 10.1152/ajpheart.00214.2002. [DOI] [PubMed] [Google Scholar]
- Cirino G, Fiorucci S, Sessa WC. Endothelial nitric oxide synthase: the Cinderella of inflammation? Trends Pharmacol Sci. 2003;24:91–95. doi: 10.1016/S0165-6147(02)00049-4. [DOI] [PubMed] [Google Scholar]
- Drew BG, Fidge NH, Gallon-Beaumier G, Kemp BE, Kingwell BA. High-density lipoprotein and apolipoprotein AI increase endothelial NO synthase activity by protein association and multisite phosphorylation. Proc Natl Acad Sci U S A. 2004;101:6999–7004. doi: 10.1073/pnas.0306266101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming I, Busse R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1–12. doi: 10.1152/ajpregu.00323.2002. [DOI] [PubMed] [Google Scholar]
- Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res. 2001;88:E68–E75. doi: 10.1161/hh1101.092677. [DOI] [PubMed] [Google Scholar]
- Fulton D, Church JE, Ruan L, Li C, Sood SG, Kemp BE, Jennings IG, Venema RC. Src kinase activates endothelial nitric-oxide synthase by phosphorylating Tyr-83. J Biol Chem. 2005;280:35943–35952. doi: 10.1074/jbc.M504606200. [DOI] [PubMed] [Google Scholar]
- Gallis B, Corthals GL, Goodlett DR, Ueba H, Kim F, Presnell SR, Figeys D, Harrison DG, Berk BC, Aebersold R, Corson MA. Identification of flow-dependent endothelial nitric-oxide synthase phosphorylation sites by mass spectrometry and regulation of phosphorylation and nitric oxide production by the phosphatidylinositol 3-kinase inhibitor LY294002. J Biol Chem. 1999;274:30101–30108. doi: 10.1074/jbc.274.42.30101. [DOI] [PubMed] [Google Scholar]
- Garcia-Cardena G, Fan R, Shah V, Sorrentino R, Cirino G, Papapetropoulos A, Sessa WC. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998;392:821–824. doi: 10.1038/33934. [DOI] [PubMed] [Google Scholar]
- Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest. 1989;83:1774–1777. doi: 10.1172/JCI114081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris MB, Bartoli M, Sood SG, Matts RL, Venema RC. Direct interaction of the cell division cycle 37 homolog inhibits endothelial nitric oxide synthase activity. Circ Res. 2006;98:335–341. doi: 10.1161/01.RES.0000203564.54250.0b. [DOI] [PubMed] [Google Scholar]
- Harris MB, Blackstone MA, Ju H, Venema VJ, Venema RC. Heat-induced increases in endothelial NO synthase expression and activity and endothelial NO release. Am J Physiol Heart Circ Physiol. 2003;285:H333–H340. doi: 10.1152/ajpheart.00726.2002. [DOI] [PubMed] [Google Scholar]
- Harris MB, Ju H, Venema VJ, Liang H, Zou R, Michell BJ, Chen ZP, Kemp BE, Venema RC. Reciprocal phosphorylation and regulation of endothelial nitric-oxide synthase in response to bradykinin stimulation. J Biol Chem. 2001;276:16587–16591. doi: 10.1074/jbc.M100229200. [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann J, Haendeler J, Aicher A, Rossig L, Vasa M, Zeiher AM, Dimmeler S. Aging enhances the sensitivity of endothelial cells toward apoptotic stimuli: important role of nitric oxide. Circ Res. 2001;89:709–715. doi: 10.1161/hh2001.097796. [DOI] [PubMed] [Google Scholar]
- Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ. Biological actions and properties of endothelium-derived nitric oxide formed and released from artery and vein. Circ Res. 1989;65:1–21. doi: 10.1161/01.res.65.1.1. [DOI] [PubMed] [Google Scholar]
- Ishii K, Sheng H, Warner TD, Forstermann U, Murad F. A simple and sensitive bioassay method for detection of EDRF with RFL-6 rat lung fibroblasts. Am J Physiol. 1991;261:H598–H603. doi: 10.1152/ajpheart.1991.261.2.H598. [DOI] [PubMed] [Google Scholar]
- Ju H, Zou R, Venema VJ, Venema RC. Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J Biol Chem. 1997;272:18522–18525. doi: 10.1074/jbc.272.30.18522. [DOI] [PubMed] [Google Scholar]
- Kou R, Greif D, Michel T. Dephosphorylation of endothelial nitric-oxide synthase by vascular endothelial growth factor. Implications for the vascular responses to cyclosporin A. J Biol Chem. 2002;277:29669–29673. doi: 10.1074/jbc.M204519200. [DOI] [PubMed] [Google Scholar]
- Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci U S A. 1991;88:4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Huang W, Harris MB, Goolsby JM, Venema RC. Interaction of the endothelial nitric oxide synthase with the CAT-1 arginine transporter enhances NO release by a mechanism not involving arginine transport. Biochem J. 2005;386:567–574. doi: 10.1042/BJ20041005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michell BJ, Chen Z, Tiganis T, Stapleton D, Katsis F, Power DA, Sim AT, Kemp BE. Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. J Biol Chem. 2001;276:17625–17628. doi: 10.1074/jbc.C100122200. [DOI] [PubMed] [Google Scholar]
- Michell BJ, Harris MB, Chen ZP, Ju H, Venema VJ, Blackstone MA, Huang W, Venema RC, Kemp BE. Identification of regulatory sites of phosphorylation of the bovine endothelial nitric-oxide synthase at serine 617 and serine 635. J Biol Chem. 2002;277:42344–42351. doi: 10.1074/jbc.M205144200. [DOI] [PubMed] [Google Scholar]
- Papapetropoulos A, Garcia-Cardena G, Madri JA, Sessa WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest. 1997;100:3131–3139. doi: 10.1172/JCI119868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radomski MW, Palmer RM, Moncada S. The anti-aggregating properties of vascular endothelium: interactions between prostacyclin and nitric oxide. Br J Pharmacol. 1987;92:639–646. doi: 10.1111/j.1476-5381.1987.tb11367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotland RS, Morales-Ruiz M, Chen Y, Yu J, Rudic RD, Fulton D, Gratton JP, Sessa WC. Functional reconstitution of endothelial nitric oxide synthase reveals the importance of serine 1179 in endothelium-dependent vasomotion. Circ Res. 2002;90:904–910. doi: 10.1161/01.res.0000016506.04193.96. [DOI] [PubMed] [Google Scholar]
- Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1996;93:13176–13181. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venema RC. Post-translational mechanisms of endothelial nitric oxide synthase regulation by bradykinin. Int Immunopharmacol. 2002;2:1755–1762. doi: 10.1016/s1567-5769(02)00185-6. [DOI] [PubMed] [Google Scholar]
- Venema RC, Sayegh HS, Kent JD, Harrison DG. Identification, characterization, and comparison of the calmodulin- binding domains of the endothelial and inducible nitric oxide synthases. J Biol Chem. 1996;271:6435–6440. doi: 10.1074/jbc.271.11.6435. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Church JE, Jagnandan D, Catravas JD, Sessa WC, Fulton D. Functional relevance of Golgi- and plasma membrane-localized endothelial NO synthase in reconstituted endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:1015–1021. doi: 10.1161/01.ATV.0000216044.49494.c4. [DOI] [PubMed] [Google Scholar]