

Abstract
Background
Atrial fibrosis is an important substrate in atrial fibrillation (AF), particularly in the setting of structural heart disease. In a canine model, congestive heart failure (CHF) produces significant atrial fibrosis and the substrate for sustained AF. This atrial remodeling is a potential therapeutic target. The objective of the present study is to evaluate the effects of the antifibrotic drug pirfenidone (PFD) on arrhythmogenic atrial remodeling in a canine CHF model.
Methods and Results
We studied 15 canines, divided equally into 3 groups: control, CHF canines not treated with PFD, and CHF canines treated with PFD. CHF was induced by ventricular tachypacing (220 bpm for 3 weeks), and oral PFD was administered for the 3-week pacing period. We performed electrophysiology and AF vulnerability studies, atrial fibrosis measurements, and atrial cytokine expression studies. Only canines in the untreated CHF group developed sustained AF (>30 minutes, 4 of 5 canines; P<0.05). Treatment of CHF canines with PFD resulted in an attenuation of arrhythmogenic left atrial remodeling, with a significant reduction in left atrial conduction heterogeneity index (median [25% to 75% interquartile range] 4.96 [3.53 to 5.64] versus 2.52 [2.11 to 2.82], P<0.01; pacing cycle length 300 ms), left atrial fibrosis (16.0% [13.0% to 17.5%] versus 8.7% [5.7% to 10.6%], P<0.01), and AF duration (1800 [1020 to 1800] seconds versus 6 [5 to 22] seconds, P<0.01). Immunoblotting studies demonstrated the drug’s effects on multiple cytokines, including a reduction in transforming growth factor-β1 expression.
Conclusions
Treatment of CHF canines with PFD results in significantly reduced arrhythmogenic atrial remodeling and AF vulnerability. Pharmacological therapy targeted at the fibrotic substrate itself may play an important role in the management of AF.
Keywords: drugs, heart failure, atrial fibrillation
Atrial fibrillation (AF) is a common clinical arrhythmia, and its management remains a challenge. Correlative data in biopsy and autopsy specimens from patients with AF have uncovered the presence of atrial fibrosis.1,2 Animal studies have shown atrial fibrosis to be an important substrate for AF, especially in the setting of structural heart disease.3-6 Pharmacological therapy targeted at the substrate may reduce AF vulnerability.
Pirfenidone (PFD) is an orally active, antifibrotic drug shown to significantly attenuate and potentially reverse collagen deposition in various experimental animal models.7-12 We hypothesize that PFD will attenuate atrial fibrosis that is characteristic of canines with congestive heart failure (CHF). The objective of the present study was to evaluate the effects of PFD on arrhythmogenic atrial remodeling and AF vulnerability in canines with ventricular tachypacing (VTP)–induced CHF.
Methods
Animal Model
This study was approved and monitored by the Laboratory Animal Resource Center at the University of California, San Francisco, and conformed to the “Guide for the Care and Use of Laboratory Animals” (National Institutes of Health publication No. 85-23, revised 1996). Fifteen adult mongrel canines (weight 25 to 32 kg) divided into 3 groups (n=5 per group) were studied: (1) control (normal animals neither paced nor treated with PFD); (2) HF (animals with VTP-induced CHF not treated with PFD); and (3) HF+PFD (animals with VTP-induced CHF treated with PFD). Canines in the paced groups underwent placement of a single-chamber pacemaker with the pacing lead positioned in the right ventricular apex, followed by radiofrequency ablation of the AV junction to create complete heart block. Canines in the HF and HF+PFD groups underwent 3 weeks of VTP at 220 bpm. For the animals in the HF+PFD group, oral PFD (800 mg 3 times per day; InterMune, Brisbane, Calif) was started 2 days before the initiation of pacing and was discontinued >6 half-lives (24 hours) before the open-chest study. On follow-up, the animals underwent electrophysiological study (EPS), as described previously.13,14 Atrial tissues were processed for immunoblotting and histology studies.
Monitoring of the Congestive Heart Failure Model
The paced groups underwent transesophageal echocardiography at the time of pacemaker implantation and at follow-up. Follow-up studies included transthoracic echocardiography, ECG monitoring, and physical examination on a weekly basis. CHF was established by clinical signs (eg, oral mucous membrane color, lethargy, and edema) and the presence of left ventricular (LV) dysfunction on echocardiography. Left atrial (LA) size was determined by measuring the LA area by planimetry from 2D echocardiographic images (2-chamber view during diastole). LV systolic function was determined by measuring LV fractional shortening (LVFS) at the level of the papillary muscle. Two repeated measurements were made for LA area and LVFS, and the mean was used for analysis.
Electrophysiological Study
During EPS, animals were anesthetized with isoflurane and mechanically ventilated. The pacemaker rate was set at 80 bpm for the EPS. The chest was opened with a midline sternotomy. A pericardial cradle was created, and 4 high-density plaques were placed on the epicardial surface of the atria (LA free wall, LA Bachmann’s bundle, right atrial [RA] free wall, and RA Bachmann’s bundle; a total of 512 unipolar electrodes with an interelectrode distance of 3.5 mm), similar to that described previously.13 Signals were sampled at 2 kHz and stored with the UnEmap system (University of Auckland, New Zealand). Neighboring plaque electrode pairs were used for bipolar stimulation at twice the diastolic threshold. Effective refractory periods (ERPs) were measured at 12 atrial sites (6 in the LA, 6 in the RA) with the single extrastimulus protocol at an 8-beat drive train with basic cycle lengths (BCLs) of 200, 300, and 400 ms. ERPs were determined in 2-ms increments. Dispersion of repolarization was taken as the coefficient of variation (SD/mean×100%) of the ERP at each BCL. During stimulation of the contralateral atrium, conduction velocity (CV) was calculated between pairs of plaque electrodes perpendicular to the activation wavefront with a customized software program.15 As described previously,15 conduction heterogeneity was quantified as the phase difference (milliseconds/millimeters), defined as the average difference in activation time between a plaque electrode and that of its neighbors normalized by the interelectrode distance. Frequency histograms were constructed for the phase differences within an atrial region. The histograms were summarized as (1) median phase (P50) and (2) 5th and 95th percentile phase, or P5 and P95 of the distribution, respectively. To quantify conduction heterogeneity, we calculated the conduction heterogeneity index, defined as the absolute conduction heterogeneity normalized by the median phase, or P95−5/P50. The same pacing signals were used to calculate CV and phase.
AF vulnerability was assessed by single-extrastimulus pacing and an atrial burst pacing protocol that entailed pacing at 1 LA site and 1 RA site. A total of 16 burst stimulations were performed for each animal, with each atrial site receiving 8 burst pacings (4 for 6 seconds, 4 for 12 seconds) at a cycle length of 50 ms and a stimulus output of 0.5 V plus twice the diastolic threshold. AF was considered sustained if the induced episode lasted >30 minutes. If sustained AF was achieved at any point in the protocol, further testing was not performed, and the longest AF duration was taken as 1800 seconds.
Histological, Immunoblotting, and Immunohistochemistry Studies
Atrial tissue samples were fixed in 10% neutral buffered formalin. The samples were embedded in paraffin and sectioned (4- to 5-μm thick) and were stained with hematoxylin and eosin, Masson’s trichrome, or Sirius red. Fibrosis was quantified from the digital photomicrographs of the Sirius red–stained sections as described previously.6 Areas containing blood vessels and perivascular interstitial cells were excluded from fibrosis quantification.
Atrial tissue samples were also frozen in liquid nitrogen for immunoblotting studies of cytokines using standard techniques. Cytokines of interest included those that have been reported to potentially play an important role in the development of AF substrate (ie, mediators of inflammation or fibrosis development).5,16-24 After homogenization in solubilizing buffer, atrial tissue that contained an equal amount of total protein (determined by DC Protein Assay, Bio-Rad, Hercules, Calif) was electrophoresed on a 4% to 20% Tris-glycine gel and then transferred onto a nitrocellulose filter. Nonspecific binding sites were blocked with 4% BSA, and the filter was incubated with diluted antibody and matched secondary antibody (Chemicon, Temecula, Calif). Protein bands were analyzed with a chemiluminescence detection method (NEN Life Science, Boston, Mass). Densitometry was used to quantify protein band signals from all animals under investigation. Quantitative protein levels were normalized to β-actin levels in each specimen run concomitantly in each lane.
Atrial distribution of the gap junction proteins connexin 43 (C×43) and 40 (C×40) was studied with immunohistochemistry techniques, as described previously.13,25 Specimens were incubated with mouse monoclonal antibody against C×40 and rabbit polyclonal antibody against C×43 (Dako, Glostrup, Denmark) overnight at 4°C. Incubation with FITC-labeled goat anti-rabbit (for C×43) and Texas Red–labeled donkey anti-mouse (for C×40) antibodies (Jackson ImmunoResearch Laboratories, West Grove, Pa) was performed. The specimens were processed and analyzed with fluorescent microscopy.
Statistical Analysis
Intragroup data variables were compared with the Friedman test. Data variables among the 3 groups (intergroup) were compared with the Kruskal-Wallis test, and if P was <0.05, follow-up comparisons of the different groups were done with the Mann-Whitney test. Proportion comparisons were performed with the Fischer exact test. Results were presented as median (25% to 75% interquartile range), and P<0.05 was deemed statistically significant.
The authors had full access to the data and take full responsibility for their integrity. All authors have read and agree to the manuscript as written.
Results
LV Function and LA Dilatation
At the time of follow-up, there was no significant difference in arterial blood pressure (BP) between the paced groups (systolic BP 95 [92–101] mm Hg for HF versus 96 [93–99] mm Hg for HF+PFD, P=NS; diastolic BP 59 [52–64] mm Hg for HF versus 61 [55–63] mm Hg for HF+PFD, P=NS).
LVFS was similar in all 3 groups at baseline (41% [38% to 43%] in control, 39% [36% to 42%] in HF, and 40% [36% to 44%] in HF+PFD, P=NS). After 3 weeks of pacing, LVFS was markedly reduced for the HF (ΔLVFS= −63% [−58% to −73%], P<0.01) and HF+PFD (ΔLVFS= −65% [−57% to −75%], P<0.01) groups compared with baseline. The LVFS at each weekly time point was similar in the paced groups.
At baseline, LA area was similar in all groups (8.6 cm2 [8.0 to 9.0 cm2] in control, 8.7 cm2 [7.9 to 9.2 cm2] in HF, and 8.9 cm2 [8.2 to 9.1 cm2] in HF+PFD, P=NS). For the paced groups, LA area (Figure 1) increased significantly after 1 week of VTP, and this increase was progressive. At each weekly time point, the increase in LA area was slightly greater in the HF+PFD group than in the HF group, although the difference between the 2 paced groups was not statistically significant.
Figure 1.
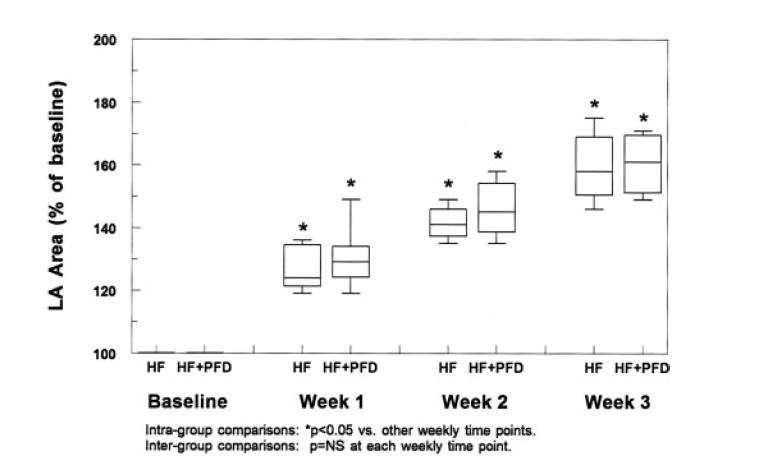
LA area measurements at baseline and percent change from baseline in the HF and HF+PFD groups. There was a progressive increase in LA area from baseline in both groups. At each weekly time point, the percent change from baseline was similar between the groups. (Box-and-whiskers plots show 5th-25th-50th-75th-95th percentile values from bottom to top.)
Clinical signs of CHF were similar between the 2 paced groups. All animals in the drug-treated group received all scheduled doses of the drug. There were no overt signs of intolerance to the drug.
Electrophysiological Parameters
Atrial ERP findings are shown in Figures 2A and 2B. In the LA, CHF resulted in a trend toward longer ERPs at all BCLs compared with controls. ERPs in the HF+PFD group were significantly longer than those in controls (P<0.04 for all BCLs) but did not reach statistical significance compared with those in the HF group. In the RA, ERPs were similar in all groups.
Figure 2.
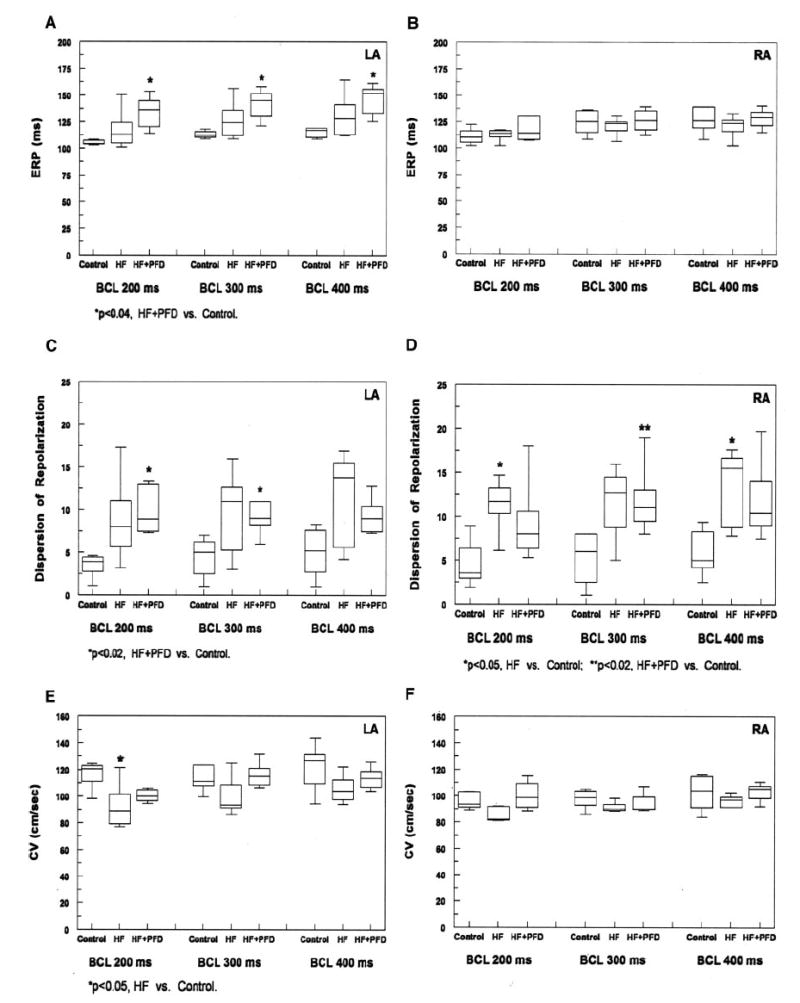
ERP (A, B), DOR (C, D), and CV (E, F) findings in both atria at 3 BCLs. A, There was a trend toward an increase in LA ERPs in the HF group compared with controls at all BCLs. PFD treatment resulted in a trend toward a further increase in LA ERPs compared with the HF group at all BCLs, and this increase reached statistical significance compared with controls (P<0.04 for all BCLs). B, RA ERPs were similar at all BCLs (P=NS). C, CHF resulted in a trend toward an increase in LA DOR compared with controls. LA DOR in the HF+PFD group was similar to that in the HF group and was significantly greater than that in controls at BCLs of 200 and 300 ms (P<0.02). D, In the RA, CHF resulted in an increase in DOR at all BCLs compared with controls, and this increase reached statistical significance at BCLs of 200 and 400 ms (P<0.05). RA DOR in the HF+PFD group was similar to that in the HF group at all BCLs (P=NS) and was significantly greater than that in controls at BCL=300 ms (P<0.02). E, Compared with those in controls, LA CVs in the HF group decreased at all BCLs and reached statistical significance at the shortest BCL of 200 ms (P<0.05). PFD treatment resulted in a trend toward an increase in LA CV at all BCLs (P=NS), and this increased CV was similar to that in controls (P=NS at all BCLs). F, RA CVs were similar for the 3 groups at all BCLs (P =NS).
Atrial dispersion of repolarization (DOR) measurements are shown in Figures 2C and 2D. In the LA, CHF resulted in a trend toward an increase in DOR compared with controls. LA DOR in the HF+PFD group was not significantly different from that in the HF group at all BCLs but was significantly greater than that in controls at BCLs of 200 and 300 ms (P<0.02). In the RA, CHF resulted in an increase in DOR at all BCLs compared with controls, and this increase reached statistical significance at BCLs of 200 and 400 ms (P<0.05). RA DOR in the HF+PFD group was similar to that in the HF group and was significantly greater than that in controls at the BCL of 300 ms (P<0.02).
Atrial CV findings are shown in Figures 2E and 2F, respectively. Compared with those in controls, LA CVs in the HF group were decreased at all BCLs but only reached statistical significance at the shortest BCL of 200 ms (P<0.05). LA CV in the HF+PFD group was generally greater than that in the HF group and was similar to that in controls (P=NS). In the RA, CVs were similar in all 3 groups.
Representative isochronal activation maps generated from each of the 4 individual atrial plaques (BCL 300 ms) are shown in Figure 3. Atrial conduction was more heterogeneous (ie, more discrete areas of slow conduction) in the HF group than in controls, and this was reduced with PFD.
Figure 3.
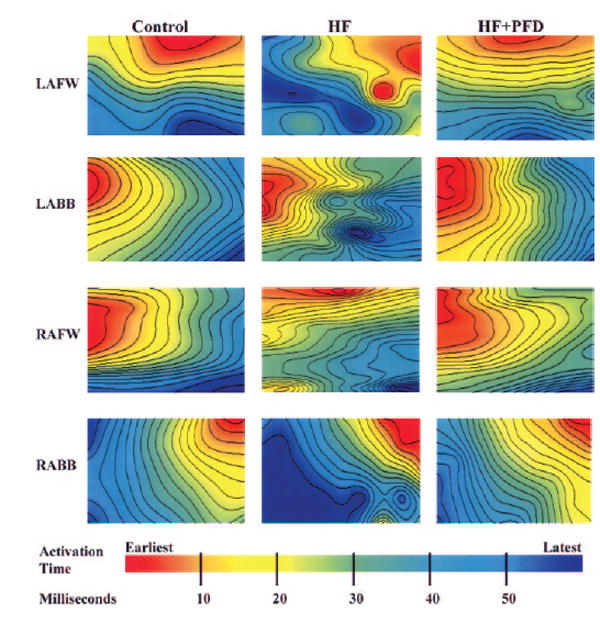
Atrial isochronal (4-ms) maps obtained when pacing from the contralateral atrium at BCL of 300 ms. CHF resulted in an increase in conduction heterogeneity (ie, more discrete areas of slow conduction) in both atria (LA greater than RA). This increase in conduction heterogeneity was reduced with PFD. (Each plot represents a 10×10 electrode portion of each plaque and is generated independently.) LAFW indicates LA free wall; LABB, LA Bachmann’s bundle; RAFW, RA free wall; and RABB, RA Bachmann’s bundle.
Atrial conduction heterogeneity was analyzed quantitatively with phase maps and derivation of the conduction heterogeneity index (CHI; Figures 4A and 4B). In the LA, CHF resulted in significantly increased CHI at all BCLs compared with controls (P<0.01). Treatment with PFD resulted in a significantly reduced CHI at all BCLs (P<0.01). The CHI in the HF+PFD group was not significantly different from that in the control group. In the RA, there was an increase in CHI in the HF group compared with controls, and this increase reached statistical significance at BCLs of 300 and 400 ms (P<0.05). This increase was less than that seen in the LA. Drug treatment resulted in a decrease in RA CHI, and this decrease reached statistical significance at a BCL of 300 ms (P<0.05). RA CHI in the control and HF+PFD groups was similar.
Figure 4.
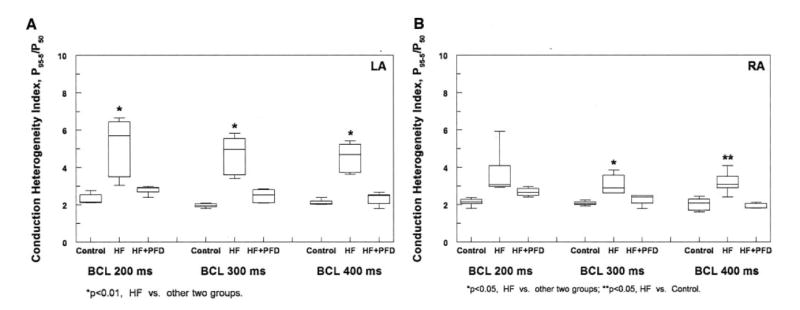
Atrial CHI findings. A, CHF resulted in a significant increase in LA CHI compared with controls at all BCLs (P<0.01). PFD treatment resulted in a significant decrease in LA CHI compared with the HF group at all BCLs (P<0.01), and this reduced CHI was similar to that seen in controls. B, CHF resulted in an increase in RA CHI at all BCLs and reached statistical significance at BCLs of 300 and 400 ms (P<0.05), although this increase was not as large as that seen in the LA. PFD resulted in a decrease in RA CHI and reached statistical significance at a BCL of 300 ms (P<0.05).
Atrial Fibrillation Vulnerability
Only canines in the HF group developed sustained AF (4/5, P<0.05 versus HF+PFD group). AF duration in the remaining untreated canine with nonsustained AF was 240 seconds (induced with 6-second RA burst pacing). CHF resulted in a significant increase in mean AF duration (Figure 5), from 7 (2 to 35) seconds in controls to 1800 [1020 to 1800] seconds in the HF group (P<0.01); PFD treatment resulted in a significant reduction in mean AF duration to 6 (5 to 22) seconds (P<0.01 versus HF), which was similar to that in controls.
Figure 5.

AF vulnerability. AF duration in the untreated CHF group (HF) was significantly longer than that in the other 2 groups. Treatment with PFD resulted in a significant reduction in AF duration to a level similar to that seen in controls.
Histological Findings
Representative LA sections stained with Sirius red are shown in Figure 6. Histological evaluations of all control atria were normal. However, LA sections of HF canines had extensive interstitial fibrosis, with prominent myocyte hypertrophy and cell loss. PFD treatment resulted in significant attenuation of interstitial fibrosis. Histological alterations were also seen in the RA (not shown) of the HF group, although they were much less extensive than those in the LA.
Figure 6.
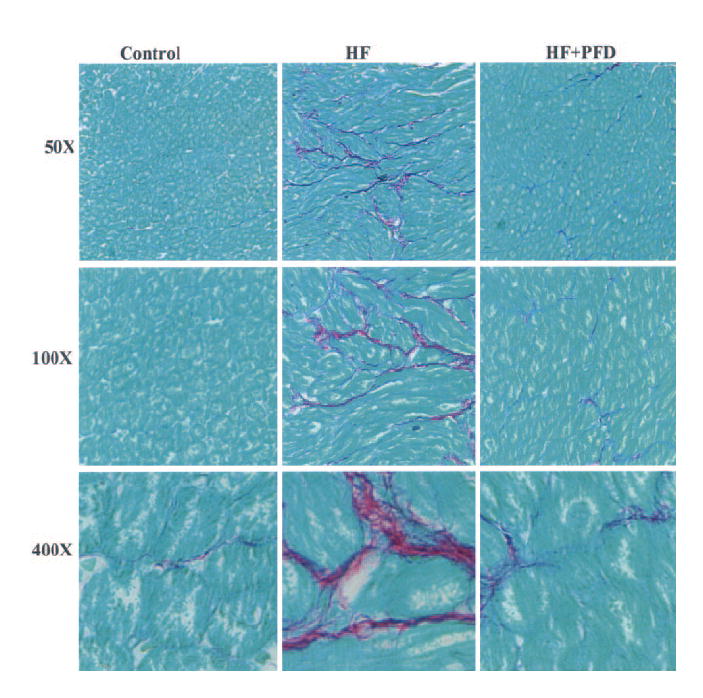
LA histological findings (Sirius red stained). A marked increase in interstitial fibrosis in the HF group was seen compared with controls. PFD resulted in markedly less fibrosis. Magnifications ×50, ×100, and ×400.
There was a significant increase in percentage LA fibrosis (Figure 7) in the HF group compared with controls (16.0% [13.0% to 17.5%] versus 3.1% [2.5% to 4.0%], P<0.01). PFD treatment resulted in a significant reduction in LA fibrosis (8.7% [5.7% to 10.6%], P<0.01 versus HF group), although it was still greater than that in controls (P<0.02).
Figure 7.

Percentage LA fibrosis. There was a significant increase in LA fibrosis in the HF group compared with controls (P<0.01); PFD resulted in a significant reduction (P<0.01), although the reduced percentage fibrosis was still greater than that in controls (P<0.02).
Shown in Figure 8 are LA cytokine immunoblotting results with corresponding protein band density measurements for transforming growth factor (TGF)-β1, total extracellular signal-regulated protein kinase-1 and -2 (ERK[1/2]), total c-Jun N-terminal kinase (JNK), total p38, tissue inhibitor of metalloproteinase (TIMP)-4, active matrix metalloproteinase (MMP)-9, and tumor necrosis factor (TNF)-α. CHF resulted in an increase in TGF-β1, JNK, MMP-9, and TNF-α (P=0.05), whereas PFD resulted in a decrease in each of these cytokines (P=0.05). CHF also resulted in a trend toward a small increase in ERK1/2 and p38 and resulted in a large decrease in TIMP-4 (P=0.05), whereas PFD resulted in a trend toward a small decrease in ERK1/2 and p38 and a large increase in TIMP-4 (P=0.05). Spatial distribution of C×43 and C×40 in the LA was similar in the 3 groups (Data Supplement).
Figure 8.
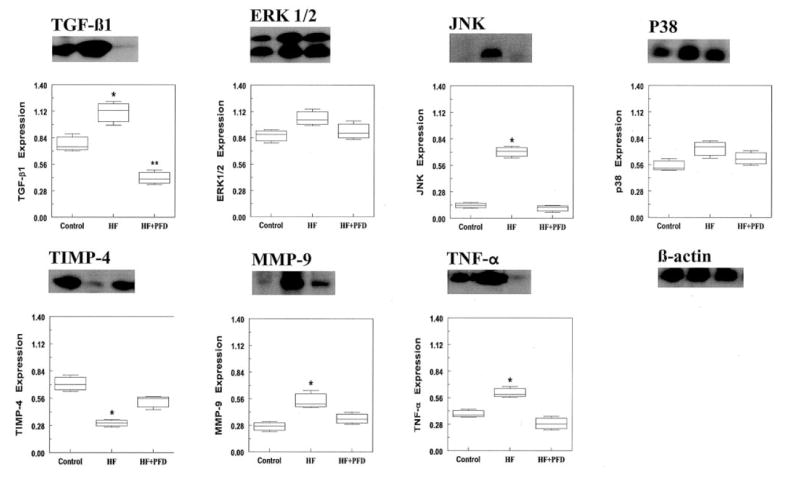
LA immunoblot findings. Shown are tissue levels of TGF-β1, total ERK[1/2] (42- and 44-kDa isoforms), total JNK (46- and 54-kDa isoforms), total p38, TIMP-4, active MMP-9 (88-kDa), and TNF-α in the control and paced groups. Below the immunoblots are corresponding band signal measurements (isoforms of ERK and JNK were averaged for quantitative analysis). CHF resulted in an increase in TGF-β1, JNK, MMP-9, and TNF-α (P=0.05), and each of these was decreased with PFD (P=0.05). CHF resulted in a trend toward a small increase in ERK[1/2] and p38 expression, and both were reduced with PFD (P=NS). TIMP-4 level was decreased with CHF (P=0.05) and increased with PFD (P=0.05). *P=0.05, HF vs other 2 groups; **P=0.05, HF+PFD vs control.
Discussion
In this study, we investigated the effects of a unique antifibrotic agent on CHF-induced atrial fibrosis and vulnerable substrate for AF. Although CHF produced marked arrhythmogenic atrial remodeling with increased AF vulnerability, PFD treatment resulted in a significant reduction in atrial fibrosis, conduction heterogeneity, and AF vulnerability.
Atrial Remodeling and Atrial Fibrillation
Atrial interstitial fibrosis can lead to increased nonuniform anisotropy in conduction26,27 and has been found to be an important substrate for AF.3,5 Canines in the present study developed significant LA fibrosis, LV dysfunction, and LA dilatation after 3 weeks of VTP, an outcome that was similar to results reported by others.3,4 These animals had a significant increase in atrial conduction heterogeneity and AF vulnerability. Although animals that were treated with PFD had similar severity of CHF and degree of LA dilatation as their untreated counterparts, the drug-treated group had a significant reduction in arrhythmogenic atrial remodeling, with reduced AF vulnerability.
In a similar canine model of pacing-induced CHF, Li et al3 observed an increase in atrial ERPs. Here, we observed a trend toward an increase in LA ERP in canines with CHF. This initial increase in LA ERP may be caused in part by an electrophysiological effect from atrial dilatation (from CHF), as has been reported.28,29 A progressive lengthening in LA ERP was observed with PFD treatment, and this lengthening may be caused by a further increase in atrial dilatation because less interstitial fibrosis is present. It is unlikely that these effects are a primary effect of PFD, because the drug was stopped >6 half-lives before the EPS, and because no alteration in RA ERP was observed.
Although CHF resulted in an increase in conduction heterogeneity and a decrease in CV, treatment with PFD prevented these changes. These effects are consistent with the histological finding of increased atrial fibrosis in the HF group that was markedly attenuated with PFD, with elimination of AF vulnerability. Li et al5 found an almost 10-fold increase in atrial fibrosis in their CHF canines (5 weeks of VTP) compared with controls. They also found that the increased atrial fibrosis was associated with increased conduction heterogeneity and increased AF vulnerability. In the present study, CHF resulted in an almost 5-fold increase in atrial fibrosis, and this was associated with increased conduction heterogeneity and AF vulnerability. Although Li et al5 found that treatment of CHF canines with enalapril resulted in a marked reduction in atrial fibrosis, with reduced conduction heterogeneity and AF vulnerability, the AF vulnerability was still significantly greater than that in controls. In contrast, we found that PFD treatment of CHF canines significantly reduced not only conduction heterogeneity but also AF vulnerability to levels similar to those in controls, although the reduced level of atrial fibrosis was still significantly greater than that in controls. This reduction in AF vulnerability was mostly accounted for by a marked reduction in the number of sustained AF episodes. We speculate that a certain threshold amount of atrial fibrosis is required to maintain AF, and when this threshold is not met, conduction heterogeneity will be insufficient, and AF episodes will not be sustained.
Effects of Pirfenidone
Mitogen-activated protein kinases (MAPKs) mediate the effects of angiotensin II on the tissue level.16 Li et al5 reported that in a canine model, VTP-induced CHF resulted in increased expression of the major MAPK subfamilies, including ERK, JNK, and p38 (total and phosphorylated). They found that treatment with enalapril resulted in a reduction in ERK and JNK (both total and phosphorylated forms). Interestingly, they also found that treatment with enalapril resulted in no effect on phosphorylated p38 and a small increase in total p38. In the present study, CHF resulted in an increase in all 3 MAPKs: a significant increase in total JNK and a trend toward a small increase in total ERK and total p38. PFD treatment resulted in a large decrease in total JNK and a trend toward a small decrease in total ERK and total p38. It has been reported that PFD decreases TGF-β1 expression,30 which we have also found in our model, and this decrease in TGF-β1 may influence MAPK expression or downstream signaling.
Atrial extracellular matrix homeostasis is regulated by a balance of MMPs and their endogenous inhibitors (TIMPs), with TIMP-4 being the most cardiospecific.31 The roles of AF and CHF in MMP and TIMP expression are complex. AF itself may differentially modulate MMP and TIMP expression.32 Furthermore, AF often occurs in the setting of CHF, which also affects MMP and TIMP expression.33 In the present study, MMP-9 was increased with CHF (with a concomitant increase in LA size) and was decreased with PFD treatment (with a concomitant decrease in AF vulnerability). Anné et al34 found no difference in MMP-9 expression in mitral valve disease patients with and without AF. Recently, Mukherjee et al32 found that LA MMP-9 was greater in CHF patients with AF than in those without AF. Nakano et al20 reported that MMP-9 was significantly increased in patients with paroxysmal or chronic AF. In that study, interestingly, LA diameter in patients with paroxysmal AF was similar to that in patients with sinus rhythm, and the authors speculated that MMP-9 itself may promote arrhythmogenic atrial remodeling, with increased AF vulnerability. Results from the present study implicate MMP-9 as a potential contributor to arrhythmogenic atrial remodeling.
The role of TIMPs in atrial remodeling and AF vulnerability has also been studied. Boixel et al21 have found that progressive CHF and LA fibrosis are not associated with an upregulation of TIMP-4. Anné et al34 found no difference in TIMP-4 expression in mitral valve disease patients with or without AF. In contrast, we found a change in TIMP-4 with CHF. Part of the discrepancy in the present findings may be accounted for by the differences in the experimental model under study. Although we observed an increase in MMP-9 with CHF, surprisingly, it was not associated with an increase in TIMP-4, and instead, we found a reduction in TIMP-4. This reduction would favor increased collagen deposition. This dysregulation in MMP-9 and TIMP-4 expression may have played a significant role in the development of marked atrial fibrosis. The effects of PFD included an attenuation of MMP-9 expression and an increase in TIMP-4 expression. These effects blunted the alteration in MMP/TIMP stoichiometry, which helped to restore atrial extracellular matrix homeostasis, resulting in less atrial fibrosis.
Recent studies suggest that inflammation may play an important role in promoting AF.35,36 However, the role of specific inflammatory cytokines in AF is not clear. Goette et al22 and Anné et al34 have found no significant increase in TNF-α in humans with AF. More recently, in a transgenic mouse model of cardiomyopathy that overexpressed TNF-α, Saba et al36 found both alterations in atrial morphology (increased fibrosis) and abnormalities in action potential propagation and calcium handling in atrial myocytes with an increased susceptibility to atrial arrhythmias. However, it is unclear from this particular study how much of the proarrhythmia risk is accounted for by TNF- α itself versus how much is accounted for by severe ventricular failure and the associated alterations in other important cytokines. In the present study, we observed an increase in TNF-α with CHF, and this increase may have played a role in elevating MMP-9 levels.24 PFD has been found to reduce TNF-α expression,37 and we observed a similar effect. The reduction in TNF-α may have contributed to the reduction in MMP-9.
Previous work from our laboratory has shown that over-expression of TGF-β1 in transgenic mice resulted in increased atrial fibrosis, conduction heterogeneity, and AF vulnerability.6 PFD has been shown to reduce expression of TGF-β1, with a significant reduction in fibrosis in multiple experimental animal models, including those of lung,30 hepatic,38 and renal8,9,39 fibrosis. We observed similar results: CHF resulted in an increase in TGF-β1 and atrial fibrosis, and PFD treatment resulted in a marked reduction in both.
Alterations in expression and distribution of gap junction proteins have been reported to play a role in promoting AF.40,41 Although PFD attenuated arrhythmogenic atrial remodeling and reduced AF vulnerability, it did not appear to alter the atrial spatial distribution of C×40 and C×43.
PFD has been reported to have broad antifibrotic and anti-inflammatory effects.7,10,30,42-44 The present results suggest that PFD attenuates CHF-related arrhythmogenic atrial remodeling via a complex set of actions, affecting multiple levels and components of the fibrosis formation and degradation signaling cascade.
Clinical Implications
In most patients, AF has traditionally been treated with antiarrhythmic drugs, with their accompanying proarrhythmia risks. Pharmacological therapy targeted at the underlying substrate has been investigated.5,45 Although angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists have been shown to be effective in attenuating arrhythmogenic atrial remodeling, these studies have only shown a blunting of AF vulnerability, perhaps because of the complex, redundant pathways for atrial fibrosis formation. Because it has the potential to attenuate increases in all 3 major cardiac MAPKs, PFD may be a more potent antifibrotic drug. We investigated this drug with no known hemodynamic effects and found it to have striking beneficial effects on arrhythmogenic atrial remodeling.
Study Limitations
Serum drug levels were not monitored during the present study. Doses were extrapolated from presumed optimal doses based on known pharmacokinetics of the compound. It is not known whether higher doses would be more effective or lower doses equally effective. The present study did not investigate whether concomitant use of other agents used to treat CHF (eg, angiotensin II type 1 receptor antagonists, angiotensin-converting enzyme inhibitors, endothelin-1 receptor antagonists, β-adrenoceptor antagonists, or aldosterone antagonists) would have additive effects against atrial fibrosis formation. It is possible that agents used to treat CHF directly (ie, those that affect hemodynamics) may augment or complement the action of PFD, because this drug has no known vasoactive (afterload reduction) properties.
Although the effects of PFD on various important cytokines are shown here, its specific actions on these proteins (ie, whether it is selective for the MAPKs or acts on other pathways responsible for fibrosis) remain to be elucidated. It is not known, and not likely, that this drug intervention would affect the substrate of primary electrical remodeling due to AF itself (“AF begets AF”), because PFD has no known direct electrophysiological effects. However, this was not studied as part of the present investigation.
Atrial fibrosis and CHF are but one substrate for AF. Other mechanisms of increased AF vulnerability (eg, delayed after-depolarization in nonischemic CHF) warrant consideration, but they were not part of the present investigation. PFD has broad antifibrotic and antiinflammatory effects, and additional effects beyond those studied here also warrant further investigation.
Conclusions
PFD treatment of canines with VTP-induced CHF results in a marked attenuation of arrhythmogenic atrial remodeling, with a reduction in AF vulnerability. This appears to be due to the drug’s broad effects on potent profibrotic mediators. Pharmacological therapy targeted at the underlying fibrotic substrate may play an important role in the management of AF.
CLINICAL PERSPECTIVE
Atrial fibrosis is an important substrate in atrial fibrillation (AF), especially in the setting of structural heart disease. In a canine model of heart failure, there is marked atrial arrhythmogenic remodeling, with increased AF vulnerability. This arrhythmogenic remodeling is a potential therapeutic target. In this investigation, we studied the effects of the antifibrotic drug pirfenidone on arrhythmogenic atrial remodeling. Drug treatment resulted in a reduction in atrial fibrosis, conduction heterogeneity, and AF vulnerability. Pharmacological therapy targeted at the underlying fibrotic substrate may play an important role in the prevention of AF.
Acknowledgments
Sources of Funding This work was supported by National Heart, Lung, and Blood Institute grant RO1-HL072854 (Dr Olgin) and American Heart Association Western States Affiliate Postdoctoral Fellowship Award (Dr Lee).
Footnotes
The online-only Data Supplement is available with this article at http://circ.ahajournals.org/cgi/content/full/CIRCULATIONAHA.106.624320/DC1.
The online version of this article, along with updated information and services, is located on the World Wide Web at: http://circ.ahajournals.org/cgi/content/full/114/16/1703
Disclosures None.
References
- 1.Kostin S, Klein G, Szalay Z, Hein S, Bauer EP, Schaper J. Structural correlate of atrial fibrillation in human patients. Cardiovasc Res. 2002;54:361–379. doi: 10.1016/s0008-6363(02)00273-0. [DOI] [PubMed] [Google Scholar]
- 2.Boldt A, Wetzel U, Lauschke J, Weigl J, Gummert J, Hindricks G, Kottkamp H, Dhein S. Fibrosis in left atrial tissue of patients with atrial fibrillation with and without underlying mitral valve disease. Heart. 2004;90:400–405. doi: 10.1136/hrt.2003.015347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li D, Fareh S, Leung TK, Nattel S. Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation. 1999;100:87–95. doi: 10.1161/01.cir.100.1.87. [DOI] [PubMed] [Google Scholar]
- 4.Shinagawa K, Shi YF, Tardif JC, Leung TK, Nattel S. Dynamic nature of atrial fibrillation substrate during development and reversal of heart failure in dogs. Circulation. 2002;105:2672–2678. doi: 10.1161/01.cir.0000016826.62813.f5. [DOI] [PubMed] [Google Scholar]
- 5.Li D, Shinagawa K, Pang L, Leung TK, Cardin S, Wang Z, Nattel S. Effects of angiotensin-converting enzyme inhibition on the development of the atrial fibrillation substrate in dogs with ventricular tachypacing-induced congestive heart failure. Circulation. 2001;104:2608–2614. doi: 10.1161/hc4601.099402. [DOI] [PubMed] [Google Scholar]
- 6.Verheule S, Sato T, Everett Tt, Engle SK, Otten D, Rubart-von der Lohe M, Nakajima HO, Nakajima H, Field LJ, Olgin JE. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-β1. Circ Res. 2004;94:1458–1465. doi: 10.1161/01.RES.0000129579.59664.9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iyer SN, Wild JS, Schiedt MJ, Hyde DM, Margolin SB, Giri SN. Dietary intake of pirfenidone ameliorates bleomycin-induced lung fibrosis in hamsters. J Lab Clin Med. 1995;125:779–785. [PubMed] [Google Scholar]
- 8.Shimizu T, Fukagawa M, Kuroda T, Hata S, Iwasaki Y, Nemoto M, Shirai K, Yamauchi S, Margolin SB, Shimizu F, Kurokawa K. Pirfenidone prevents collagen accumulation in the remnant kidney in rats with partial nephrectomy. Kidney Int Suppl. 1997;63:S239–S243. [PubMed] [Google Scholar]
- 9.Shimizu T, Kuroda T, Hata S, Fukagawa M, Margolin SB, Kurokawa K. Pirfenidone improves renal function and fibrosis in the post-obstructed kidney. Kidney Int. 1998;54:99–109. doi: 10.1046/j.1523-1755.1998.00962.x. [DOI] [PubMed] [Google Scholar]
- 10.Iyer SN, Gurujeyalakshmi G, Giri SN. Effects of pirfenidone on procollagen gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J Pharmacol Exp Ther. 1999;289:211–218. [PubMed] [Google Scholar]
- 11.Miric G, Dallemagne C, Endre Z, Margolin S, Taylor SM, Brown L. Reversal of cardiac and renal fibrosis by pirfenidone and spirono-lactone in streptozotocin-diabetic rats. Br J Pharmacol. 2001;133:687–694. doi: 10.1038/sj.bjp.0704131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mirkovic S, Seymour AM, Fenning A, Strachan A, Margolin SB, Taylor SM, Brown L. Attenuation of cardiac fibrosis by pirfenidone and amiloride in DOCA-salt hypertensive rats. Br J Pharmacol. 2002;135:961–968. doi: 10.1038/sj.bjp.0704539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verheule S, Wilson E, Everett Tt, Shanbhag S, Golden C, Olgin J. Alterations in atrial electrophysiology and tissue structure in a canine model of chronic atrial dilatation due to mitral regurgitation. Circulation. 2003;107:2615–2622. doi: 10.1161/01.CIR.0000066915.15187.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sih HJ, Zipes DP, Berbari EJ, Adams DE, Olgin JE. Differences in organization between acute and chronic atrial fibrillation in dogs. J Am Coll Cardiol. 2000;36:924–931. doi: 10.1016/s0735-1097(00)00788-9. [DOI] [PubMed] [Google Scholar]
- 15.Verheule S, Wilson E, Banthia S, Everett THT, Shanbhag S, Sih HJ, Olgin J. Direction-dependent conduction abnormalities in a canine model of atrial fibrillation due to chronic atrial dilatation. Am J Physiol Heart Circ Physiol. 2004;287:H634–H644. doi: 10.1152/ajpheart.00014.2004. [DOI] [PubMed] [Google Scholar]
- 16.Yano M, Kim S, Izumi Y, Yamanaka S, Iwao H. Differential activation of cardiac c-jun amino-terminal kinase and extracellular signal-regulated kinase in angiotensin II-mediated hypertension. Circ Res. 1998;83:752–760. doi: 10.1161/01.res.83.7.752. [DOI] [PubMed] [Google Scholar]
- 17.Thomas CV, Coker ML, Zellner JL, Handy JR, Crumbley AJ, III, Spinale FG. Increased matrix metalloproteinase activity and selective upregulation in LV myocardium from patients with end-stage dilated cardiomyopathy. Circulation. 1998;97:1708–1715. doi: 10.1161/01.cir.97.17.1708. [DOI] [PubMed] [Google Scholar]
- 18.Spinale FG, Coker ML, Krombach SR, Mukherjee R, Hallak H, Houck WV, Clair MJ, Kribbs SB, Johnson LL, Peterson JT, Zile MR. Matrix metalloproteinase inhibition during the development of congestive heart failure: effects on left ventricular dimensions and function. Circ Res. 1999;85:364–376. doi: 10.1161/01.res.85.4.364. [DOI] [PubMed] [Google Scholar]
- 19.Spinale FG, Coker ML, Bond BR, Zellner JL. Myocardial matrix degradation and metalloproteinase activation in the failing heart: a potential therapeutic target. Cardiovasc Res. 2000;46:225–238. doi: 10.1016/s0008-6363(99)00431-9. [DOI] [PubMed] [Google Scholar]
- 20.Nakano Y, Niida S, Dote K, Takenaka S, Hirao H, Miura F, Ishida M, Shingu T, Sueda T, Yoshizumi M, Chayama K. Matrix metalloproteinase-9 contributes to human atrial remodeling during atrial fibrillation. J Am Coll Cardiol. 2004;43:818–825. doi: 10.1016/j.jacc.2003.08.060. [DOI] [PubMed] [Google Scholar]
- 21.Boixel C, Fontaine V, Rucker-Martin C, Milliez P, Louedec L, Michel JB, Jacob MP, Hatem SN. Fibrosis of the left atria during progression of heart failure is associated with increased matrix metalloproteinases in the rat. J Am Coll Cardiol. 2003;42:336–344. doi: 10.1016/s0735-1097(03)00578-3. [DOI] [PubMed] [Google Scholar]
- 22.Goette A, Arndt M, Rocken C, Staack T, Bechtloff R, Reinhold D, Huth C, Ansorge S, Klein HU, Lendeckel U. Calpains and cytokines in fibrillating human atria. Am J Physiol Heart Circ Physiol. 2002;283:H264–H272. doi: 10.1152/ajpheart.00505.2001. [DOI] [PubMed] [Google Scholar]
- 23.Bradham WS, Bozkurt B, Gunasinghe H, Mann D, Spinale FG. Tumor necrosis factor-alpha and myocardial remodeling in progression of heart failure: a current perspective. Cardiovasc Res. 2002;53:822–830. doi: 10.1016/s0008-6363(01)00503-x. [DOI] [PubMed] [Google Scholar]
- 24.Bradham WS, Moe G, Wendt KA, Scott AA, Konig A, Romanova M, Naik G, Spinale FG. TNF-alpha and myocardial matrix metalloproteinases in heart failure: relationship to LV remodeling. Am J Physiol Heart Circ Physiol. 2002;282:H1288–H1295. doi: 10.1152/ajpheart.00526.2001. [DOI] [PubMed] [Google Scholar]
- 25.Verheule S, Wilson EE, Arora R, Engle SK, Scott LR, Olgin JE. Tissue structure and connexin expression of canine pulmonary veins. Cardiovasc Res. 2002;55:727–738. doi: 10.1016/s0008-6363(02)00490-x. [DOI] [PubMed] [Google Scholar]
- 26.Spach MS, Boineau JP. Microfibrosis produces electrical load variations due to loss of side-to-side cell connections: a major mechanism of structural heart disease arrhythmias. Pacing Clin Electrophysiol. 1997;20:397–413. doi: 10.1111/j.1540-8159.1997.tb06199.x. [DOI] [PubMed] [Google Scholar]
- 27.Spach MS, Miller WT, III, Dolber PC, Kootsey JM, Sommer JR, Mosher CE., Jr The functional role of structural complexities in the propagation of depolarization in the atrium of the dog: cardiac conduction disturbances due to discontinuities of effective axial resistivity. Circ Res. 1982;50:175–191. doi: 10.1161/01.res.50.2.175. [DOI] [PubMed] [Google Scholar]
- 28.Boyden PA, Tilley LP, Albala A, Liu SK, Fenoglio JJ, Jr, Wit AL. Mechanisms for atrial arrhythmias associated with cardiomyopathy: a study of feline hearts with primary myocardial disease. Circulation. 1984;69:1036–1047. doi: 10.1161/01.cir.69.5.1036. [DOI] [PubMed] [Google Scholar]
- 29.Chen YJ, Chen SA, Tai CT, Yu WC, Feng AN, Ding YA, Chang MS. Electrophysiologic characteristics of a dilated atrium in patients with paroxysmal atrial fibrillation and atrial flutter. J Interv Card Electrophysiol. 1998;2:181–186. doi: 10.1023/a:1009759717250. [DOI] [PubMed] [Google Scholar]
- 30.Iyer SN, Gurujeyalakshmi G, Giri SN. Effects of pirfenidone on transforming growth factor-beta gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J Pharmacol Exp Ther. 1999;291:367–373. [PubMed] [Google Scholar]
- 31.Greene J, Wang M, Liu YE, Raymond LA, Rosen C, Shi YE. Molecular cloning and characterization of human tissue inhibitor of metalloproteinase 4. J Biol Chem. 1996;271:30375–30380. doi: 10.1074/jbc.271.48.30375. [DOI] [PubMed] [Google Scholar]
- 32.Mukherjee R, Herron AR, Lowry AS, Stroud RE, Stroud MR, Wharton JM, Ikonomidis JS, Crumbley AJ, III, Spinale FG, Gold MR. Selective induction of matrix metalloproteinases and tissue inhibitor of metalloproteinases in atrial and ventricular myocardium in patients with atrial fibrillation. Am J Cardiol. 2006;97:532–537. doi: 10.1016/j.amjcard.2005.08.073. [DOI] [PubMed] [Google Scholar]
- 33.Li YY, Feldman AM, Sun Y, McTiernan CF. Differential expression of tissue inhibitors of metalloproteinases in the failing human heart. Circulation. 1998;98:1728–1734. doi: 10.1161/01.cir.98.17.1728. [DOI] [PubMed] [Google Scholar]
- 34.Anné W, Willems R, Roskams T, Sergant P, Herijers P, Holemans P, Ector H, Heidbuchel H. Matrix metalloproteinases and atrial remodeling in patients with mitral valve disease and atrial fibrillation. Cardiovasc Res. 2005;67:655–666. doi: 10.1016/j.cardiores.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 35.Chung MK, Martin DO, Sprecher D, Wazni O, Kanderian A, Carnes CA, Bauer JA, Tchou PJ, Niebauer MJ, Natale A, Van Wagoner DR. C-reactive protein elevation in patients with atrial arrhythmias: inflammatory mechanisms and persistence of atrial fibrillation. Circulation. 2001;104:2886–2891. doi: 10.1161/hc4901.101760. [DOI] [PubMed] [Google Scholar]
- 36.Saba S, Janczewski AM, Baker LC, Shusterman V, Gursoy EC, Feldman AM, Salama G, McTiernan CF, London B. Atrial contractile dysfunction, fibrosis, and arrhythmias in a mouse model of cardiomyopathy secondary to cardiac-specific overexpression of tumor necrosis factor-α. Am J Physiol Heart Circ Physiol. 2005;289:H1456–H1467. doi: 10.1152/ajpheart.00733.2004. [DOI] [PubMed] [Google Scholar]
- 37.Nakazano H, Oku H, Yamane S, Tsuruta Y, Suzuki R. A novel anti-fibrotic agent pirfenidone suppresses tumor necrosis factor-alpha at the translational level. Eur J Pharmacol. 2002;446:177–185. doi: 10.1016/s0014-2999(02)01758-2. [DOI] [PubMed] [Google Scholar]
- 38.Garcia L, Hernandez I, Sandoval A, Salazar A, Garcia J, Vera J, Grijalva G, Muriel P, Margolin S, Armendariz-Borunda J. Pirfenidone effectively reverses experimental liver fibrosis. J Hepatol. 2002;37:797–805. doi: 10.1016/s0168-8278(02)00272-6. [DOI] [PubMed] [Google Scholar]
- 39.Shihab FS, Bennett WM, Yi H, Andoh TF. Pirfenidone treatment decreases transforming growth factor-beta1 and matrix proteins and ameliorates fibrosis in chronic cyclosporine nephrotoxicity. Am J Transplant. 2002;2:111–119. doi: 10.1034/j.1600-6143.2002.020201.x. [DOI] [PubMed] [Google Scholar]
- 40.van der Velden HM, Ausma J, Rook MB, Hellemons AJ, van Veen TA, Allessie MA, Jongsma HJ. Gap junctional remodeling in relation to stabilization of atrial fibrillation in the goat. Cardiovasc Res. 2000;46:476–486. doi: 10.1016/s0008-6363(00)00026-2. [DOI] [PubMed] [Google Scholar]
- 41.Polontchouk L, Haefliger JA, Ebelt B, Schaefer T, Stuhlmann D, Mehlhorn U, Kuhn-Regnier F, De Vivie ER, Dhein S. Effects of chronic atrial fibrillation on gap junction distribution in human and rat atria. J Am Coll Cardiol. 2001;38:883–891. doi: 10.1016/s0735-1097(01)01443-7. [DOI] [PubMed] [Google Scholar]
- 42.Iyer SN, Margolin SB, Hyde DM, Giri SN. Lung fibrosis is ameliorated by pirfenidone fed in diet after the second dose in a three-dose bleomycin-hamster model. Exp Lung Res. 1998;24:119–132. doi: 10.3109/01902149809046058. [DOI] [PubMed] [Google Scholar]
- 43.Cain WC, Stuart RW, Lefkowitz DL, Starnes JD, Margolin S, Lefkowitz SS. Inhibition of tumor necrosis factor and subsequent endotoxin shock by pirfenidone. Int J Immunopharmacol. 1998;20:685–695. doi: 10.1016/s0192-0561(98)00042-3. [DOI] [PubMed] [Google Scholar]
- 44.Kaneko M, Inoue H, Nakazawa R, Azuma N, Suzuki M, Yamauchi S, Margolin SB, Tsubota K, Saito I. Pirfenidone induces intercellular adhesion molecule-1 (ICAM-1) down-regulation on cultured human synovial fibroblasts. Clin Exp Immunol. 1998;113:72–76. doi: 10.1046/j.1365-2249.1998.00618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumagai K, Nakashima H, Urata H, Gondo N, Arakawa K, Saku K. Effects of angiotensin II type 1 receptor antagonist on electrical and structural remodeling in atrial fibrillation. J Am Coll Cardiol. 2003;41:2197–2204. doi: 10.1016/s0735-1097(03)00464-9. [DOI] [PubMed] [Google Scholar]