

Abstract
Previously we demonstrated the ability of ethyl glycinato substituted polyphosphazenes to neutralize the acidic degradation products and control the degradation rate of poly(lactic acid-glycolic acid) by blending. In this study, blends of high strength poly[(50% ethyl alanato) (50% p-phenyl phenoxy) phosphazene] (PNEA50PhPh50) and 85:15 poly(lactic acid-glycolic acid) (PLAGA) were prepared using a mutual solvent approach. Three different solvents, methylene chloride (MC), chloroform (CF) and tetrahydrofuran (THF) were studied to investigate solvent effects on blend miscibility. Three different blends were then fabricated at various weight ratios namely 25:75 (BLEND25), 50:50 (BLEND50), and 75:25 (BLEND75) using THF as the mutual solvent. The miscibility of the blends was evaluated by scanning electron microscopy (SEM), differential scanning calorimetry (DSC), and Fourier transform infrared spectroscopy (FTIR). Among these, BLEND25 was miscible while BLEND50 and BLEND75 were partially miscible. Furthermore, BLEND25 formed apatite layers on its surface as evidenced in a biomimetic study performed. These novel blends showed cell adhesion and proliferation comparable to PLAGA. However, the PNEA50PhPh50 component in the blends was able to increase the phenotypic expression and mineralized matrix synthesis of the primary rat osteoblasts (PRO) in vitro. Blends of high strength poly[(50% ethyl alanato) (50% p-phenyl phenoxy) phosphazene] (PNEA50PhPh50) and 85:15 poly(lactic acid-glycolic acid) (PLAGA) are promising biomaterials for a variety of musculoskeletal applications.
Keywords: Polyphosphazenes, poly(lactic acid-glycolic acid), polymer blends, osteocompatibility, bone tissue engineering
Introduction
Biomaterials are central to tissue engineering as they provide necessary biophysical and biochemical cues that regulate cellular behavior [1,2]. Efforts have been made to develop novel biomaterials with improved biological and mechanical properties utilizing the recent developments in biology and tissue engineering [3]. A great deal of research has been devoted to synthesize novel biomaterials using various chemical methods, biological methods or fine-tuning the existing materials for certain applications [4]. The success of tissue engineering as an alternative therapeutic strategy toward the repair, restoration or regeneration of living tissues depends to a great extent on the development of novel biodegradable polymers [5]. Biodegradable polymers as scaffold materials need to possess a number of properties including non-toxic degradation products, mechanical properties that match the surrounding tissue and appropriate degradation rate to match new tissue formation under physiological conditions [6].
Currently, some major classes of synthetic biodegradable polymers that have been investigated for tissue engineering applications include polyesters, polyorthoesters, poly(α-amino acids), and polyanhydrides [6]. Among them, poly(lactic acid-glycolic acid) (PLAGA) is one of the most commonly used biodegradable polymers for biomedical applications due to its commercial availability, biocompatibility, controlled degradation rate and excellent mechanical properties [7,8]. However, it degrades by bulk erosion and the acidic degradation products can lead to a catastrophic failure of the structural integrity of the scaffold. Furthermore, acidic degradation products showed clinical complications such as inflammatory and foreign body reactions in a study involving 1,000 patients over a period of 9 years [9]. These studies have prompted researchers to develop novel biodegradable polymers with non-toxic and neutral degradation products, with appropriate degradation kinetics and mechanical propertiessuitable for tissue engineering applications.
Polyphosphazenes constitute a unique class of high molecular weight polymers with an inorganic backbone consisting of alternating phosphorous and nitrogen atoms linked by alternating single and double bonds, with two organic or organometallic side groups on each phosphorous atom. The synthetic flexibility of polyphosphazenes has led to the development of a wide variety of polymers with diverse physical, chemical and biological properties [10]. Among them, amino acid ester polyphosphazenes are known to be hydrolytically sensitive and degrade into non-toxic and neutral products [11–14]. Also the degradation rates can be modified by varying the nature and ratio of the side groups [11–14]. Thus, amino acid ester polyphosphazenes have been found to be attractive candidates for biomedical applications [15–17]. We have recently demonstrated relatively good mechanical properties, tunable degradation rate, and excellent osteocompatibility of alanine ethyl ester and p-phenylphenoxy cosubstituted polyphosphazenes both in vitro and in vivo [14,18,19].
In order to develop novel biomaterials with excellent properties, the physical blending of polyphosphazenes with PLAGA serves as a practical method that synergistically combines the excellent properties of two classes of biocompatible polymers [20–23]. We previously developed biodegradable ethyl glycinato substituted polyphosphazene (EG-PPHOS)/PLAGA blends and demonstrated that diverse properties could be obtained via compositional changes of the pure polymers [20]. It was shown that the acidic degradation products of PLAGA could be effectively neutralized by the degradation products of polyphosphazenes [21]. However, the mechanical performance of EG-PPHOS due to the lower glass transition temperature (Tg) value makes this polymer system less optimal for bone tissue engineering applications [11,14,20]. On the other hand, poly[(50% ethyl alanato) (50% p-phenyl phenoxy) phosphazene has a higher Tg value due to the presence of the bulky aromatic side groups. It is further expected to improve its mechanical properties by blending with PLAGA, and hence can serve as a better scaffold material for bone tissue engineering applications [14].
The objective of the present study was to develop novel biomaterials through physicallyblending PLAGA 85:15 (LA:GA; 85:15) with poly[(50% ethyl alanato) (50% p-phenyl phenoxy) phosphazene] (PNEA50PhPh50) at three different compositions namely 25:75 (BLEND25), 50:50 (BLEND50), and 75:25 (BLEND75). The miscibility, physico-chemical properties, osteointegrativity, and osteocompatibilty of these prepared blends were characterized.
Materials and methods
Materials
Poly(lactic acid-glycolic acid), PLAGA 85:15 (LA:GA is of 85:15, = 117 kDa) was purchased from Alkermes (Wilmington, OH). Hexachlorocyclotriphosphazene (HCCTP) was procured from Nippon Fine Chemicals, Japan. HCCTP was purified by recrystallization from heptane and sublimation at 50°C. p-Phenylphenol and L-Alanine ethyl ester hydrochloride were obtained from Sigma (St. Louis, MO) and Aldrich (Milwaukee, WI), respectively. The polyphosphazene was synthesized with the use of standard Schlenk line techniques under an argon atmosphere. Methylene chloride (MC), chloroform (CF), and tetrahydrofuran (THF), all from Fisher Scientific (Hampton, NH), were used as received. The 31P NMR (145 MHz) and 1H NMR (360 MHz) spectra of the polymers were obtained using a Bruker 360 MHz NMR spectrometer in order to confirm the structure and side-group ratios. 31P NMR chemical shifts are reported in ppm relative to 85% phosphoric acid as an external reference. Molecular weights of the polymers were measured by gel permeation chromatography (GPC) using a Hewlett Packard LC 1100 series. Samples for GPC analysis were prepared in concentrations of approximately 1% (w/v) in THF and eluted at a rate of 1ml/min through a size exclusion column (Phenomenex, Phenogel, USA) calibrated using a polystyrene standard (Polysciences, Inc., Warrington, PA).
Synthesis of polyphosphazene
Poly[(50% ethyl alanato) (50% p-phenyl phenoxy) phosphazene] (PNEA50PhPh50) was synthesized according to a reported procedure [10,14,18,19]. Briefly, poly(dichlorophosphazene) was dissolved in dry THF and first allowed to react with sodium salt of p-phenylphenol in dry THF in order to replace 50% of the chlorine atoms. Then a large excess of L-alanine ethyl ester hydrochloride and triethyl amine in dry THF was allowed to react with the partially substituted polymers to replace the remaining chlorine atoms. The reaction mixture was refluxed for 48 h when complete substitution of the chlorine atoms was confirmed using 31P NMR. The polymer solution was filtered and concentrated using a rotary evaporator, then precipitated in hexanes. Subsequent precipitations into hexanes were used to purify the polymer. The polymers were dried under vacuum and stored under argon before use.
Polymer blend fabrication
Films of PNEA50PhPh50, BLEND25 (PNEA50PhPh50:PLAGA 25:75), BLEND50 (PNEA50PhPh50:PLAGA 50:50), BLEND75 (PNEA50PhPh50:PLAGA 75:25) and PLAGA were prepared using the mutual solvent method [20]. Briefly, PNEA50PhPh50 and PLAGA with the following weight ratios of PNEA50PhPh50 to PLAGA: 25:75, 50:50, and 75:25, total weight 1 g, were dissolved separately in 7 mL of mutual solvent (MC, CF, or THF). Then the polymer solutions were mixed and stirred at room temperature for 24 h. The mixed solution in the total volume of 14 mL was subsequently poured into petri dishes lined with Bytac paper and the solvent was allowed to evaporate slowly at 4°C for 48 h followed by freeze drying. Finally, circular discs of 10 mm in diameter and 0.1 mm in thickness were bored from films using a cork borer.
Solvent effect on the blend miscibility
Since there is potential hydrogen bonding between carbonyl group in PLAGA and amino group in PNEA50PhPh50, blend miscibility should be achieved, at least at some compositions if a suitable solvent has been chosen. Different mutual solvents such as methylene chloride (MC), chloroform (CF), and tetrahydrofuran (THF) were used to investigate the solvent effect on the blend miscibility. These blends were characterized for their miscibility by scanning electron microscopy (SEM) and differential scanning calorimetry (DSC).
Characterization of Polyphosphazene/PLAGA blends
Surface morphology of the blends
The surfaces of the blend films were sputter coated with gold using a Hummer V sputtering system (Technics Inc., Baltimore, MD) and examined under a JEOL 6700F scanning electron microscope (SEM) (JEOL, Boston, MA, USA).
Differential scanning calorimetry (DSC) measurements
The glass transition temperatures (Tg) were determined by differential scanning calorimetry using TA instruments DSC Q1000 with Thermal Analysis software. Polymer samples were heated from −70°C to 100°C at a heating rate of 3°C/min under a nitrogen atmosphere. The Tg was determined from the half height point of the heat capacity change in the thermogram.
Fourier transform infrared (FTIR) analysis
The FTIR spectra of thin films of polymers and blends were recorded using a Brucker Vector 22 FTIR spectrophotometer at a resolution of 4 cm−1 and with an average of 50 scans. The blend films were cast on KBr plates and were sufficiently thin to be within an absorbance range where the Beer-Lambert law was obeyed.
Biomimetic process
Simulated body fluid (SBF) 1.5x was prepared by dissolving NaCl, NaHCO3, KCl, K2HPO4·3H2O, MgCl2·6H2O, CaCl2 and Na2SO4 (Aldrich, Milwaukee, WI) in distilled deionized water, and buffered at pH 7.25 with Tris-HCl as reported previously [24]. The concentration of the ions were 213.0 mM Na+, 7.5 mM K+, 3.8 mM Ca2+, 2.3 mM Mg2+, 6.3 mM HCO3−, 223.0 mM Cl−, 1.5 mM HPO42−, 0.75 mM SO42−. The apatite deposition on the surface of PNEA50PhPh50 and BLEND25 was evaluated by incubating the 10 mm circular polymer films in 15 mL 1.5x SBF with an immersion ratio of 10 (apparent surface area of specimen (mm2) to the volume of SBF (mL)) for 21 days at 37°C. 1.5x SBF solution was changed every 24 h during this study. At days 7, 14, and 21, the films (n=3) were removed from SBF and washed gently with distilled water for 3 times followed by drying at room temperature for at least 24 h. The surface morphology and calcium and phosphorus content of apatite layer were examined with SEM and energy dispersive X-ray spectroscopy (EDX) respectively (JEOL 6700F coupled with EDX detector).
Cell isolation
Primary rat osteoblast (PRO) cells were isolated from calvarias of 2-day-old neonatal Sprague-Dawley rats according to a standard procedure [25]. Briefly, the calvarias were dissected, minced and rinsed in phosphate-buffered saline (PBS). Four digestions were performed using the digesting solution of collagenase and trypsin. The cells from the first 45-min digestion were discarded to minimize fibroblast contamination. The cells from the last three consecutive 30-min digestions were collected and the supernatant from these digestions was centrifuged at 1300 rpm for 9 min. The resulting cell pellet was suspended in Ham’s F-12 media (Gibco) supplemented with 12% FBS and 1% penicillin/streptomycin. Passage-two cells were used for the current study. Prior to cell seeding, all polymer films were sterilized with ultraviolet (UV) light for 15 min after being treated with 70% ethanol for 30 min and distilled water twice for 15 min on each side.
Cell seeding
The polymer films were pre-incubated in Ham’s F-12 media for 1 h prior to cell seeding. PRO cells were seeded onto the polymer films at a density of 6 × 104 cells/cm2 after removing the pre-incubation media. The films with the cells were incubated at 37°C for 2 h to promote cell attachment. The cells were cultured at 37°C in a 95% humidified air and 5% CO2 for various periods of time after adding 1 mL supplemented Ham’s F-12 media to each well. The media were changed every 2 days. Two types of media were used for cell culture: normal media (Ham’s F-12 media supplemented with 12% FBS and 1% penicillin/streptomycin) and mineralized media (Ham’s F-12 media supplemented with 12% FBS, 1% penicillin/streptomycin, 3mM of β-glycerophosphate (Sigma) and 10μg/ml of ascorbic acid (Fisher)). The cultures were maintained for 21 days. At days 1, 3, 7, 14, and 21, polymer films cultured in the normal media were removed and characterized for cell proliferation and morphological analysis. At days 7, 14, and 21, cells cultured in the mineralized media were characterized for cell differentiation and mineralized matrix synthesis.
Cell morphology
At days 1, 3, 7, 14, and 21, the polymer films were gently washed with PBS to remove any unattached cells. Specimens were fixed at 4°C in 1% and 3% glutaraldehyde for 1 and 24 h, respectively. After being dehydrated with increasing concentration of ethanol (10, 30, 50, 70, 90, 95, and 100%) and washed with distilled water, the films were allowed to air dry overnight at room temperature. SEM samples were sputter coated with Au/Pd and examined under a JEOL 6700F SEM.
Cell proliferation
The proliferation of PRO cells on the polymer films was analyzed with the use of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl-2-(4-sulfophenyl)-2H-tetrazolium) (MTS, Promega) mitochondrial reduction. This assay is based on the ability of metabolically active cells to bioreduce a tetrazolium-based compound, MTS, to a purple formazan product. The intensity of the resulting colored solution as measured by the absorbance at 490 nm is directly proportional to the number of viable cells. Briefly, PRO cells were seeded onto the polymer films at a seeding density of 50,000 cells per scaffold. Cells seeded onto tissue-culture polystyrene (TCPS) were used as a control. At days 1, 3, 7, 14, and 21, the films with cultured cells were washed with PBS, transferred into a new 48-well tissue-culture plate containing 500 μL culture medium and 100 μL MTS substrate and incubated for 2 h in a humidified atmosphere with 5% CO2 at 37°C. At the end of the incubation period, the reaction was stopped by adding 125 μL of 10% sodium dodecyl sulfate (SDS) solution. The resulting solution was diluted in a 4:1 ratio using distilled water and the absorbance was read at 490 nm using a Tecan SpectroFluo Plus reader (TECAN USA). Cell numbers were calculated based on a standard curve established with the incubation of different concentrations of PRO cells in 500 μL culture medium and 100 μL MTS for 2 h in a humidified atmosphere with 5% CO2 at 37°C.
Alkaline phosphatase activity
Alkaline phosphatase (ALP) activity of PRO cells on the polymer films was evaluated as an early marker of the retention of osteoblastic phenotype using an ALP substrate kit (Bio-Rad). The colorimetric method is based on the conversion of p-nitrophenyl phosphate (P-NPP) into p-nitrophenol (P-NP) in the presence of ALP. The osteoblastic phenotype was stimulated by supplementing the culture medium with 3mM of β-glycerophosphate and 10μg/mL of ascorbic acid. In brief, at days 7, 14, and 21, the media were removed, the cells were washed with PBS, and then lysed with 1 mL of 1% Triton X-100 solution for 30 min. The cell lysates were collected and stored in a −70°C freezer. Three freeze-thaw cycles were performed before analysis. After the third thawing, 100 mL sample was added to 400 mL of P-NPP substrate and buffer solution mixture and incubated at 37°C for 30 min. At the end of the incubation time, the reaction was stopped by adding 500 mL of 0.4 N of sodium hydroxide. The optical density of the solution was determined by the absorbance at 405nm using the TECAN. The intensity of the color formed is proportional to ALP activity. The results for ALP activity were further normalized by the amount of proteins on the scaffolds as determined in a companion protein assay. The protein concentration of cell lysates was determined with a BCA Protein Assay Reagent kit (Pierce) which is based on bicinchoninic acid (BCA) for the colorimetric detection and quantitation of total protein. Proteins of the cell suspension in an alkaline medium can reduce the cupric ions to cuprous ions which can form a chelation complex with BCA. The purple color of this complex is directly proportional to the protein concentration and the absorbance is read at 562 nm using TECAN.
Mineralization assay
Mineralized matrix synthesis was evaluated using Alizarin Red staining method for calcium deposition [26]. This colorimetric analysis is based on solubilizing the red matrix precipitate with cetylpyridinium chloride (CPC) to yield a purple solution. In brief, at days 14 and 21, polymer films were fixed in 70% ethanol at 4°C for 1 h and then stained with 40 mM Alizarin Red (Sigma) solution for 10 min at room temperature. After washing five times with distilled water and once with PBS, the red matrix precipitate was solubilized in 1 mL of 10% CPC (Sigma) until color was stable. The optical density of the solution was read at 562 nm using TECAN. The results for calcium deposition were also normalized with total protein content as determined in BCA assay.
Statistical analysis
All the experiments were run in triplicate per sample. Quantitative data were reported as mean ± standard deviation (SD). Statistical analysis was performed using a one-way anlaysis of variance (one-way ANOVA). Comparison between the two means was determined using the Tukey test with a minimum confidence level of p< 0.05 for statistical significance.
Results
Polymer characterization
The PNEA50PhPh50 polymer was found to have a moderate molecular weight ( = 1,570±20 kDa) with a polydispersity of 2.41±0.07, which is typical for polyphosphazenes obtained by the ring-opening polymerization of hexachlorocyclotriphosphazene [27]. The polymer had a high glass transition temperature of 24.6°C, which is due to the steric bulk and the possibility of π − π stacking of the biphenyl units [14].
The structure and composition of the PNEA50PhPh50 (scheme I) were confirmed by 31P NMR and 1H NMR. 31P NMR peaks were obtained at −1.8, −4.3, −15.6 ppm; 1H NMR peaks were obtained at 1.4 (3H), 4.1 (4H), 7.3–7.5 (9H) ppm.
Scheme I.
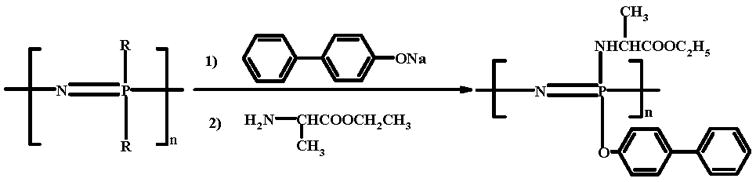
Synthesis of poly[(50% ethyl alanato) (50% p-phenyl phenoxy) phosphazene] (PNEA50PhPh50).
Solvent effect on the blend miscibility
Fig. 1 shows the surface morphology of BLEND25 thin films using different casting solvents. The films of BLEND25 using MC as the solvent are shown in Fig. 1A and B. Fig. 1C and D present that the films fabricated using CF as the solvent. All these films have a non-uniform surface with two different domains visible. On the other hand, BLEND25 presented in Fig. 1E and F using THF as the solvent shows a smooth uniform surface with no evidence of phase separation.
Figure 1.

SEM micrographs of BLEND25 films fabricated using three different solvents, where (A, B) methylene chloride, (C, D) chloroform, and (E, F) tetrahydrofuran were presented in two different magnifications of ×1000 and ×5000 respectively. BLEND25 fabricated using THF resulted in a uniform and smooth surface without any visual phase separation.
Fig. 2 represents the DSC thermogram for the BLEND25 films fabricated using three different solvents. Blend films fabricated with solvents MC and CF showed the presence of two distinct transitions corresponding to two Tg values for each film indicating phase separation. Whereas films fabricated using THF as the solvent showed a single Tg and its value was intermediate between those of the two parent polymers.
Figure 2.

Differential scanning calorimetry thermograms for BLEND25 films fabricated using three different solvents namely methylene chloride (MC), chloroform (CF) and tetrahydrofuran (THF). Blends fabricated using THF presented a single Tg value ~33.5°C indicating the blend miscibility, while two temperature transitions (indicated by the arrows) were observed for BLEND25 fabricated using MC and CF.
Characterization of Polyphosphazene/PLAGA blends
Surface morphology of the blends
The morphology of the PLAGA-PNEA50PhPh50 blend films at various compositions fabricated using THF as a casting solvent is shown in Fig. 3. The SEM micrograph presented in Fig. 3A showed that BLEND25 had a smooth uniform surface with no evidence of phase separation. Whereas micrographs for blends namely BLEND50 and BLEND75 presented in Fig. 3B and C showed the visible phase separation.
Figure 3.
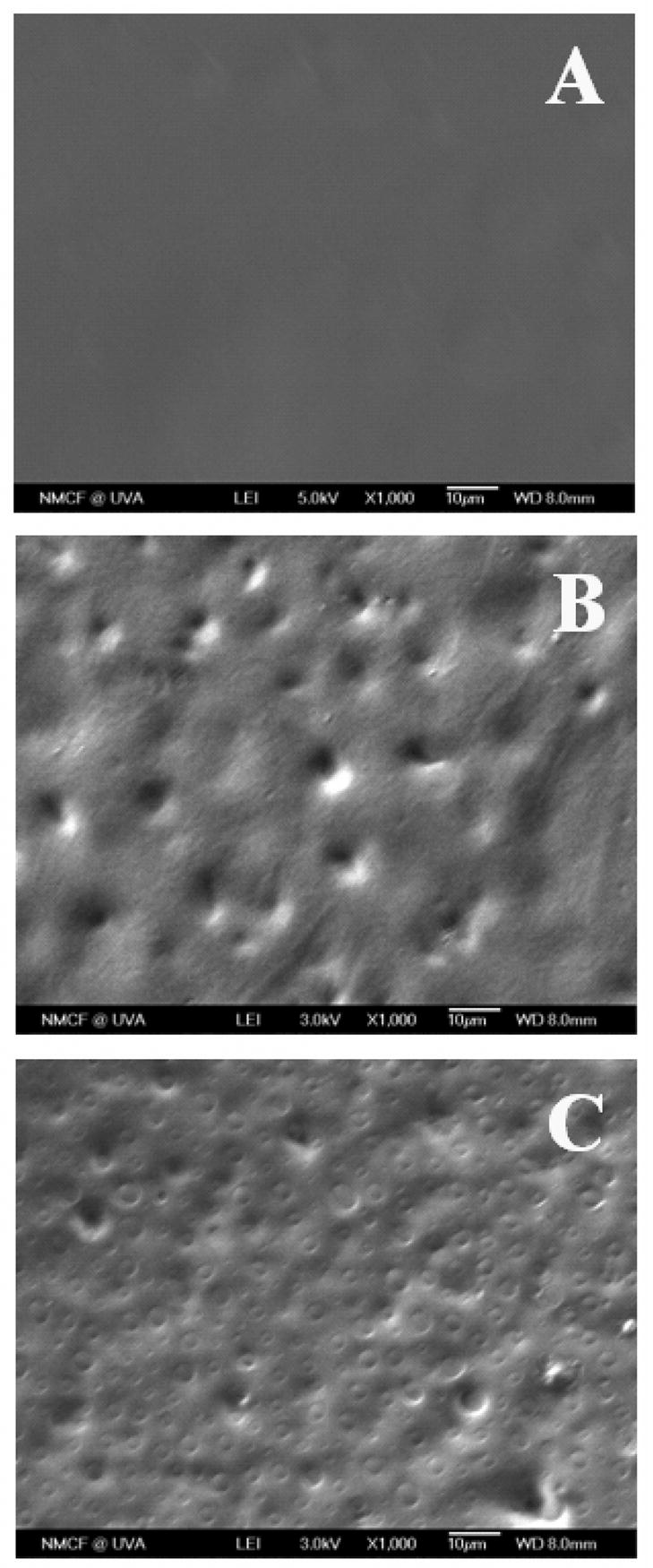
SEM micrographs of (A) BLEND25, (B) BLEND50, and (C) BLEND75 fabricated using THF as solvent at a magnification of ×1000. BLEND25 showed uniform morphology indicating blend miscibility whereas other two blends showed visible phase separation.
Differential scanning calorimetry (DSC) measurements
Glass transition temperatures for the parent polymers and the blends are listed in Table 1. BLEND25 showed a single Tg ~33.5°C and whereas PLAGA and PNEA50PhPh50 showed Tg ~50.7°C and ~24.6°C, respectively. The single Tg value for BLEND25 was intermediate between those of the two parent polymers indicating polymer miscibility at that composition. Blends at two other compositions namely BLEND50 and BLEND75 presented two Tg values at 27.4°C and 38.8°C, 26.1°C and 47.2°C respectively. The upper Tg for these two blends was found to be 3–12°C lower than that of PLAGA and the lower Tg for the blends was found to be 2–3°C higher than that of PNEA50PhPh50, indicating partial miscibility.
Table 1.
The glass transition temperatures (Tg) of PNEA50PhPh50-PLAGA blends as determined by DSC studies and apparent weight fraction (ω) of PNEA50PhPh50 and PLAGA in the PNEA50PhPh50-rich phase and PLAGA-rich phase of blends.
| Sample | PNEA50PhPh50 (%) | Tg′a(K) | Tg″a(K) | ΔTg(K) | ω1′b | ω2′b | ω1″b | ω2″b | Δωc |
|---|---|---|---|---|---|---|---|---|---|
| PNEA50PhPh50 | 100 | 297.8 | |||||||
| BLEND75 | 75 | 299.3 | 320.4 | 21.1 | 0.943 (0.938) | 0.057 (0.062) | 0.134 (0.125) | 0.866 (0.875) | 16.25 (14.94) |
| BLEND50 | 50 | 300.6 | 312.0 | 11.4 | 0.893 (0.884) | 0.107 (0.116) | 0.456 (0.435) | 0.544 (0.565) | 7.48 (6.88) |
| BLEND25 | 25 | 306.7 | 0 | ||||||
| PLAGA | 0 | 323.9 | |||||||
Tg′ and Tg″ represented the glass transition temperature of PNEA50PhPh50-rich and PLAGA-rich phase, respectively.
The apparent weight fraction obtained by using Wood method and those in bracket using Fox method.
Δω = ω1′/ω2′− ω1″/ω2″.
Furthermore, for BLEND50 and BLEND75, we can estimate the apparent weight fractions of PNEA50PhPh50 and PLAGA in PNEA50PhPh50-rich phase and PLAGA-rich phase, respectively. For a miscible blend system, the Tg of the blend can be described by the Wood [28] equation: Tg=w1 Tg1+ w2 Tg2 where wi and Tgi are the weight fraction and the Tg of polymer i (1 and 2 designate PNEA50PhPh50 and PLAGA, respectively). For a partially miscible blend, the Wood equation can be rearranged as ω1=(Tg−Tg2)/(Tg1−Tg2) where ω1 is the apparent weight fraction of PNEA50PhPh50 in either the PNEA50PhPh50-rich phase or the PLAGA-rich phase denoted by ω1′ and ω1″ and Tg represents the glass transition temperature of the corresponding phase. Similarly, for Fox equation [29], we can have ω1= [Tg1 (Tg−Tg2)]/[ Tg( Tg1−Tg2)]. The miscibility of PNEA50PhPh50 and PLAGA can be evaluated by ΔTg(=Tg″−Tg′) as well as the value of Δω (=ω1′/ω2′−ω1″/ω2″). The smaller the Δω, the better miscibility the blend system has. Among these three blends, BLEND25 has zero ΔTg, whereas BLEND75 has the biggest ΔTg and Δω as shown in Table 1. Therefore, the order of miscibility of the blends is BLEND25>BLEND50>BLEND75.
Fourier transform infrared (FTIR) analysis
Fig. 4 shows the FTIR spectra of the polymers and the corresponding blends in the carbonyl stretching frequency region. As it is evident from FTIR, the C=O stretching vibrations of PNEA50PhPh50 and PLAGA occur at ~1739 and ~1750 cm−1, respectively. The carbonyl hydrogen bonding was confirmed with a shift of the carbonyl peak from ~1750 to ~1739 cm−1. For BLEND25, an additional band was observed at ~1678 cm−1.and such IR bands of hydrogen bonded carbonyl groups could have a redshift depending on the chemical structure of the polymers [23,30].
Figure 4.
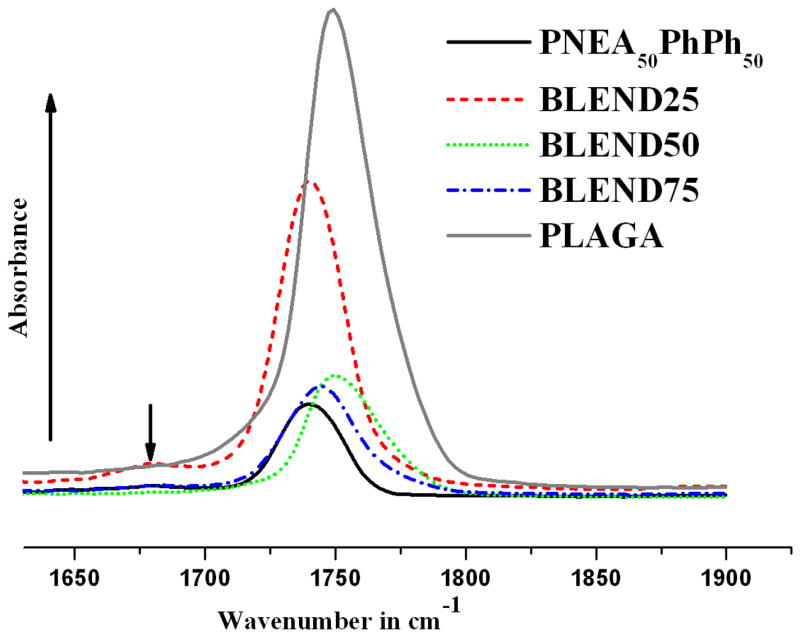
FTIR spectrum presenting carbonyl absorption region for PNEA50PhPh50, PLAGA and their blends. Carbonyl stretching frequencies shifted from ~1750 to ~1739 cm−1 indicating intermolecular hydrogen bonding that cause the miscibility of these blends.
Biomimetic process
Fig. 5A–F showed SEM micrographs of polymer films after incubation in 1.5x SBF at 37°C for three weeks. At day 21, a layer of spherical nano-apatite was observed on the surface of PNEA50PhPh50 (Fig. 5A–C) and BLEND25 (Fig. 5D–F). Furthermore, EDX confirms the presence of calcium and phosphorus with Ca/P atomic ratio around 1.67 (Fig. 5G), which is same as the stoichiometric ratio of 1.67 for hydroxyapatite [Ca10(PO4)6(OH)2].
Figure 5.
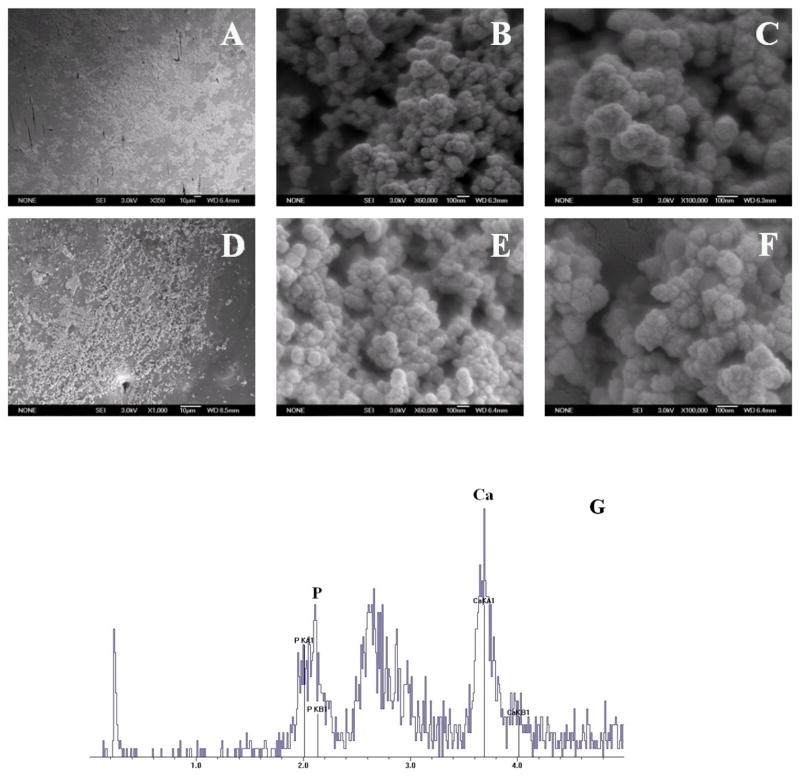
SEM micrographs of PNEA50PhPh50 and BLEND25 films incubated in 1.5x SBF at 37°C for 21 days, where (A–C) represented PNEA50PhPh50 after 21 days of incubation at a magnification of ×350, ×60,000 and ×100,000 respectively; (D–F) represented BLEND25 after 21 days of incubation at a magnification of ×1000, ×60,000 and ×100,000 respectively; (G) confirmed the presence of calcium and phosphorus with Ca/P atomic ratio around 1.67.
In vitro cellular behavior on blend surfaces
Cell morphology and proliferation
The behavior of osteoblasts in contact with polymer films was visualized using SEM. The morphological sequences reported represent the average cellular morphology found at each time point, because all individual cells do not spread at the same rate. PRO showed a normal morphological sequence of proliferation on the polymer blends. At day 7, cells with filopodial projections started to spread out on the surface of BLEND75 as shown in Fig. 6A and B. At day 14, spindle-like cells completely covered the surface of the BLEND75 as evident from Fig. 6C and D. After 21 days of culture, completely spread interconnected osteoblast cells formed multilayers on the surface of BLEND75 as evident from Fig. 6E and F.
Figure 6.
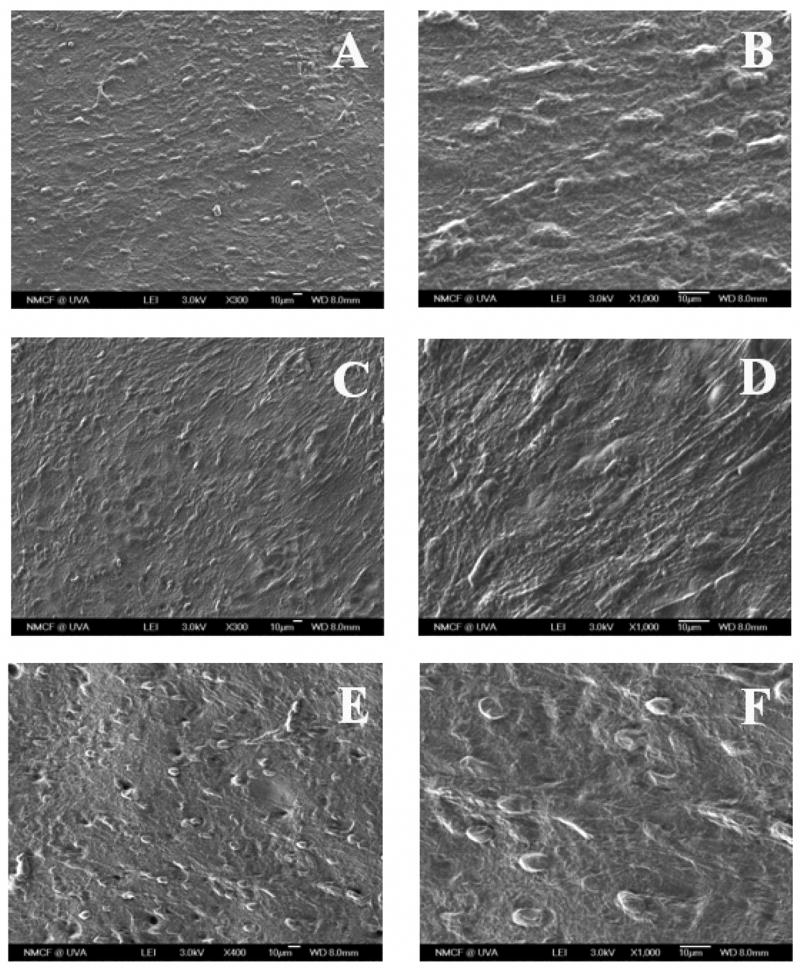
SEM micrographs of primary rat osteoblasts cultured on the films of BLEND75 at different time points, where (A, B) 7 day, (C, D) 14 day, and (E, F) 21 day were presented at a magnification of ×300 and ×1000 respectively for each pair. PRO showed a normal morphological sequence of proliferation with time and by day 21 completely covered cell multilayers were observed.
PRO proliferation on the surfaces of PNEA50PhPh50, BLEND25, BLEND50, BLEND75 and PLAGA films were evaluated by SEM and using the MTS assay. At day 21, a multilayer of PRO could be found on the surfaces of polymer films as evident from Fig. 6F and 7. Fig. 8 presented the PRO proliferation expressed as the number of cells attached to the polymer films and TCPS controls as a function of culture time. The polymer films and TCPS were seeded with 50,000 cells per well, which was consistent with the results at day 1 as shown in Fig. 8. The cell numbers on the polymer films increased with culture time, which confirmed that the cells were able to proliferate on the polyphosphazene and corresponding blends. It was seen that the cell adhesion and growth was significantly higher on tissue culture polystyrene (TCPS) than on all the polymeric substrates after day 7. Furthermore, the comparison of cell number between different polymeric groups yielded no significant differences at early time points (days 1, 3, 7, and 14). By day 21, cell numbers on BLEND25 and BLEND50 were significantly higher than PNEA50PhPh50. The amount of cells on all the blends was comparable to PLAGA at all time points.
Figure 7.
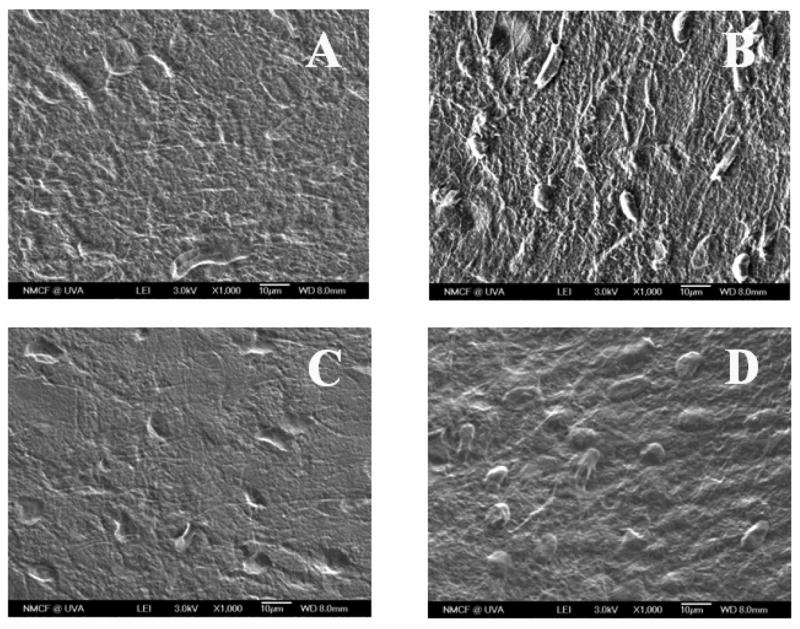
SEM micrographs of primary rat osteoblasts cultured on different polymer films for a period of 21 days, where (A) PNEA50PhPh50, (B) BLEND25, (C) BLEND50, and (D) PLAGA surfaces were presented at a magnification of ×1000. Surfaces of all the polymer and blend films were completely covered by PRO multilayers.
Figure 8.
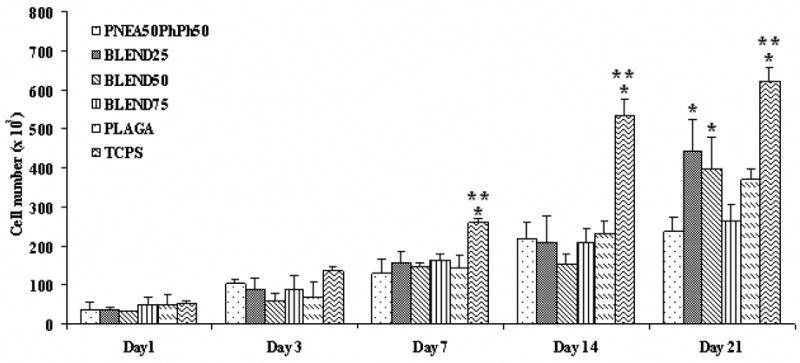
Cell proliferation measured by MTS assay. Cell numbers on these blend films at all the time points were comparable to PLAGA. BLEND25 and BLEND50 showed significantly higher cell number than PNEA50PhPh50 at day 21. (*) denotes significant difference (p<0.05) as compared to PNEA50PhPh50 matrices while (**) indicates significant difference (p<0.05) as compared to PLAGA matrices.
Alkaline phosphatase activity
The phenotypic expression of PRO cultured on the polymer films was evaluated by measuring the ALP activity. It was found that ALP activity of the cells on PNEA50PhPh50 and BLEND75 was significantly enhanced (p<0.05) compared to PLAGA at all time points as evident from Fig. 9. Moreover, the expression of ALP in cells cultured on PNEA50PhPh50 was significantly higher than TCPS at day 21. This demonstrated that bone cell phenotype expression could be stimulated by the PNEA50PhPh50 component.
Figure 9.
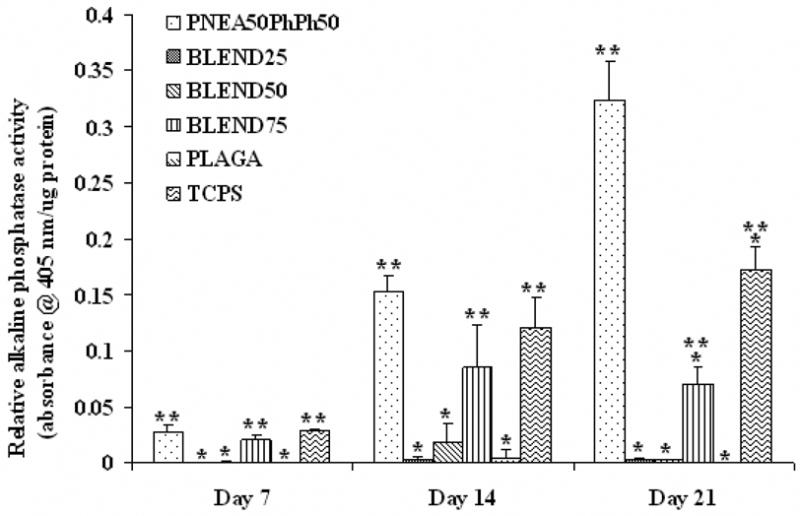
Alkaline phosphatase activity for osteoblastic phenotype expression. At all time points, ALP activity was significantly enhanced on PNEA50PhPh50 and BLEND75 surfaces as compared to PLAGA. (*) denotes significant difference (p<0.05) as compared to PNEA50PhPh50 matrices while (**) indicates significant difference (p<0.05) as compared to PLAGA matrices.
Mineralized matrix formation
The production of calcified matrix was assessed by Alizarin Red histochemical staining. Alizarin Red analysis showed increase in calcium accumulation on all the substrates with culture time, which is evident from Fig. 10. Comparable level of calcium deposition was also found on all the substrates at day 14. However, after 21 days of culture, calcium deposition on all the blends was improved compared to the parent polymers, and the increase for BLEND50 and BLEND75 was significant (p<0.05).
Figure 10.
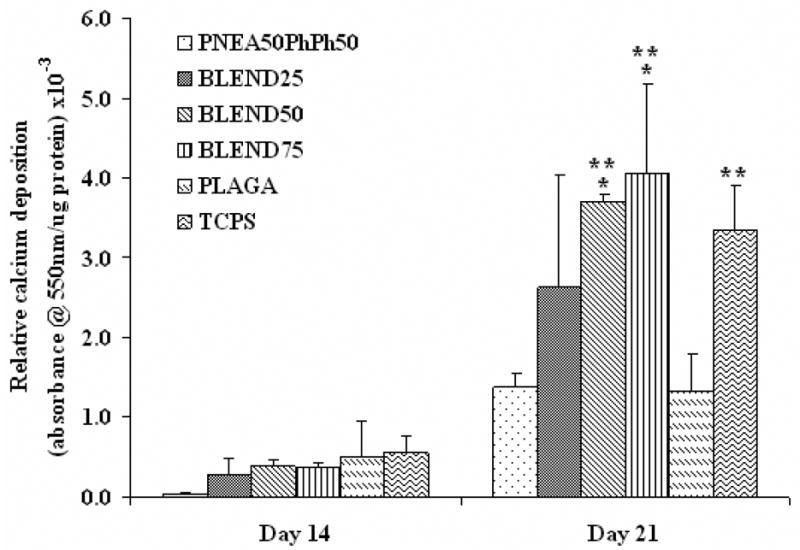
Synthesis of mineralized matrix was assessed by Alizarin Red assay. All the blends showed higher amount of calcium deposition as compared to the parent polymers at day 21. (*) denotes significant difference (p<0.05) as compared to PNEA50PhPh50 matrices while (**) indicates significant difference (p<0.05) as compared to PLAGA matrices.
Discussion
The structure of the PNEA50PhPh50 polymer was confirmed by multinuclear NMR, DSC and molecular-weight determination. The glass transition temperature of this mixed-substituent polymer was 34.6°C higher than that of the single-substituent polymer poly[bis(ethyl alanato)phosphazene] (PNEA) (Tg ~−10°C) due to the incorporation of the bulky phenyl phenoxy groups [14]. This makes the mixed-substitutent polymer possess higher mechanical properties and thus more suitable for bone tissue engineering applications. Moreover, the polymers with hydrolytically less sensitive phenyl phenoxy groups was shown to degrade at a slower rate than amino-acid ester single-substituent polymer in vivo [19].
Previous studies have shown the feasibility of developing miscible blends combining glycine ethyl ester co-substituted polyphosphazenes with PLAGA and showed controllable degradation rates with neutral degradation products [20,21]. We have also evaluated the osteoconductivity of PNEA-PLAGA blend systems and demonstrated the higher osteoblast adhesion on the blend films than the parent polymers [22]. The current study aimed to develop novel biodegradable blends of PNEA50PhPh50 and PLAGA as potential scaffold materials for biomedical purposes. It was hypothesized that the blends can potentially combine the proven biocompatibility and good mechanical properties of PNEA50PhPh50 with the good properties of PLAGA and could lead to the development of osteocompatible, osteointegrative, non-cytotoxic and biodegradable biomaterials for orthopaedic applications.
Blend compatibility or miscibility is very important in applications that require uniform properties [31,32]. For example, two polymers with different degradation profiles can result in a uniform and predictable degradation profile only if they form miscible blends [20,21]. Also, polymer miscibility is also very important in blends that are designed for bone tissue engineering applications [22]. Therefore, the miscibility of PNEA50PhPh50-PLAGA blends has been investigated in order to develop novel blend systems that can incorporate the beneficial features of the parent polymers.
Solvents used to fabricate blends have a significant effect on their miscibility and morphology as reported extensively in the recent literatures [33–36]. For example, Wu et al. [33] characterized the poly(vinyl acetate)-poly(ethylene oxide) (PVAc-PEO) blends and for their study they used chloroform and benzene as solvents. Non-polar benzene resulted in more homogeneous blends for the PVAc-PEO system. In this present study, we investigated three mutual solvents, MC, CF, and THF to study the effect on blend miscibility. The surface morphology of BLEND25 fabricated using these solvents was visualized using SEM as presented in Fig. 1. BLEND25 fabricated using MC or CF showed a rough and phase separated morphology as evident from Fig. 1A–D. While the solvent THF resulted in a smooth surface with no indication of phase separation as seen from Fig. 1E and F. The miscibility of the blends was further confirmed from the DSC thermograms. BLEND25 prepared from THF showed a single transition temperature in the DSC thermogram while two transition temperatures observed for the films fabricated using MC or CF as seen from Fig. 2. Observed single Tg value indicates the blend miscibility and this result is consistent with surface morphology for BLEND25 fabricated using THF as solvent.
Different blend systems were fabricated with different blend compositions using THF as a solvent to study the effect of PNEA50PhPh50 content in the blend system. Three blends namely BLEND25, BLEND50 and BLEND75 were evaluated for their surface morphology, miscibility, osteointegrativity and osteocompatibility. Two blend systems BLEND50 and BLEND75 showed evidence of phase separation visualized by SEM (Fig. 3B and C). DSC thermograms of BLEND50 and BLEND75 showed presence of two Tg values which are slightly different from the Tg values of the parent polymers showing partial miscibility as seen from Table 1. However BLEND25 had a single Tg value around 33.5°C which is intermediate to the Tg values of the parent polymers indicating blend miscibility.
The miscibility of any two polymers in a blend is generally a result of strong intermolecular interactions between the polymers such as through hydrogen bonding, dipole-dipole interactions and/or van der Waals forces [20]. In the present study, hydrogen bonding between amino groups of PNEA50PhPh50 and ester groups of PLAGA is predominant over dipole-dipole and van der Waals forces of attraction that might potentially improve blend miscibility [22]. FTIR measurements support the extent of hydrogen bonding resulting from carbonyl and amino group interactions of two polymers. It is evident from FTIR spectrum presented in Fig. 4, that the carbonyl hydrogen bonding was confirmed with a shift of the carbonyl band from ~1750 to ~1739 cm−1. BLEND25 showed an additional band ~ 1678 cm−1 which might be due to the redshift as a result of hydrogen bonded carbonyl groups [23,30]. A similar hydrogen bonding effect was reported for the polyurethane-polyester blend system resulting from the interaction of –C(=O)-NH- and carbonyl groups [30]. The present blend system PNEA50PhPh50-PLAGA also has structural similarity and might form hydrogen bonding between -N=P(NH-)- and carbonyl groups if the steric hindrance of the α-CH3 groups of the alanine and lactide units, and bulky aromatic side groups has been overcomed [22]. In the present study, it is evident that PNEA50PhPh50 can form a miscible blend with PLAGA at a weight ratio of 1:3 (BLEND25) via the hydrogen bonding when using THF as the solvent. THF has definitely played an important role to increase the probability of hydrogen bonding between the PNEA50PhPh50 and PLAGA. However, with an increasing amount of PNEA50PhPh50 in the blend composition, the steric hindrance from the bulky aromatic side groups has increased so dramatically that might possibly limit the blend miscibility as observed in BLEND50 and BLEND75.
The films of PNEA50PhPh50 and blends also showed the ability to nucleate inorganic minerals in 1.5x SBF at 37°C. A multilayer of nano-apatite was formed on the surface of PNEA50PhPh50 and BLEND25 after 21 days (Fig. 5). Similar apatite layers were also found on BLEND50 and BLEND75. Nucleation of inorganic salts on the blend surface is induced by the carboxylic groups resulting from the hydrolysis of PLAGA and alanine side groups fromPNEA50PhPh50, which possibly chelate calcium ions in the SBF solution [37,38]. Such an apatite deposition might be able to promote osteoblast proliferation and differentiation, that makes our blend systems very promising for bone tissue engineering applications [24].
Our previous studies have demonstrated better osteoblast responses to PNEA-PLAGA blend systems than parent polymers. The highest osteoblast adhesion was observed for blends [22]. In this study, PRO cells were used, since they have properties closer to the in vivo cells and might be more indicative of further in vivo studies. PRO showed a normal morphological sequence of adhesion and proliferation on all the blends as reported on the surface of other osteocompatible biodegradable polymers (Fig. 6). A multilayer of osteoblast cells could be found on the surfaces of the polymer and blend films after 21 days post cell seeding as evident from Fig. 6F and 7. This observation was further supported by viability and proliferation results of osteoblast cells on these films as presented in Fig. 8. Cell numbers on these blend films at all the time points were comparable to PLAGA films, which indicated good osteocompatibility of the novel blends. Furthermore, BLEND25 and BLEND50 showed significantly higher cell number than the PNEA50PhPh50 after 21 days. Direct comparison of the cell number on different blends at day 21 showed that with the increase of PNEA50PhPh50 in the blend, the cell number decreased accordingly from BLEND25 to BLEND75, indicating that better miscibility might induce better cell growth [22].
Alkaline phosphatase expression, an indicator of the osteoblastic phenotype, on all the blends peaked at day 14 as seen from Fig. 9 and the gradual elevation of ALP expression from BLEND25 to BLEND75 suggested that the ALP activity could be enhanced by increasing the PNEA50PhPh50 content in the blend. Furthermore, ALP activity of the cells on PNEA50PhPh50 and BLEND75 was significantly higher than on PLAGA at all time points, which again indicated that the phenotypic expression of osteoblasts was enhanced due to the presence of PNEA50PhPh50. When comparing the results of cell proliferation and ALP activity, cell number stopped increasing and ALP maintained a high level on PNEA50PhPh50, BLEND75 and TCPS from day 14 to day 21, whereas BLEND25, BLEND50 and PLAGA showed minimal ALP and continued cell proliferation during the same period. In general, the cells of regenerating tissues have an initial period of proliferation, which diminishes when certain threshold of cell density is reached that contributes to contact inhibition as well as differentiation signaling cascades [39]. These sequential proliferation and differentiation phases explain the above findings. Alizarin Red analysis showed that calcium deposition had similar trend as ALP expression. At day 21, BLEND75 had the highest calcium deposition, whereas BLEND25 had the least as seen from Fig. 10, which again demonstrated the biological importance of adding the PNEA50PhPh50 component. In addition, the calcium deposition on all the blends was enhanced compared to the parent polymers which demonstrated the benefits of blending. In the present study, the expression of alkaline phosphatase by PRO started at day 7, and matrix synthesis at day 14 as evident from Fig. 9 and 10. Similar observations were also made earlier using 2D cultures [39]. Furthermore, with the onset of mineralization after day 14, there is an increased expression of an ordered deposition of calcium and phosphate as seen from Fig. 10. At day 21, expression of alkaline phosphatase tended to plateau for all the blends, indicating a later stage of osteoblast differentiation which is evident from Fig. 9. This presents a developmental sequence of osteoblast maturation similar to the one observed in calvaria in vivo [40].
Based on the composition, these blends possess different physical, chemical and biological properties. BLEND25 is more miscible and has more cell attachment and proliferation; whereas BLEND75 has highest ALP activity and calcium deposition. Different blends can be adopted depending on the application purposes. For the bone scaffold materials, although BLEND75 showed highest ALP activity and calcium deposition, miscible BLEND25 is preferred because of its buffering capacity to have neutral degradation products, since miscibility determines the efficacy of the blends to buffer the acidic degradation products of PLAGA.
Conclusions
In this study, an amino acid ester phenyl phenoxy phosphazene (PNEA50PhPh50) has been synthesized, characterized. Blends of PNEA50PhPh50 and PLAGA were prepared using the mutual solvent approach at various compositions. THF produced a miscible blend at a weight ratio of 1:3 (BLEND25) and other compositions namely BLEND50 and BLEND75 were partially miscible. The biomimetic study showed the feasibility of nucleating bone-like apatite on the blend surface. Furthermore, the in vitro study on the blends indicated that the cell adhesion and proliferation on novel blends was comparable to PLAGA and better than PNEA50PhPh50. The presence of PNEA50PhPh50 also contributed to the enhancement of the phenotypic expression and mineralized matrix synthesis of the primary rat osteoblast cells in vitro. Blends of alanine containing polyphosphazenes and poly(lactic acid-glycolic acid) are promising biomaterials for a variety of musculoskeletal applications.
Acknowledgments
This work was supported by NIH RO1 EB004051. Dr. Laurencin was the recipient of Presidential Faculty Fellow Award from the National Science Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Langer R, Tirrell DA. Designing materials for biology and medicine. Nature. 2004;428:487–492. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- 2.Hubbell JA. Biomaterials in Tissue Engineering. Nat Biotech. 1995;13:565–576. doi: 10.1038/nbt0695-565. [DOI] [PubMed] [Google Scholar]
- 3.Burg KJL, Porter S, Kellam JF. Biomaterial developments for bone tissue engineering. Biomaterials. 2000;21:2347–2359. doi: 10.1016/s0142-9612(00)00102-2. [DOI] [PubMed] [Google Scholar]
- 4.Ratner BD, Bryant SJ. BIOMATERIALS: Where We Have Been and Where We are Going. Annu Rev Biomed Eng. 2004;6:41–75. doi: 10.1146/annurev.bioeng.6.040803.140027. [DOI] [PubMed] [Google Scholar]
- 5.Vacanti JP, Vacanti CA. The history and scope of tissue engineering. In: Lanza RP, Langer R, Vacanti JP, editors. Principles of Tissue Engineering. New York: Academic Press; 2000. pp. 3–8. [Google Scholar]
- 6.Gunatillake PA, Adhikari R. Biodegradable synthetic polymers for tissue engineering. Eur Cell Mater. 2003;5:1–16. doi: 10.22203/ecm.v005a01. [DOI] [PubMed] [Google Scholar]
- 7.Middleton JC, Tipton AJ. Synthetic biodegradable polymers as orthopedic devices. Biomaterials. 2000;21:2335–2346. doi: 10.1016/s0142-9612(00)00101-0. [DOI] [PubMed] [Google Scholar]
- 8.Cordewener FW, Schmitz JP. The Future of Biodegradable Osteosyntheses. Tissue Eng. 2000;6:413–424. doi: 10.1089/107632700418119. [DOI] [PubMed] [Google Scholar]
- 9.Bostman OM. Osteoarthritis of the ankle after foreign-body reaction to absorbable pins and screws: A THREE- TO NINE-YEAR FOLLOW-UP STUDY. J Bone Joint Surg Br. 1998;80-B:333–338. doi: 10.1302/0301-620x.80b2.8302. [DOI] [PubMed] [Google Scholar]
- 10.Allcock HR. Chemistry and Applications of Polyphosphazenes. Hoboken, New Jersey: John Wiley & Sons; 2003. [Google Scholar]
- 11.Allcock HR, Fuller TJ, Mack DP, Matsumura K, Smeltz KM. Synthesis of Poly[(amino acid alkyl ester)phosphazenes] Macromolecules. 1977;10:824–830. [Google Scholar]
- 12.Allcock HR, Fuller TJ, Matsumura K. Hydrolysis pathways for aminophosphazenes. Inorg Chem. 1982;21:515–521. [Google Scholar]
- 13.Allcock HR, Pucher SR, Scopelianos AG. Poly[(amino acid ester)phosphazenes] as substrates for the controlled release of small molecules. Biomaterials. 1994;15:563–569. doi: 10.1016/0142-9612(94)90205-4. [DOI] [PubMed] [Google Scholar]
- 14.Singh A, Krogman NR, Sethuraman S, et al. Effect of Side Group Chemistry on the Properties of Biodegradable L-Alanine Cosubstituted Polyphosphazenes. Biomacromolecules. 2006;7:914–918. doi: 10.1021/bm050752r. [DOI] [PubMed] [Google Scholar]
- 15.Nair S, Katti DS, Laurencin CT. Biodegradable polyphosphazenes for drug delivery applications. Adv Drug Deliv Rev. 2003;55:467–482. doi: 10.1016/s0169-409x(03)00039-5. [DOI] [PubMed] [Google Scholar]
- 16.Laurencin CT, Norman ME, Elgendy HM, et al. Use of polyphosphazenes for skeletal tissue regeneration. J Biomed Mater Res. 1993;27:963–973. doi: 10.1002/jbm.820270716. [DOI] [PubMed] [Google Scholar]
- 17.Laurencin CT, El-Amin SF, Ibim SE, et al. A highly porous 3-dimensional polyphosphazene polymer matrix for skeletal tissue regeneration. J Biomed Mater Res. 1996;30:133–138. doi: 10.1002/(SICI)1097-4636(199602)30:2<133::AID-JBM1>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 18.Sethuraman S, Nair LS, Singh A, et al. Synthesis and evaluation of novel amino acid ester phenyl phenoxy polyphosphazene for bone tissue engineering. Transactions of the 5th Combined Meeting of the Orthopaedic Research Societies of Canada; USA, Japan, and Europe. October 2004; p. 5. [Google Scholar]
- 19.Sethuraman S, Nair LS, Laurencin CT. In vivo biodegradability and biocompatibility evaluation of novel alanine ester based polyphosphazenes in a rat model. J Biomed Mater Res A. 2006;77A:679–687. doi: 10.1002/jbm.a.30620. [DOI] [PubMed] [Google Scholar]
- 20.Ibim SEM, Ambrosio AMA, Kwon MS, El-Amin SF, Allcock HR, Laurencin CT. Novel polyphosphazene/poly(lactide-co-glycolide) blends: miscibility and degradation studies. Biomaterials. 1997;18:1565–1569. doi: 10.1016/s0142-9612(97)80009-9. [DOI] [PubMed] [Google Scholar]
- 21.Ambrosio AMA, Allcock HR, Katti DS, Laurencin CT. Degradable polyphosphazene/poly([alpha]-hydroxyester) blends: degradation studies. Biomaterials. 2002;23:1667–1672. doi: 10.1016/s0142-9612(01)00293-9. [DOI] [PubMed] [Google Scholar]
- 22.Nair LS, Allcock HR, Laurencin CT. Biodegradable Poly[bis(ethyl alanato)phosphazene]-Poly(lactide-co-glycolide) Blends: Miscibility and Osteocompatibility Evaluations. Mater Res Soc Symp Proc. 2005 [Google Scholar]
- 23.Qiu LY, Zhu KJ. Novel blends of poly[bis(glycine ethyl ester) phosphazene] and polyesters or polyanhydrides: compatibility and degradation characteristics in vitro. Polymer Int. 2000;49:1283–1288. [Google Scholar]
- 24.Chou Y-F, Huang W, Dunn JCY, Miller TA, Wu BM. The effect of biomimetic apatite structure on osteoblast viability, proliferation, and gene expression. Biomaterials. 2005;26:285–295. doi: 10.1016/j.biomaterials.2004.02.030. [DOI] [PubMed] [Google Scholar]
- 25.Schwartz E. Culture of animal cells. New York: Wiley; 1987. [Google Scholar]
- 26.Botchwey EA, Pollack SR, Levine EM, Laurencin CT. Bone tissue engineering in a rotating bioreactor using a microcarrier matrix system. J Biomed Mater Res. 2001;55:242–253. doi: 10.1002/1097-4636(200105)55:2<242::aid-jbm1011>3.0.co;2-d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nair LS, Lee DA, Bender JD, et al. Synthesis, characterization, and osteocompatibility evaluation of novel alanine-based polyphosphazenes. J Biomed Mater Res A. 2006;76A:206–213. doi: 10.1002/jbm.a.30532. [DOI] [PubMed] [Google Scholar]
- 28.Lawrence A. Wood. Glass transition temperatures of copolymers. J Polymer Sci. 1958;28:319–330. [Google Scholar]
- 29.Fox T. Influence of diluent and of copolymer composition on the glass temperature of a polymer system. Bull Am Phys Soc. 1956;1:123. [Google Scholar]
- 30.Coleman MM, Skrovanek DJ, Hu J, Painter PC. Hydrogen bonding in polymer blends. 1. FTIR studies of urethane-ether blends. Macromolecules. 1988;21:59–65. [Google Scholar]
- 31.Su CC, Woo EM. Cure kinetics and morphology of amine-cured tetraglycidyl-4,4′-diaminodiphenylmethane epoxy blends with poly(ether imide) Polymer. 1995;36:2883–2894. [Google Scholar]
- 32.Hourston DJ, Lane S, Zhang HX. Toughened thermoplastics: 3. Blends of poly(butylene terephthalate) with (butadiene-co-acrylonitrile) rubbers. Polymer. 1995;36:3051–3054. [Google Scholar]
- 33.Wu WB, Chiu WY, Liau WB. Casting solvent effect on crystallization behavior of poly(vinyl acetate)/poly(ethylene oxide) blends: DSC study. J Appl Polymer Sci. 1997;64:411–421. [Google Scholar]
- 34.Tang M, Liau W-R. Solvent effect on the miscibility of poly(4-hydroxystyrene)-poly(ethylene oxide) blends. Eur Polymer J. 2000;36:2597–2603. [Google Scholar]
- 35.Radhakrishnan S, Venkatachalapathy PD. Effect of casting solvent on the crystallization in PEO/PMMA blends. Polymer. 1996;37:3749–3752. [Google Scholar]
- 36.He Y, Asakawa N, Inoue Y. Biodegradable blends of high molecular weight poly(ethylene oxide) with poly(3-hydroxypropionic acid) and poly(3-hydroxybutyric acid): a miscibility study by DSC, DMTA and NMR spectroscopy. Polymer Int. 2000;49:609–617. [Google Scholar]
- 37.Murphy WL, Mooney DJ. Bioinspired Growth of Crystalline Carbonate Apatite on Biodegradable Polymer Substrata. J Am Chem Soc. 2002;124:1910–1917. doi: 10.1021/ja012433n. [DOI] [PubMed] [Google Scholar]
- 38.Masami Tanahashi TM. Surface functional group dependence on apatite formation on self-assembled monolayers in a simulated body fluid. J Biomed Mater Res. 1997;34:305–315. doi: 10.1002/(sici)1097-4636(19970305)34:3<305::aid-jbm5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 39.Stein GS, Lian J, Stein J. Principles of Bone Biology. San Diego: Academic; 1996. [Google Scholar]
- 40.Dworetzky S, Fey E, Penman S, Lian J, Stein J, Stein G. Progressive Changes in the Protein Composition of the Nuclear Matrix During Rat Osteoblast Differentiation. Proc Natl Acad Sci U S A. 1990;87:4605–4609. doi: 10.1073/pnas.87.12.4605. [DOI] [PMC free article] [PubMed] [Google Scholar]