

Summary
Intramembrane proteases are important enzymes in biology. The recently solved crystal structures of rhomboid protease GlpG have provided useful insights into the mechanism of these membrane proteins. Besides revealing an internal water-filled cavity that harbored the Ser-His catalytic dyad, the crystal structure identified a novel structural domain (L1 loop) that lies on the side of the transmembrane helices. Here using site-directed mutagenesis we confirmed that L1 loop is partially embedded in the membrane, and showed that alanine substitution of a highly preferred tryptophan (Trp-136) at the distal tip of L1 loop near lipid:water interface reduced GlpG proteolytic activity. Crystallographic analysis showed that W136A mutation did not modify the structure of the protease. Instead, the polarity for a small and lipid-exposed protein surface, at the site of the mutation, has changed. The crystal structure, now refined at 1.7 Å resolution, also clearly defined a 20 Å wide hydrophobic belt around the protease, which likely corresponded to the thickness of the compressed membrane bilayer around the protein. This improved structural model predicts that all critical elements of the catalysis, including the catalytic serine and the L5 cap, need to be positioned within a few angstroms of the membrane surface, and may explain why the protease activity is sensitive to changes in the protein:lipid interaction. Based on these findings, we propose a model where the end of substrate transmembrane helix first partitions out of the hydrophobic core region of the membrane before it bends into the protease active site for cleavage.
Keywords: intramembrane proteolysis, rhomboid protease, GlpG, membrane protein, x-ray crystallography
Introduction
Intramembrane proteases are highly specialized membrane proteins that cleave transmembrane peptide substrate within the plane of cell membranes. They play important roles in many processes that are of great interest in biology and medicine1–5. Of the three known mechanistic classes, the rhomboid family of serine intramembrane proteases is now the best understood in terms of structure and function6,7. The crystal structure of E. coli GlpG, a bacterial rhomboid protease, was the first structure solved for any intramembrane protease8, and together with a series of similar structures that followed9–12, represent at this time the only structures that are known to high-resolution for these membrane proteins. The crystal structure not only validated the hypothesis that peptide hydrolysis can occur within cell membranes, but also raised new questions regarding the details of its mechanism.
One intriguing aspect of the crystal structure is about a conserved “loop” (L1) that lies to the side of the transmembrane (TM) helices (Figure 1(a))8. The position of this little domain indicates that its lower portion may be inserted in the hydrocarbon region of the lipid bilayer, an insertion very rarely observed in structures of other integral or peripheral membrane proteins. Furthermore, the possibility that L1 may have a functional role in rhomboid protease activity was suggested by the pattern of sequence conservation in the rhomboid proteins, and by earlier mutagenesis studies. Sequence similarities among active rhomboid proteases are very low in general (see Figure 1 legend), and the few invariant residues mostly cluster around the active site. The only exception to this conservation pattern is a highly preferred WR sequence motif (E. coli GlpG Trp-136 and Arg-137) located near the tip of L1, which is separate from the main body of the protease and its active site (Figure 1(a)). The uniqueness of the motif was recognized early, and mutagenesis experiments suggested that it was important for activity13,14. However, it was not previously clear how the WR motif, and the loop itself, could affect the function of rhomboid proteases.
Figure 1.

Cartoon illustrations of GlpG highlighting the catalytic dyad, L1 loop and L5 cap. (a) and (b) correspond to the side and back views, respectively. A previous alignment of 87 rhomboid sequences indicated that a large fraction of the sequence (67%) have a tryptophan or tyrosine at position 136 (and most followed by an arginine)34. This level of conservation is significant especially in light of the facts that sequence identities among functional rhomboid proteases are extremely low, and that only eight residues, six at the active site plus two glycines involved in helix packing, are invariant. Tryptophan and tyrosine are the most preferred residues at membrane:water interface20.
Other outstanding mechanistic questions relate to how the protease interacts with TM substrates, and whether (and how) lipids may influence the enzyme mechanism16. The observed conformational plasticity near L5 cap (Figure 1(b)), which normally blocks a side portal that leads to the active site8,11, raised one possibility that opening of L5 cap would allow substrate to bend into the active site between TM helices S2 and S5 of GlpG12. A different model has been proposed where an entire TM helix (S5) tilts 35° away from the rest of the protein to open up a gap in the membrane-embedded portion of the protease for substrate entry9,15. Here we evaluate these models by re-calculating the precise locations of protease active site and the scissile bond in the substrate TM helix in relation to the lipid bilayer.
Results
L1 loop is half-inserted in the membrane
Although our initial observation regarding the position of L1 loop in relation to the TM helices was confirmed by subsequent crystal structures9–11, a limitation of any x-ray analysis is that the membrane protein has to be extracted from cell membranes by detergents. In order to examine the position of L1 loop in a native membranous environment, we introduced cysteines to 11 selected locations that span the length of the loop, and studied their susceptibilities to chemical modification by AMS, a membrane-impermeable alkylating agent, in intact cells that express the single cysteine mutants (Figure 2(a)). All cysteine mutants were proteolytically active, indicating that the gross conformation of the protease was not affected by the mutations (Figure 2(b)). Among these, only 4 mutants (D116C, F127C, L131C and R137C) could be modified by AMS, whereas the rest were not, suggesting that the latter group was either protected by the membrane, or folded inside the protein. Upon addition of detergent, which dissolved the membrane, all 11 cysteine mutants became modifiable by AMS, indicating that they were exposed on the protein surface (Figure 2(c)). When combined with the crystallographic data, these results clearly demonstrated that L1 loop is partially embedded in the membrane, and is laterally attached to the bundle of TM helices, which enables it to interact directly with the lipid (Figure 2(d)).
Figure 2. AMS modification of Cys residues introduced into L1 loop.
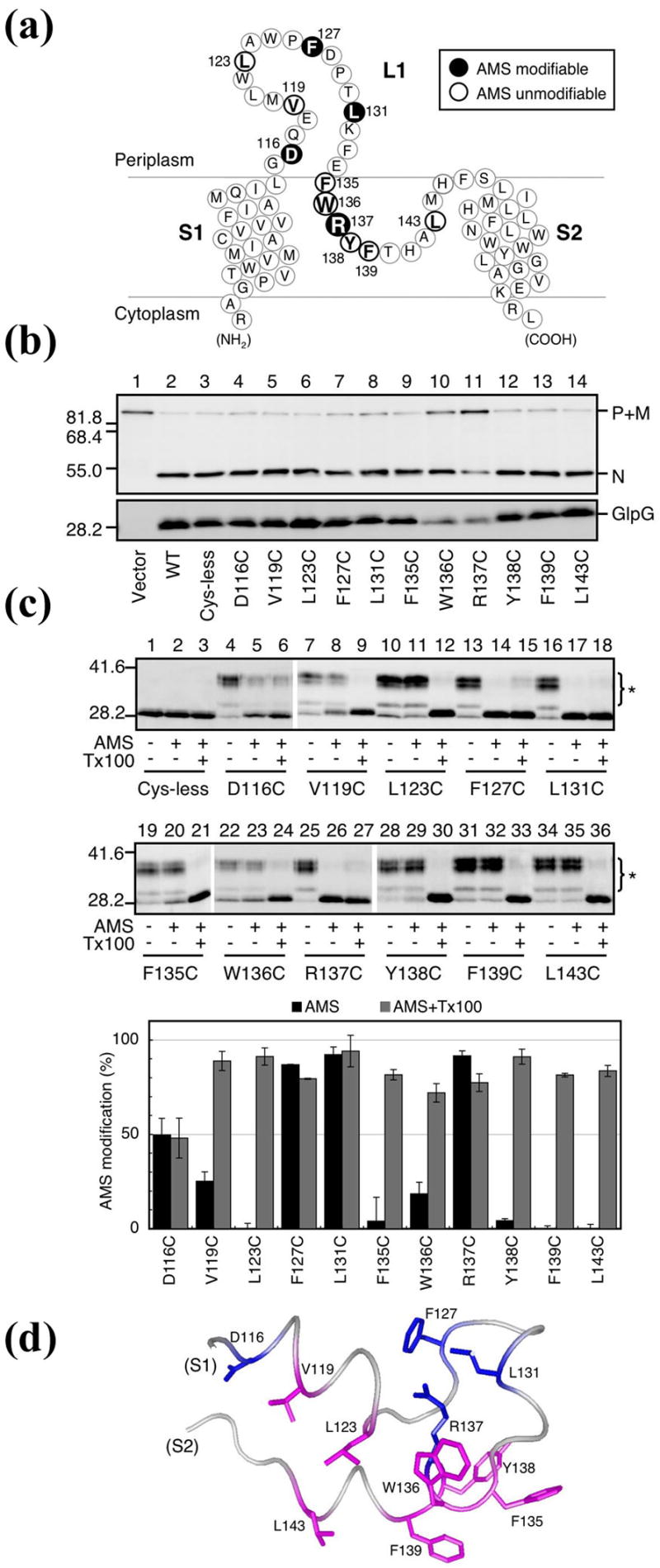
(a) A schematic representation of L1 loop showing the positions of Cys substitutions (bold).
(b) In vivo proteolytic activities of the single Cys derivatives of GlpG-His6-Myc in strain GW345 (ΔglpG)/pSTD1076 (PhoA-LY2-MBP-His6)17. Plasmid-bearing cells were grown at 30 °C in LB containing 1mM IPTG and 1mM cAMP for 15min. Proteins were analyzed by western blot using either anti-PhoA (top) or anti-Myc (bottom) antibodies. P, M and N indicate the precursor form, the mature form (with signal sequence removed) and the N-terminal fragment (after GlpG cleavage) of the model substrate, respectively. Molecular size marker positions (in kDa) are shown on the left.
(c) AMS modification of Cys mutants. Spheroplasts were prepared from strain GW338 (ΔglpG) expressing Cys-less GlpG-His6-Myc or its single Cys derivatives, and treated with 1mM AMS at 24°C for 15min in the presence or absence of 1% Triton X-100 (Tx100). Proteins were precipitated with trichloroacetic acid, solubilized in 1% SDS and 5mM malPEG, and analyzed by western blot (anti-Myc). Asterisks indicate malPEG-modified GlpG-His6-Myc (up-shifted). Prior cysteine alkylation by AMS in intact spheroplasts would prevent malPEG modification of GlpG. The proportions of the AMS-modified form in total GlpG-His6-Myc are graphically depicted (lower panel). Average values from at least two independent experiments are shown with standard deviations. (d) Correlation with the crystal structure8. Residues modified by AMS are shown in blue, and those not modified in magenta.
The WR motif and L1 loop are important for protease activity
To help explain the role of L1 loop in the function of GlpG, we mutated the conserved WR motif near its distal tip individually to alanines (W136A, R137A), and studied the proteolytic activities of these two mutants in intact cells against a model TM substrate (the TM domain 2 of LacY)17. In contrast to results reported previously for drosophila rhomboid-113, both GlpG mutants were proteolytically active (the cysteine mutants described above, W136C and R137C, were also active; Figure 2(b)), indicating that neither Trp-136 nor Arg-137 is essential for the proteolytic reaction in cell membranes (Figure 3(a)). However, when we “titrated” down the amount of expressed GlpG in the cell by using a low copy expression vector, or used a less efficient substrate (the TM domain of drosophila rhomboid-1 substrate, gurken)18, it became also apparent that both W136A and R137A were much less active than the wildtype, suggesting that these residues play a role in fine-tuning the activity of the protease (Figure 3(b)). In both experimental systems, W136A and R137A had a similar level of activity reduction when compared to the wildtype.
Figure 3. Proteolytic activity of the W136A, R137A, ΔWR, and ΔL1 mutants.
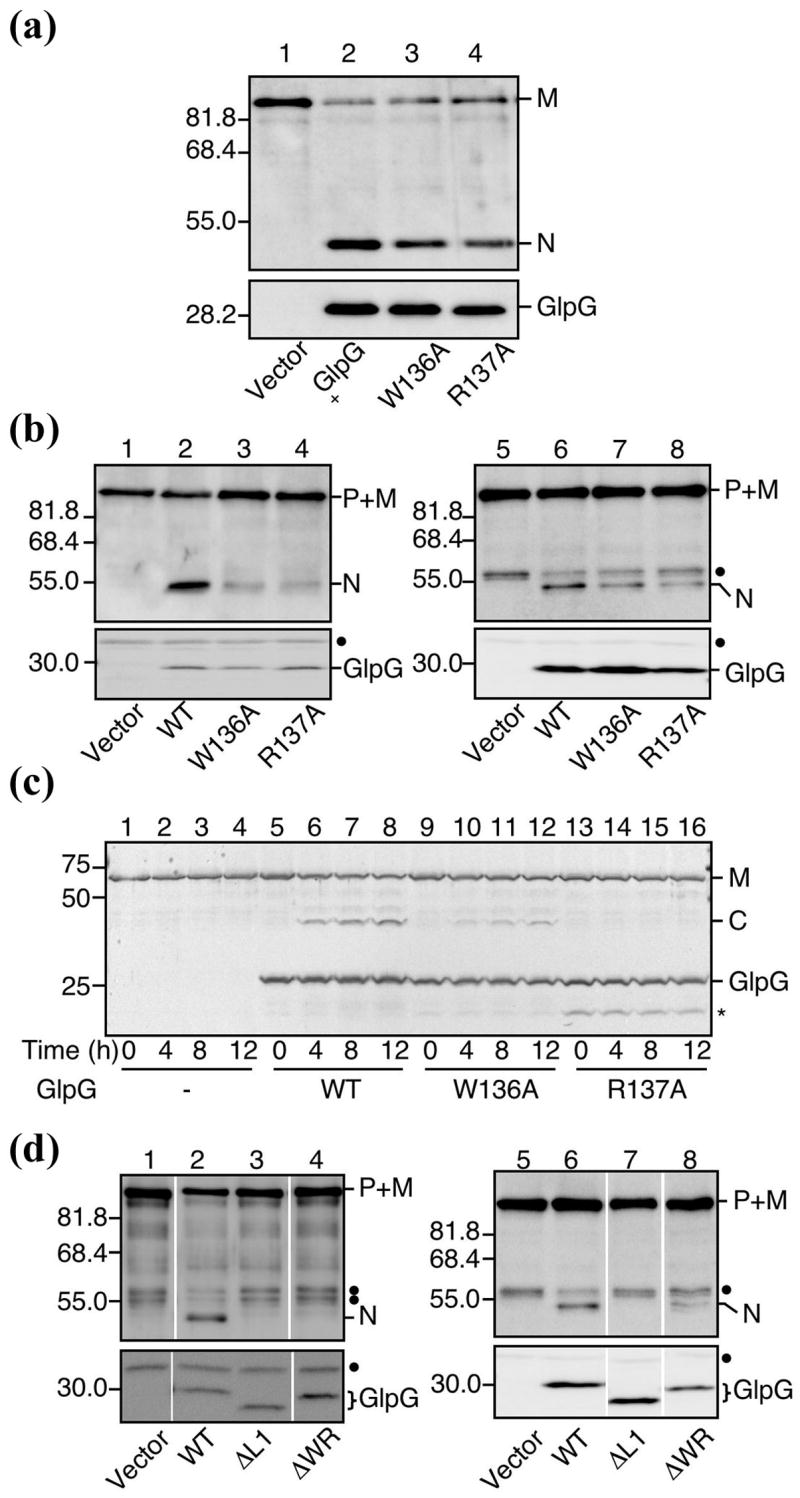
(a) In vivo proteolytic activity of W136A and R137A expressed from a high-copy vector. Strain GW345 (ΔglpG)/pSTD1086 (PhoA-LY2-Spc) was transformed with pTYE007 (Vector, lane 1), pGW55 (GlpG+-His6-Myc, lane 2), pGW366 (GlpG(W136A)-His6- Myc, lane 3) or pGW367 (GlpG(R137A)-His6-Myc, lane 4). Plasmid-bearing cells were grown at 25°C in LB containing 1mM IPTG and 1mM cAMP for 30min, and analyzed by western blot using either anti-PhoA (top) or anti-Myc (bottom).
(b) In vivo activity of W136A and R137A under a low GlpG expression condition (left panel) or with a less efficient substrate (gurken; right panel). Strains GW345 (ΔglpG)/pSTD1076 (PhoA-LY2-MBP-His6) (lanes 1–4) and GW345 (ΔglpG)/pSTD1278 (PhoA-GurkenTM-MBP-His6) (lanes 5–8) were transformed with pMW118 (Vector, lane 1), pSTD1273 (GlpG-His6-Myc, lane 2), pGW380 (GlpG(W136A)-His6-Myc, lane 3), pGW381 (GlpG(R137A)-His6-Myc, lane 4), pTYE007 (Vector, lane 5), pGW55 (GlpG- His6-Myc, lane 6), pGW366 (GlpG(W136A)-His6-Myc, lane 7) or pGW367 (GlpG(R137A)-His6-Myc, lane 8). Plasmid-bearing cells were grown at 30°C and analyzed as in (A). Closed circles indicate non-specific bands.
(c) In vitro activities of purified W136A and R137A. Purified Bla-LY2-MBP-His6 (0.5μM) was mixed with purified GlpG-His6-Myc (2.5 μM, lanes 5–8), GlpG(W136A)-His6-Myc (2.5 μM, lanes 9–12), GlpG(R137A)-His6-Myc (2.5 μM, lanes 13–16), or buffer alone (lanes 1–4) and incubated at 37°C. A portion of the reaction mixture was withdrawn at the indicated time points and subjected to SDS-PAGE and coomassie brilliant blue staining. Asterisk indicates a presumable degradation fragment of GlpG.
(d) In vivo activities of ΔL1 and ΔWR. Strains GW345 (ΔglpG)/pSTD1076 (PhoA-LY2-MBP-His6) (lanes 1–4) and GW345 (ΔglpG)/pSTD1278 (PhoA-GurkenTM-MBP-His6) (lanes 5–8) were transformed with pMW118 (Vector, lane 1), pSTD1273 (GlpG-His6- Myc, lane 2), pSTD1274 (GlpG(ΔL1)-His6-Myc, lane 3), pSTD1276 (GlpG(ΔWR)-His6- Myc, lane 4), pTYE007 (Vector, lane 5), pGW55 (GlpG-His6-Myc, lane 6), pSTD1220 (GlpG(ΔL1)-His6-Myc, lane 7) or pSTD1221 (GlpG(ΔWR)-His6-Myc, lane 8).
We also tried to study and compare their activities in vitro. Unfortunately, the protocol previously developed for purifying the wildtype was apparently not suitable for the mutants17: the purified R137A was completely inactive in detergent solution, whereas W136A retained some activity (Figure 3(c)). We can not reliably compare these activities to that of the wildtype because the activity loss could reflect differences in how each particular protein responds to solubilization by detergents.
We also constructed a deletion mutant (ΔWR) where Trp-136 and Arg-137 have been removed from the protein sequence all together. This is expected to alter the structure of L1 loop more significantly. Surprisingly, the mutant retained some, albeit much less, proteolytic activity as judged by our cell based assays, even when expressed from a low copy vector (Figure 3(d)). Although it demonstrated that the WR motif is not required for a functional protease, this result did reinforce the view that a properly structured L1 loop is needed to achieve optimal activity. When we deleted the entire L1 loop from residues 116 to 142, the mutant protein (ΔL1) was no longer active (Figure 3(d)).
The crystal structure of W136A mutant
To help explain the effect of the mutations, and to gain further insight into the role of L1 loop in protease function, we determined the structure of W136A by x-ray diffraction, at a resolution of 1.7 Å (we have unfortunately not been able to crystallize R137A). To generate an accurate comparison with the wildtype structure, we have crystallized wildtype GlpG from similar conditions and solved its structure at 1.9 Å resolution (Table 1).
Table 1.
Data collection and refinement statistics.
| Data collection | W136A | Wildtype |
|---|---|---|
| Cell dimensions (Å) | a=110.7, c=127.5 | a=111.0, c=129.0 |
| Wavelength (Å) | 1.0809 | 1.0809 |
| a Resolution (Å) | 40.0-1.7 (1.76-1.70) | 40.0-1.9 (1.97-1.90) |
| Observed reflections | 350,382 | 362,789 |
| Unique reflections | 33,056 | 23,768 |
| Redundancy | 10.6 | 15.3 |
| a Completeness (%) | 99.6 (99.5) | 97.9 (89.8) |
| a <I/σ> | 17.2 (4.1) | 18.6 (1.2) |
| a,b Rmerge | 0.064 (0.284) | 0.066 (0.657) |
|
| ||
| Refinement | ||
|
| ||
| Resolution (Å) | 40.0-1.7 | 40.0-1.9 |
| c Rwork/Rfree | 0.200/0.217 | 0.215/0.228 |
| Number of atoms | ||
| Protein | 1,422 | 1,431 |
| Detergent | 175 | 169 |
| Water | 127 | 79 |
| B-factors | ||
| Protein | 24.2 | 32.2 |
| Detergent | 49.2 | 59.6 |
| Water | 42.1 | 46.9 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.006 | 0.007 |
| Bond angle (°) | 1.19 | 1.15 |
Highest resolution shell is shown in parentheses.
Rmerge = Σ | Ii − <I> |/Σ |Ii
Rwork = Σ | Fo − Fc|/Σ Fo. Rfree is the cross-validation R factor for the test set of reflections (10% of the total) omitted in model refinement.
The mutant structure was largely identical to that of the wildtype: their backbones were superimposable, with an r.m.s. deviation of less than 0.1 Å; the active site, the L5 cap, and their surroundings were also similar. The conformation of the L1 loop was not affected by the mutation either, suggesting that, although Trp-136 appears to occupy a strategically important position, and participates in several hydrogen bonding and Van der Waals interactions with the rest of the loop, it does not appear to be required for the folding and maintenance of the loop structure itself. Our current finding also indicates that the structure of L1 is quite rigid, which would seem unlikely if L1 functions as a movable gate to control the lateral entry of TM substrates8,11. Examples of other dynamic protein structures show that such movable elements are usually quite flexible by themselves, and that their conformations are often sensitive to external perturbations such as crystal packing or mutation. The conformational rigidity of the L1 loop is in clear contrast with the flexibilities observed for the L5 cap and TM helix S59,10,12, which further reinforces the view that substrate access to active site is probably from the side opposite of the L1 loop, likely through the side portal blocked by the cap. Recent mutagenesis experiments also seemed to be consistent with these models15.
With the indole ring of Trp-136 removed, the void left behind attracted six water molecules (Figure 4(a)), which were stabilized by a chain of hydrogen bonds with each other, and with exposed polar atoms on the protein (Figure 4(b)). This situation has similarities to internal cavities created by mutations in the lysozyme that formed new solvent binding sites19. The presence of new water molecules indicated that the protein structure underneath the fused aromatic ring of Trp-136 is hydrophilic, and only after being masked by the tryptophan, could it be positioned deeper into the hydrophobic region of the membrane (Figure 4(c) and (d)). This is consistent with biochemical data showing that cysteine introduced to position 137 (behind the tryptophan) can react with AMS (a water-requiring reaction), whereas cysteine at position 136 (exposed to lipid) can not (Figure 2(c)). The upper edge of Trp-136 indole ring (with its amide group hydrogen bonded to a carbonyl oxygen located higher) defines the boundary for the hydrophobic core region of the membrane (see below) (Figure 4(c)). With the indole group removed, the exposed polar surface is no longer compatible with its buried location (Figure 4(d)). Based on these observations, we believe that the reduced activity of W136A was caused indirectly by a change in interactions between the mutant protein and the surrounding lipid (see further discussion below).
Figure 4. The crystal structure of W136A mutant.
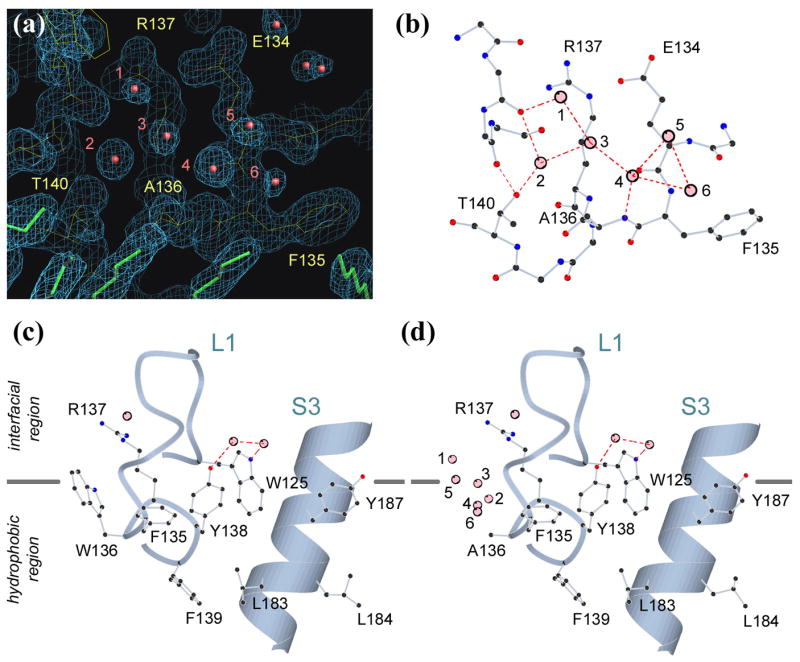
(a) Electron density, contoured at 1.5σ level, at the site of mutation. Water molecules are shown in red, and detergents in green.
(b) The water molecules (numbered 1 to 6) substituting the indole ring of Trp-136 are stabilized by a network of hydrogen bonds.
(c) The structure of the wildtype showing the interfacial location of Trp-136.
(d) After mutation, the protein surface is no longer compatible with its buried location.
The hydrophobic belt around GlpG is only 20 Å wide
A slight advantage achieved by the higher resolution study is probably the better distinction between non-protein electron densities that corresponded to either water molecules or detergents: the density for water is usually roundish, and always near a polar atom for hydrogen-bonding, whereas detergents have well-defined long shapes, with a few clearly showing the sugar ring at one end. The polar surfaces of the membrane protein are almost completely covered with immobilized waters. Based on the high-resolution structures generated in this study, we attempted to construct a picture of GlpG in natural membranes, and our final model has the following features: (i) the two layers of externally-bound water are roughly parallel to the assumed membrane plane (Figure 5(a)); (ii) all the polar side chains, as well as the externally-bound water molecules, are located outside the hydrophobic core of the membrane; (iii) the number of tryptophan and tyrosine residues at the membrane:water interface is maximized with most of them oriented such that their aromatic rings are below the hydrophobic boundary, and the polar atoms (amide nitrogen, phenol oxygen) surface to the interfacial region to form hydrogen bonds (e.g., Trp-125, 136 and Tyr-138, 187 in Figure 4(c))20.
Figure 5. The model of membrane-embedded GlpG.

(a) The Cα trace of the protein is shown in yellow, externally bound water shown in red, water inside the membrane protein in blue. The water molecules substituting Trp-136 in the mutant are shown in white. The two horizontal lines mark the boundaries of the hydrophobic core of the membrane around the protease.
(b) A histogram of the number of water molecules observed across the bilayer in the same color scheme as in (a).
(c) The location of various structural elements important for protease function within the lipid bilayer. Parts of L1 (marked by *) and TM helix S2 (**) are omitted to show the internal active site.
We drew several structural conclusions from the refined model. First, the hydrophobic TM area of GlpG is quite narrow. The distance between the two hydration shells, which corresponds roughly to the width of the hydrophobic belt around the membrane protein, is only 20 Å (Figure 5(b)). Therefore, the lipid bilayer, whose hydrocarbon region usually spans a distance of 27 Å, must deform significantly to match the “hydrophobic thickness” of GlpG21,22. Secondly, the lower portion of L1 loop is completely buried in the membrane, with Trp-136 snorkeling up to the surface, and with Phe-139 reaching the midline of the bilayer (Figure 5(c)). The buried part of L1 will probably displace lipid in the outer leaflet of the bilayer, complicating the tension within the deformed membrane. Thirdly, the catalytic serine (Ser-201) is merely 3 Å below the hydrophobic boundary, which implies that catalysis takes place very close to the interface. Finally, the backbone of the L5 cap is above the boundary, with Phe-245 dipping into the hydrophobic region and blocking the side portal to the active site. It should be obvious that L5 cap, as well as other parts of the protein that protrude out of the hydrophobic boundaries, is still within the interfacial region of the membrane, which has an additional thickness of about 10–15 Å, and is enriched with polar head groups of the lipid.
Discussion
In this study we combined biochemical and functional characterizations with structural analysis to investigate the mechanism of rhomboid intramembrane protease GlpG. A major surprise of our findings was that the alanine substitution of a conserved tryptophan (Trp-136) in L1 loop, which significantly reduced the proteolytic activity of GlpG in intact cells, did not appear to alter the structure of the protease at all. The only visible difference between the wildtype and mutant enzymes was confined to a small surface area, at the distal tip of L1 loop away from protease active site and its cap. The difference was expected to influence how protease interacts with lipids, but how this effect caused a reduction in protease activity was not immediately clear. In order to explain this result, we re-analyzed the crystal structure, now better refined beyond 2 Å resolution. We discovered that the hydrophobic belt around the protease was only 20 Å wide, which is probably a common feature for all rhomboid proteases because their TM segments are of similar lengths. We examine below the mechanistic implications of this structural finding, and try to rationalize why the enzyme mechanism of GlpG is sensitive to perturbations in protein:lipid interactions.
Lipid bilayers are dynamic structures. Although it is generally assumed that their hydrophobic core region is about 30 Å thick22, they deform significantly around membrane proteins so that their core can match the hydrophobic TM portion of the protein21. Our refined structural analysis indicated that, immediately surrounding GlpG, the lipid bilayers are probably thinner (20 Å). When substrate diffuses into these compressed regions of the bilayer to make contact with the protease, its membrane-embedded position will become destabilized and the ends of its TM helix will be under pressure to stick out of the membrane. It is therefore tempting to speculate that this may provide the physical forces to push (and unfold) the cleavage site, which is normally just a few residues below the membrane surface, to the polar interfacial region of the membrane or directly into the active site of the protease. The fact that rhomboid proteases can apparently cleave substrates not only within the TM domains14,15, but also in the juxtamembrane region17,18,23, or at the membrane:water boundary24, seems to suggest a universal mechanism where the cleavage site, regardless of its original location, enters the protease primarily from the interfacial region of the membrane, instead of from within its hydrophobic core (Figure 6). This possibility would reinforce the general importance of L5 cap in controlling substrate access to the active site (Figure 5(c))12. Our previous observation that the cleavage site in the TM domain of gurken can spontaneously partition into aqueous solution also raises the possibility that such a property could be an important factor determining which TM helices are likely to be recognized and cleaved by rhomboid intramembrane proteases18.
Figure 6.
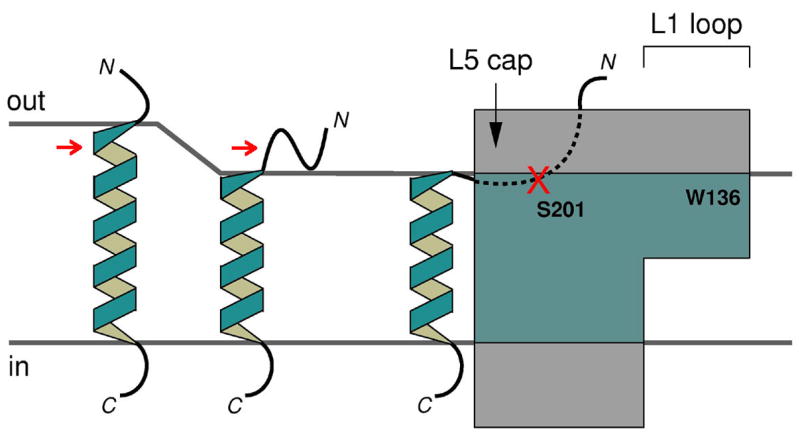
Proposed model for substrate entry into rhomboid protease from the interfacial region of the membrane. The portions of substrate and protease that are embedded in the hydrophobic core region of the membrane are shown in blue. The red arrows point at the scissile bond, initially buried, but exposed after substrate gets close to the protease.
In contrast to soluble proteases that operate in a mostly homogeneous environment, intramembrane proteases are restricted in cell membranes whose physical properties change drastically across the bilayer. Our analysis raises the possibility that various structural elements of GlpG, evolved to execute specific functions, need to be kept at the right depth in the bilayer in order to function properly (Figure 5(c)), and that active site capping and catalysis both need to occur within a very narrow zone at the interface. Therefore, it may be extremely critical for this enzyme to be able to precisely maintain, and probably adjust, its orientation during the processes of substrate binding and product release. The unique property and half-submerged location of L1 loop, as well as its rigid attachment to the main body of the protease, all seem to suggest that L1 could play a major role in fulfilling this requirement, a role that would also explain why the enzyme activity is very sensitive to mutations within L1 loop despite their distance from the active site.
Like rhomboid proteases, S2P cleaves substrate also near the end of the TM helices. Although the structure and mechanism of S2P are not yet known, sequence analysis and mutagenesis studies have identified a conserved asparagine-proline motif within substrate TM region to be essential for cleavage25. This discovery has led to the proposal that the Asn-Pro motif may cause part of the substrate helix to unwind, pushing the scissile bond to membrane surface for cleavage25. Therefore, if their proposal on S2P and our current model of GlpG are correct, there seems to be a common mechanistic pattern, at least for those that cleave at the end of TM helices, where substrate TM helices need to first partition out of the membrane before being cleaved. For rhomboid proteases, this process is probably facilitated by three structural factors: first, the active site of the protease is located very close to membrane:water interface; second, the surface property of the protease is such that the lipid bilayer around the protein is compressed, which would force the end of substrate helix out of the membrane; third, the cleavage site within the substrate TM region may also have an intrinsic propensity to partition into an aqueous environment.
Materials and Methods
Bacterial strains and plasmids
GW345 is a ΔglpG derivative of CU14126 and constructed by introduction of glpG::kan from JWK5687 (obtained from NBRP (NIG, Japan):E. coli) into CU141 by P1 transduction and following removal of the kan cassette according to the method described in reference 27. GW338 was described previously17.
pGW55, pSTD1086, pTYE007 were described previously17,23,28. Plasmids encoding a single Cys derivative of GlpG-His6-Myc were constructed essentially as described previously18. pSTD1076 was constructed by ligating a KpnI-HindIII lacYTM2-malE-his6 fragment of pGW86 with pSTD1075 that had been digested with these enzymes23. pSTD1273 was constructed by cloning a SacI-SpeI glpG-his6-myc fragment of pGW55 into a pSC101-based vector, pMW118 (Nippon Gene). pSTD1278 was constructed as follows. First, a KpnI-SpeI lacYTM2 fragment of pSTD1076 was replaced by a KpnI-SpeI gurkenTM fragment of pGW14017. Then, a SacI-HindIII fragment of the resulting plasmid encoding pPhoA-GurkenTM-MBP-His6 was cloned into the same sites of pSTD68929. Plasmids encoding a GlpG derivative carrying a mutation or deletion in the L1 region were constructed by site-directed mutagenesis and recloning as described previously17.
Biochemical and functional analysis
AMS and malPEG modification of single Cys derivatives of GlpG-His6-Myc was carried out essentially as described previously18. To assay in vivo GlpG activity, cells of GW345 carrying an appropriate plasmid were grown at 25 or 30°C in LB to an early log phase and induced with 1mM IPTG and 1mM cAMP for the indicated time periods. Proteins were analyzed by western blot with anti-Myc or anti-PhoA antibodies. To study the in vitro activity, GlpG-His6-Myc and its derivatives were purified from cells of SN140/pGW55, SN140/pGW366 and SN140/pGW367, and Bla-LacYTM2-MBP-His6 was purified from cells of GW408/pGW88. In vitro activities of the GlpG derivatives were assayed as described previously17.
Crystallographic analysis
The TM core domains of GlpG were purified as described previously8. The W136A mutant was crystallized at room temperature by hanging-drop vapor diffusion method from a 5mg/ml membrane protein solution in 10mM Tris-HCl (pH 7.6) and 20mM nonylglucoside, over a reservoir solution of 3.5M NH4Cl and 100mM Tris-HCl (pH 8.5). The wildtype was crystallized from a similar condition (1.5M NH4Cl, 100mM Bis-Tris at pH 7.0). The crystals were cryo-protected by step-wise transferring them to an artificial mother liquor containing 25% glycerol before flash-freezing in liquid nitrogen. X-ray diffraction data were collected at 100K on NSLS beamline X29 at Brookhaven National Laboratory. All diffraction images were processed by HKL200030. Since these crystals (space group R32) were isomorphous to a previous crystal form (obtained from a different condition)8, the published wildtype structure (PDB: 2IC8), stripped of detergent and water molecules and rigid-body refined, was directly used as the starting model for fo-fc and 2fo-fc map calculations. The resolution and quality of the current data, 1.7 Å for W136A and 1.9 Å for the wildtype, produced very clear electron density features for the protein, and for the bound solvent molecules (detergent, water). Based on these maps, detergents were added using O31. The protein:detergent models were improved by iterative rounds of map inspection, manual adjustment, and conjugate gradient minimization and B-factor refinement by CNS32. Waters were automatically picked by CNS during the last two rounds of improvement, and were individually examined (some were deleted because they fell into short stretches of density that corresponded to disordered detergents).
The illustrations in Figure 1, Figure 4(b), Figure 4(c), Figure 4(d) and Figure 5(c) were generated by MOLSCRIPT33.
Acknowledgments
We thank H. Robinson and A. Saxena at NSLS X29 for assistance during x-ray diffraction data collection. Financial support for the beamline comes principally from US Department of Energy, and from the National Institutes of Health. This work was supported by a New Scholar Award in Aging from the Ellison Medical Foundation (Y.H.), a gift from the Neuroscience Education and Research Foundation (Y.H.), NIH GM082839 (Y.H.), and by grants from CREST, Japan Science and Technology Corporation (Y.A.), and from Japan Society for the Promotion of Science (JSPS) (Y.A.).
Abbreviations used
- TM
transmembrane
Footnotes
Data deposition: The atomic coordinates and structure factors will be deposited in the protein data bank.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100:391–398. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- 2.Haass C, Steiner H. Alzheimer disease gamma-secretase: a complex story of GxGD-type presenilin proteases. Trends Cell Biol. 2002;12:556–562. doi: 10.1016/s0962-8924(02)02394-2. [DOI] [PubMed] [Google Scholar]
- 3.Weihofen A, Martoglio B. Intramembrane-cleaving proteases: controlled liberation of proteins and bioactive peptides. Trends Cell Biol. 2003;13:71–78. doi: 10.1016/s0962-8924(02)00041-7. [DOI] [PubMed] [Google Scholar]
- 4.Freeman M. Proteolysis within the membrane: rhomboids revealed. Nat Rev Mol Cell Biol. 2004;5:188–197. doi: 10.1038/nrm1334. [DOI] [PubMed] [Google Scholar]
- 5.Wolfe MS, Kopan R. Intramembrane proteolysis: theme and variations. Science. 2004;305:1119–1123. doi: 10.1126/science.1096187. [DOI] [PubMed] [Google Scholar]
- 6.Ha Y. Structural principles of intramembrane proteases. Curr Opin Struct Biol. 2007 doi: 10.1016/j.sbi.2007.06.010. in the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lemberg MK, Freeman M. Mol Cell. 2007 doi: 10.1016/j.molcel.2007.12.003. in the press. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Zhang Y, Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature. 2006;444:179–183. doi: 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- 9.Wu Z, Yan N, Feng L, Oberstein A, Yan H, Baker RP, Gu L, Jeffrey PD, Urban S, Shi Y. Structural analysis of a rhomboid family intramembrane protease reveals a gating mechanism for substrate entry. Nat Struct Mol Biol. 2006;13:1084–1091. doi: 10.1038/nsmb1179. [DOI] [PubMed] [Google Scholar]
- 10.Ben-Shem A, Fass D, Bibi E. Structural basis for intramembrane proteolysis by rhomboid serine proteases. Proc Natl Acad Sci USA. 2007;104:462–466. doi: 10.1073/pnas.0609773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lemieux MJ, Fischer SJ, Cherney MM, Bateman KS, James MN. The crystal structure of the rhomboid peptidase from Haemophilus influenzae provides insight into intramembrane proteolysis. Proc Natl Acad Sci USA. 2007;104:750–754. doi: 10.1073/pnas.0609981104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Ha Y. Open-cap conformation of intramembrane protease GlpG. Proc Natl Acad Sci USA. 2007;104:2098–2102. doi: 10.1073/pnas.0611080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Urban S, Lee JR, Freeman M. Drosophila rhomboid-1 defines a family of putative intramembrane serine proteases. Cell. 2001;107:173–182. doi: 10.1016/s0092-8674(01)00525-6. [DOI] [PubMed] [Google Scholar]
- 14.Lemberg MK, Menendez J, Misik A, Garcia M, Koth CM, Freeman M. Mechanism of intramembrane proteolysis investigated with purified rhomboid proteases. EMBO J. 2005;24:464–472. doi: 10.1038/sj.emboj.7600537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baker RP, Young K, Feng L, Shi Y, Urban S. Enzymatic analysis of a rhomboid intramembrane protease implicates transmembrane helix 5 as the lateral substrate gate. Proc Natl Acad Sci USA. 2007;104:8257–8262. doi: 10.1073/pnas.0700814104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Urban S, Wolfe MS. Reconstitution of intramembrane proteolysis in vitro reveals that pure rhomboid is sufficient for catalysis and specificity. Proc Natl Acad Sci USA. 2005;102:1883–1888. doi: 10.1073/pnas.0408306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maegawa S, Ito K, Akiyama Y. Proteolytic action of GlpG, a rhomboid protease in the Escherichia coli cytoplasmic membrane. Biochemistry. 2005;44:13543–13552. doi: 10.1021/bi051363k. [DOI] [PubMed] [Google Scholar]
- 18.Maegawa S, Koide K, Ito K, Akiyama Y. The intramembrane active site of GlpG, an E. coli rhomboid protease, is accessible to water and hydrolyses an extramembrane peptide bond of substrates. Mol Microbiol. 2007;64:435–447. doi: 10.1111/j.1365-2958.2007.05679.x. [DOI] [PubMed] [Google Scholar]
- 19.Xu J, Baase WA, Quillin ML, Baldwin EP, Matthews BW. Structural and thermodynamic analysis of the binding of solvent at internal sites in T4 lysozyme. Protein Sci. 2001;10:1067–1078. doi: 10.1110/ps.02101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Killian JA, von Heijne G. How proteins adapt to a membrane-water interface. Trends Biochem Sci. 2000;25:429–434. doi: 10.1016/s0968-0004(00)01626-1. [DOI] [PubMed] [Google Scholar]
- 21.Engelman DM. Membranes are more mosaic than fluid. Nature. 2005;438:578–580. doi: 10.1038/nature04394. [DOI] [PubMed] [Google Scholar]
- 22.Bretscher MS, Munro S. Cholesterol and the Golgi apparatus. Science. 1993;261:1280–1281. doi: 10.1126/science.8362242. [DOI] [PubMed] [Google Scholar]
- 23.Akiyama Y, Maegawa S. Sequence features of substrates required for cleavage by GlpG, an Escherichia coli rhomboid protease. Mol Microbiol. 2007;64:1028–1037. doi: 10.1111/j.1365-2958.2007.05715.x. [DOI] [PubMed] [Google Scholar]
- 24.Stevenson LG, Strisovsky K, Clemmer KM, Bhatt S, Freeman M, Rather PN. Rhomboid protease AarA mediates quorum-sensing in Providencia stuartii by activating TatA of the twin-arginine translocase. Proc Natl Acad Sci USA. 2007;104:1003–1008. doi: 10.1073/pnas.0608140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ye J, Davé UP, Grishin NV, Goldstein JL, Brown MS. Asparagine-proline sequence within membrane-spanning segment of SREBP triggers intramembrane cleavage by site-2 protease. Proc Natl Acad Sci USA. 2000;97:5123–5128. doi: 10.1073/pnas.97.10.5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akiyama Y, Ogura T, Ito K. Involvement of FtsH in protein assembly into and through the membrane. I. Mutations that reduce retention efficiency of a cytoplasmic reporter. J Biol Chem. 1994;269:5218–5224. [PubMed] [Google Scholar]
- 27.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akiyama Y, Yoshihisa T, Ito K. FtsH, a membrane-bound ATPase, forms a complex in the cytoplasmic membrane of Escherichia coli. J Biol Chem. 1995;270:23485–23490. doi: 10.1074/jbc.270.40.23485. [DOI] [PubMed] [Google Scholar]
- 29.Kanehara K, Ito K, Akiyama Y. YaeL proteolysis of RseA is controlled by the PDZ domain of YaeL and a Gln-rich region of RseA. EMBO J. 2003;22:6389–6398. doi: 10.1093/emboj/cdg602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 31.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 32.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 33.Kraulis PJ. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 34.Koonin EV, Makarova KS, Rogozin IB, Laetitia D, Letellier MC, Pellegrini L. The rhomboids: a nearly ubiquitous family of intramembrane serine proteases that probably evolved by multiple ancient horizontal gene transfers. Genome Biol. 2003;4:R19. doi: 10.1186/gb-2003-4-3-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]