

Abstract
Certain highly soluble proteins, such as Escherichia coli maltose-binding protein (MBP), have the ability to enhance the solubility of their fusion partners, making them attractive vehicles for the production of recombinant proteins, yet the mechanism of solubility enhancement remains poorly understood. Here, we report that the solubility-enhancing properties of MBP are dramatically affected by amino acid substitutions that alter the equilibrium between its “open” and “closed” conformations. Our findings indicate that the solubility-enhancing activity of MBP is mediated by its open conformation and point to a likely role for the ligand-binding cleft in the mechanism of solubility enhancement.
Keywords: maltose binding protein, solubility enhancer, affinity tag, fusion protein, solubility tag
Proteins that normally accumulate in the form of insoluble aggregates when they are overproduced in E. coli can sometimes be recovered in a soluble, properly folded form if they are fused to a solubility-enhancing partner [1]. Consequently, the use of solubility-enhancing fusion partners has become an attractive alternative to the refolding of proteins. Many proteins have been reported to exhibit solubility-enhancing characteristics, including MBP, NusA, DsbA, thioredoxin, T7PK, Skp, SUMO, GB1 and the ZZ domain [2], although the evidence supporting these claims is stronger in some instances than in others. Yet the mechanism(s) by which certain soluble proteins enhance the solubility of their fusion partners remains poorly understood.
E. coli MBP is an excellent solubility enhancer [3,4,5,6], but it is not the most effective affinity tag from the standpoint of protein purification [7,8]. Some fusion proteins do not bind efficiently to amylose resin, and even when they do, the purity of proteins after amylose affinity chromatography is usually inadequate. Two groups recently described mutations in MBP that increase its affinity for maltose [9,10]. Using a variety of experimental techniques, both groups reached the conclusion that these mutations exert their effect by altering the equilibrium between the “open” and “closed” conformations of MBP so as to favor the latter. In the open conformation, the ligand-binding cleft of MBP is exposed to solvent, whereas the closed conformation resembles that of the ligand-bound protein in which the cleft is largely buried. We originally set out to determine whether these mutations would improve the performance of MBP as an affinity tag for protein purification but serendipituously discovered that they have a profound impact on the ability of MBP to promote the solubility of its fusion partners, indicating that the solubilizing properties of MBP are mediated by its open conformation. Moreover, we have found that mutations of conserved hydrophobic residues in the ligand-binding cleft of MBP give rise to a similar phenotype, implicating the cleft in the mechanism of solubility enhancement.
Materials and methods
Construction of mutant His-MBP destination vectors
A plasmid encoding the DM mutant of E. coli MBP (pLH1DM2) was provided by Patrick Telmer and Brian Shilton [9]. This plasmid was transformed into an expression vector that was designed to overproduce the DM mutant MBP in the cytoplasm of E. coli as follows. First, pLH1DM2, which carries a combination of two amino acid substitutions (M321A and Q325A) and in-frame deletion of residues 172-176 in MBP, was used as the template for polymerase chain reaction (PCR) with oligodeoxribonucleotide primers PE-42 (5′-GGC ACA CGA CCG CTT TGG TGG CTA C-3′) and PE-1424 (5′-CTG ACG ACC GCT GGC GGC GTT GAT CAC C-3′). This PCR amplicon included the amino acid substitutions and deletion that define the DM mutant. In a second PCR, the C-terminal portion of the MBP coding sequence, along with some DNA from the downstream att recombination site, was amplified from pKM596 [11] with the primers PE-1423 (5′-GGT GAT CAA CGC CGC CAG CGG TCG TCA G-3′) and PE-1425 (5′-CAT TGA GCA ACT GAC TGA AAT GCC TC-3′). Next, the amplicons from these two PCRs were combined and used as the template for another PCR with primers PE-42 and PE-1425. The resulting PCR amplicon was digested with BglII and NotI, and then inserted between the unique BglII and NotI sites in pDEST-HisMBP [12] to generate an otherwise idential derivative of pDEST-HisMBP encoding the DM mutant MBP. Derivatives of pDEST-HisMBP encoding the I329W, W62E/Y155E, and W230E/W340E mutant MBPs were constructed with a QuickChange Site-Directed Mutagenesis kit (Stratagene). The nucleotide sequences of all HisMBP destination vectors were confirmed experimentally.
Construction of fusion protein expression vectors
Various wild-type and mutant HisMBP fusion protein expression vectors were constructed by Gateway recombinational cloning (Invitrogen), using the five destination vectors described above in combination with three entry clones (E6, p16, and GFP) described previously [3]. The standard LR protocol was employed [13].
Protein expression and evaluation of solubility
The expression and solubility of various HisMBP fusion proteins was evaluated by standard procedures, using E. coli BL21(DE3) CodonPlus-RIL cells (Stratagene), as described [3,14,12]. All cultures were grown at 37 °C. Preparation of total and soluble intracellular protein samples and SDS-PAGE were performed as described [13].
Overproduction and purification of wild-type and mutant HisMBP proteins
Escherichia coli DB3.1 cells (Invitrogen, Carlsbad, CA, USA) containing wild-type or mutant HisMBP destination vectors were grown separately to saturation at 37 °C in Luria broth supplemented with 100 g/ml ampicillin. The saturated cultures were diluted in 1 L of the same medium (1:50) and grown to mid-log phase (A600nm ~ 0.4-0.5) at 37 °C. At this point, IPTG was added to a final concentration of 1mM to initiate production of the recombinant proteins and the cultures were grown for an additional 3-4 h at the same temperature. The cells were harvested by centrifugation and the cell pellets were stored at -80 °C.
All chromatography steps were carried out at 4 °C. E. coli cell paste was suspended in ice-cold 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 25 mM imidazole (buffer A) containing complete EDTA-free protease-inhibitor cocktail (Roche Molecular Biochemicals). The cells were lysed with an APV Gaulin Model G1000 homogenizer at 69 Mpa and centrifuged at 30,000g for 30 min at 4 °C. The supernatant was filtered through a 0.45 μM polyethersulfone membrane and then loaded onto three tandem 5 mL HisTrap affinity columns (Amersham Biosciences) equilibrated in buffer A. The column was washed to baseline with buffer A and then eluted with linear gradient from 25 to 250 mM imidazole in buffer A. The fractions containing MBP were pooled and concentrated to a volume of 5 mL. This sample was next applied to a 26/60 Hiprep Sephacryl S100 prep-grade column (Amersham Biosciences) equilibrated with 25 mM Tris-HCl (pH 8.0), 150 mM NaCl, 2mM TCEP. The peak fractions containing pure MBP were pooled and concentrated. Aliquots were flash frozen with liquid nitrogen and stored at -80 °C. The final product was judged to be >95% pure by SDS-PAGE (data not shown).
Thermal unfolding
Denaturation of wild-type and mutant MBPs was performed as a function of increasing temperature. Protein samples with and without 50 mM maltose were incubated at the desired temperature for 15 min before measurement by CD. The actual temperature of the sample in the cuvette was monitored continuously. Occasionally, samples were also checked for any possible aggregations due to heat by light scattering measurements. CD measurements were recorded on an Aviv Model 202 spectrophotometer. Conformational changes were monitored at 222 nm. All unfolding transitions were analyzed as described [15,16]. For comparison of the results, the data were normalized and expressed as the fraction of protein unfolded (Fun). Fun was calculated from the equation Fun = (Fobs-Fn) / (Fu-Fn), where Fobs is the observed signal at a given temperature while Fn and Fu are the values of native and unfolded proteins, respectively.
Results
Design and construction of expression vectors
Seeking to improve the performance of MBP as an affinity tag, we incorporated amino acid substitutions that reportedly gave rise to a “high affintiy” maltose-binding phenotype [9,10] into vectors designed to overproduce MBP in the cytoplasm of E. coli. All of the mutations were introduced into the Gateway destination vector pDEST-HisMBP [11]. pDEST-HisMBP and its mutant derivatives produce the mature form of MBP with a hexahistidine tag joined to its N-terminus. The hexahistidine tag does not interfere with the ability of MBP to promote the solubility of its fusion partners [12]. However, its presence enabled us to purify all of the mutant MBPs examined in this study, some of which do not bind to amylose resin (see below), in precisely the same manner for biophysical characterization, using immobilized metal affinity chromatography. The MBPs produced from these destination vectors also have a short peptide sequence appended to their C-termini that is derived from the translation product of the att recombination site. We refer to the products expressed by these destination vectors as “unfused” forms of MBP (despite the short extensions of their termini), whereas we use the term “fusion proteins” to describe MBPs to which passenger proteins have been fused by recombinational cloning.
Impact of the DM and I329W mutations on the solubility of MBP fusion proteins
We initially elected to work with the DM mutant, which is a combination of two amino acid substitutions (M321A and Q325A) and an in-frame deletion of residues 172-176 (Fig 1). The combination of these mutations reportedly increases the affinity of MBP for maltose by approximately 100-fold [9]. We found that, in the unfused state, the DM mutant was abundantly produced in the cytosol of E. coli and just as soluble as wild-type MBP (Fig 2). Therefore, we next constructed three fusion proteins in order to evaluate the performance of the DM mutant during amylose affinity chromatography, using human papilloma virus E6, human p16 and Green Fluorescent Protein (GFP) as passenger proteins. Although poorly soluble in an unfused state, these three passenger proteins can be rendered soluble by fusing them to MBP [3]. Unexpectedly, however, we found that the DM mutation(s) dramatically reduced the solubility of all three fusion proteins (Fig 3).
Fig 1.
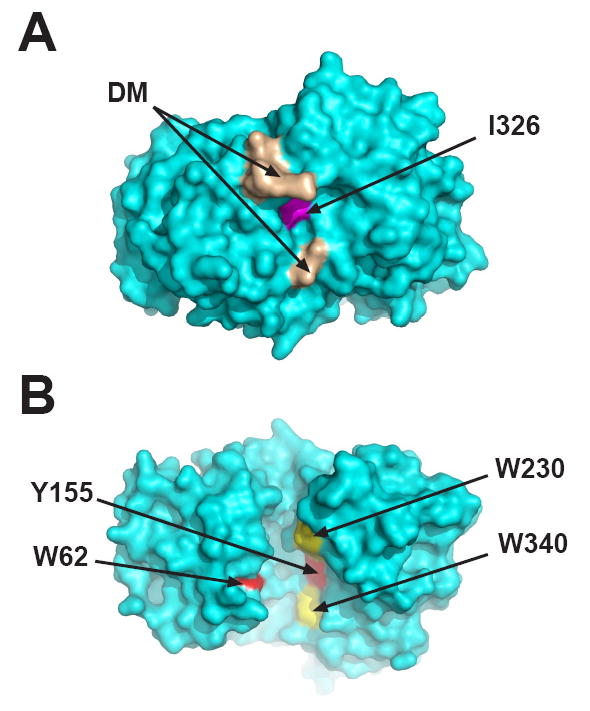
Locations of mutated residues in MBP. E. coli MBP is shown in the open (ligand-free) conformation (Sharff et al., 1992); PDB code 1OMP. (A) Space-filling view of the “hinge” region on the opposite side of the molecule from the ligand-binding cleft. Residues comprising the DM mutant are colored brown and I329W is colored magenta. (B) Space-filling view of the ligand-binding cleft (180° rotation along the X axis relative to the image in panel A). The sites of the double mutations in the cleft W62/Y155 and W230/W340 are colored red and yellow, respectively. These images were generated with PyMOL (DeLano Scientific LLC, Palo Alto, CA., USA).
Fig 2.
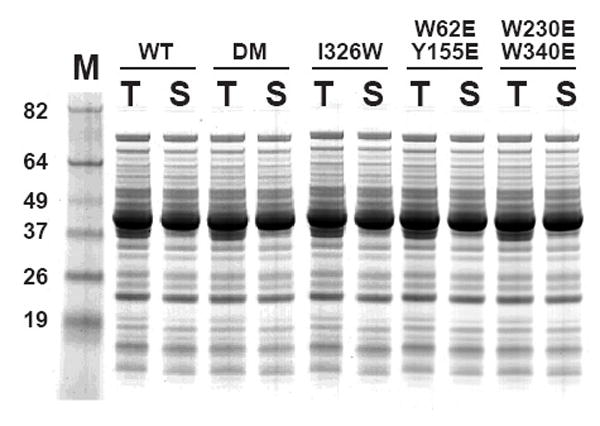
Overproduction of unfused HisMBPs in E. coli. Samples of the total (T) and soluble (S) protein from DB3.1 cells containing expression vectors producing wild-type HisMBP (WT), the I329W mutant, the DM mutant, the W62E/Y155E mutant and the W230E/W340E mutant were prepared as described in Materials and Methods and analyzed by SDS-PAGE and Commassie Blue staining. All cultures were grown at 37 °C and induced with IPTG at mid-log phase. M, broad-range molecular weight markers (Invitrogen).
Fig 3.
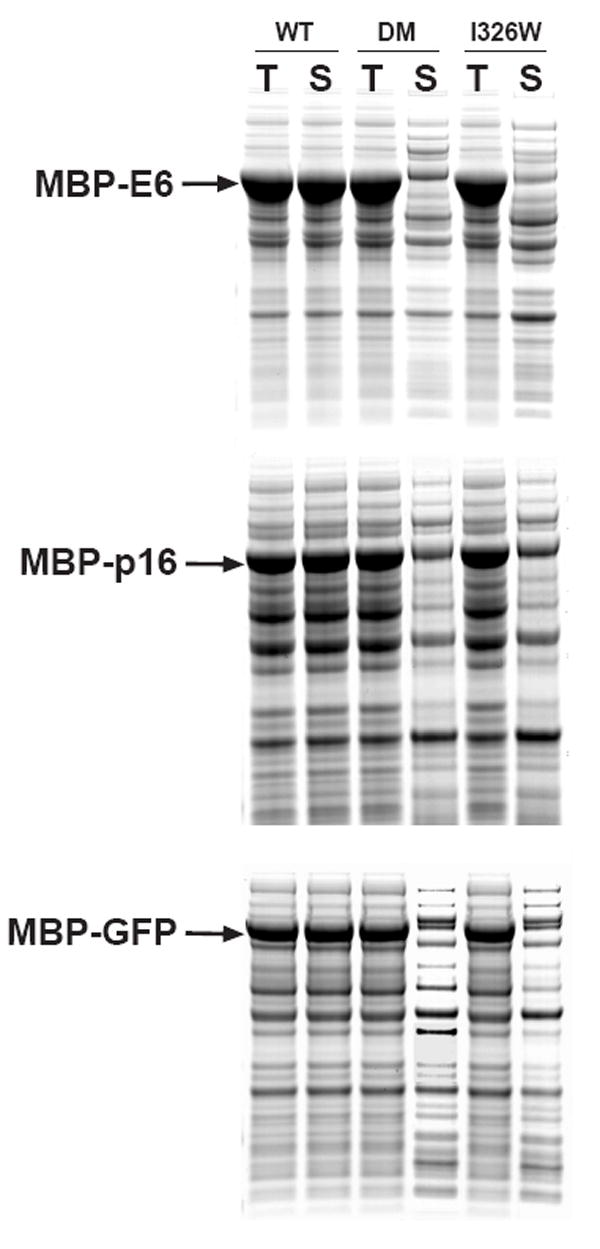
Expression and solubility of wild-type and mutant MBP fusion proteins. The passenger proteins were E6 (top), p16 (middle) and GFP (bottom). All fusion proteins were expressed at 37 °C, induced at mid-log phase with IPTG, and analyzed by SDS-PAGE and Coomassie Blue staining. T, total intracellular protein; S, soluble intracellular protein.
The impact of these mutations on the solubility of MBP fusion proteins could be manifested on a global level, by altering the equilibrium between open and closed conformations of the protein, or on a local level by disrupting an interaction site on the surface of the protein. To distinguish between these possibilities, we also tested the effect of another mutation in MBP (I329W; Fig 1) that had also been shown to alter the equilibrium between open and closed conformations [10]. Like the DM mutation(s), the I329W substitution did not alter the solubility of MBP in its unfused state (Fig 2) but dramatically impeded the solubility of the same MBP fusion proteins (Fig 3). Hence, the insolubility phenotype is not linked to a specific amino acid substitution but rather is correlated with the global effect of the mutations on the conformation of MBP in solution.
Stability of the DM and I329W mutants
To determine whether the DM and I329W mutations destabilize the global fold of MBP, we conducted temperature-induced equilibrium unfolding experiments monitored by circular dichroism. As shown in Fig. 4, the stability of the I329W mutant is indistinguishable from that of wild-type MBP, either in the presence or absence of maltose, and the DM mutant is only slightly less stable. As might be expected, the binding of maltose resulted in an elevation of the transition midpoint. It is known from the crystallographic data that when maltose is bound, MBP undergoes a substantial conformational change in which the angle between the two domains changes by 35 degrees and is accompanied by closure of the cleft. The binding of maltose in a cleft between the two domains causes them to close around the ligand, increasing the number of electrostatic and hydrophobic interactions between them [17].
Fig 4.
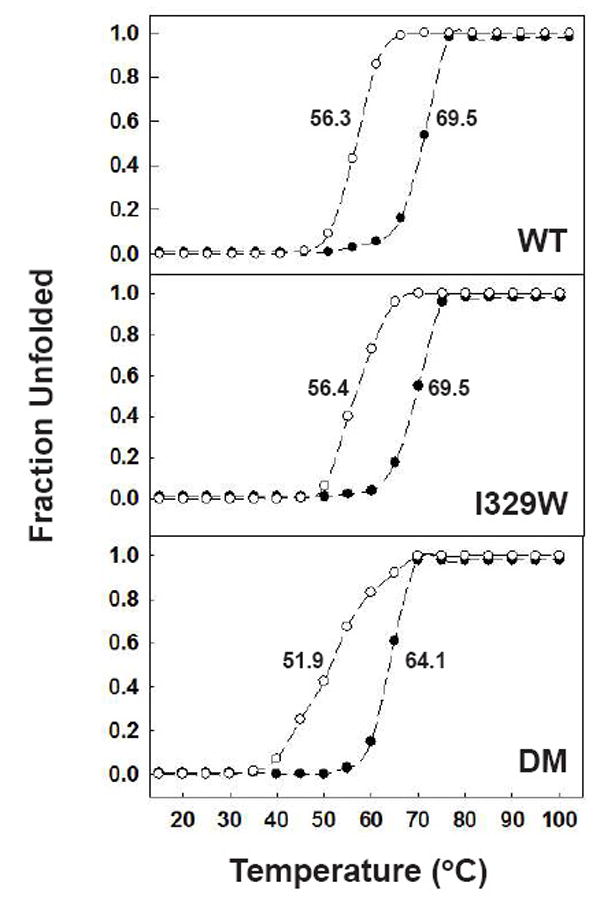
Temperature-induced unfolding of wild-type MBP (WT), the I329W mutant and the DM mutant. Experiments were performed at pH 7.2 in 50 mM sodium phosphate buffer in the absence (○) and presence (●) of 50 mM maltose and monitored by changes in ellipticity at 222 nm. The resulting data were normalized to calculate the fraction of unfolded protein and plotted as a function of temperature. The spectra were recorded after 15 min incubation at each temperature.
Performance of the DM and I329W mutants during amylose affinity chromatography
As stated above, our original objective was to ascertain whether or not the DM and I329W mutations, which were reported to increase the affinity of MBP for maltose, would improve the performance of MBP during amylose affinity chromatography. Because they are unable to function as effective solubilizing agents, we recognized that these mutant MBPs would be of little practical value for the production of recombinant proteins in general. Nevertheless, for the record, we compared their behavior on an amylose affinity column using the unfused forms of the proteins. No significant differences were observed (data not shown).
Effect of mutations in the ligand-binding cleft of MBP
Apart from the relative orientation of the two lobes that cradle the carbohydrate ligand, the structural differences between the open and closed conformations of MBP are confined to two regions of the protein: the ligand-binding cleft, which is exposed in the open conformation, and the surface of the “hinge” region on the opposite side of the protein. The amino acids that comprise the hinge regions of various maltodextrin-binding proteins that have the ability to function as general solubility enhancers [18] are not highly conserved whereas the hydrophobic nature of the ligand binding clefts is. Hence, the present results are consistent with an earlier proposal that the ligand-binding cleft in MBP may act as a transient binding site for hydrophobic folding intermediates of passenger proteins and prevent their self-association and aggregation [3,19]. Yet, when Fox et al. examined the effect of replacing individual hydrophobic amino acids in the cleft with glutamic acid, they failed to observe any effect of these substitutions on the solubility of MBP fusion proteins [19].
Reasoning that perhaps single amino acid substitutions were not enough to disrupt interactions that may occur between the passenger proteins and the ligand-binding cleft in MBP, we decided to revisit the issue of site-directed mutagenesis of the cleft residues. Accordingly, we constructed two double mutants (W62E/Y155E and W230E/W340E) from individual cleft mutations that exhibited no adverse effect on the solubility of MBP fusion proteins in the study by Fox et al. (Fig. 1) As shown in Fig. 2, the W62E/Y155E and W230E/W340E mutants were just as soluble as wild-type MBP, the DM mutant, and the I329W mutant in their unfused state. However, the W62E/Y155E and W230E/W340E mutations markedly reduced the solubility of several different MBP fusion proteins under the same conditions (Fig. 5).
Fig 5.
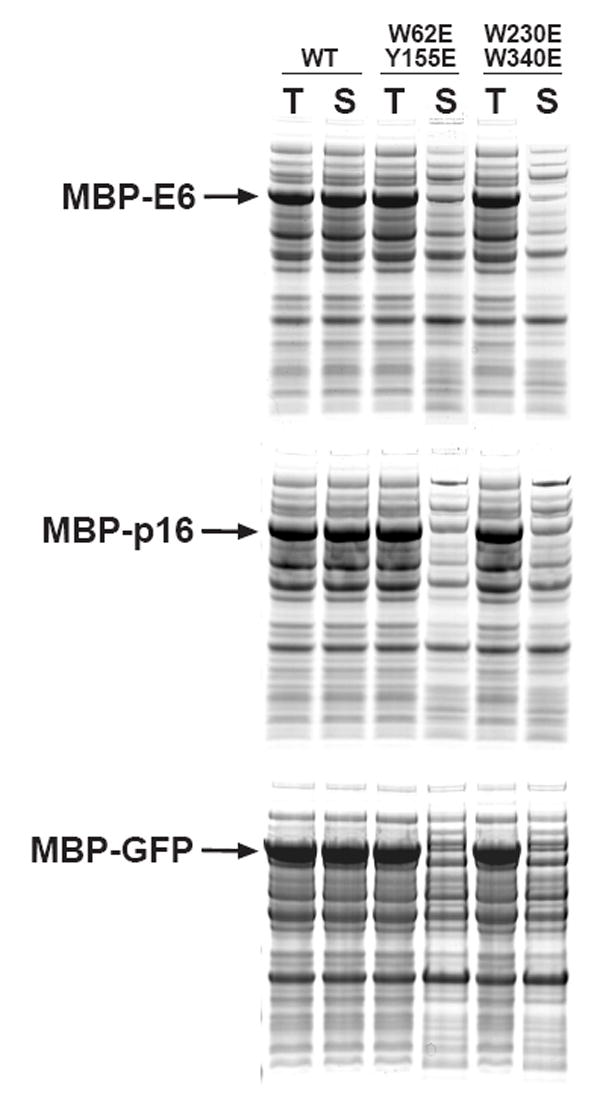
Effect of double mutations in the ligand-binding cleft (W62E/Y155E and W230E/W340E) on the solubility of MBP fusion proteins. The passenger proteins were E6 (top), p16 (middle) and GFP (bottom). Data for wild-type MBP (WT) fusion proteins are also included for comparison. All fusion proteins were expressed at 37 °C, induced at mid-log phase with IPTG, and analyzed by SDS-PAGE and Coomassie Blue staining. T, total intracellular protein; S, soluble intracellular protein.
Discussion
Several mechanisms have been proposed to explain how some soluble proteins are able to enhance the solubility of their fusion partners. According to one model, the fusion proteins form soluble micelle-like structures in which the aggregation-prone passenger proteins are sequestered on the inside, away from the solvent, while the soluble protein domains face outward. Indeed, there is evidence to suggest that such structures do form in some cases [20]. When they occur, however, these structures represent dead ends because they do not lead to the recovery of properly folded passenger proteins, although they may co-exist with a population of monodisperse fusion protein in which the passenger is folded. Yet, in many cases passenger proteins can be recovered in a properly folded form with high efficiency when they are fused to a solubility-enhancing partner. In these cases, some other mechanism must be operating.
Another way in which solubility-enhancing fusion partners may promote the proper folding of their passenger proteins is by acting as “chaperone magnets” the soluble protein initiates its fusion partner into a chaperone-mediated folding pathway. In fact, Douette et al. have presented evidence that MBP and NusA fusion proteins interact with GroEL in E. coli [21]. It is not entirely clear, however, how these rather large fusion proteins could engage the GroEL/GroES chaperone apparatus in the same manner as its natural substrates do, because the size of the chaperone cavity would seem to limit substrates to approximately 50 kDa [22].
A third possibility is that solubility-enhancing proteins possess an intrinsic chaperone-like activity that manifests itself in the context of a fusion protein [3]. According to this model, transient and interactive interactions between partially folded passenger proteins and hydrophobic patches on the solubility enhancer prevent their self association and thereby guide them toward their native structures. Solubility enhancers would not necessarily play an active role in the folding of their fusion partners, but might serve instead to reduce unproductive off-pathway aggregation [13]. This could explain why only certain highly soluble proteins can function as solubility enhancers.
Another proposal is that the ability of proteins to enhance the solubility of their fusion partners is correlated with their net charges. For instance, the highly acidic, solubility-enhancing SET tag [23] has been proposed to exert its effect by inhibiting aggregation through electrostatic repulsion and may not adopt a globular conformation at all. Su et al. observed that globular proteins with increasingly higher net acidity exhibited a greater capacity to enhance the solubility of two passenger proteins [24]. However, in a comparative study of MBPs from a variety of microorganisms, no correlation between net charge and solubility-enhancing ability was observed [18].
It is possible that different solubility-enhancing proteins work by different mechanisms, or even by utilizing more than one mechanism, although comparative studies with MBP and E. coli NusA seem to suggest otherwise [12]. In any case, the results presented here are most consistent with the proposal that MBP posesses intrinsic chaperone-like qualities in the context of a fusion protein, and that the hydrophobic ligand-binding cleft plays a central role in its mechanism of action.
Acknowledgments
We thank Patrick Telmer and Brian Shilton for kindly providing us with the DM mutant of MBP, and Ping Sun for assistance with the preparation of Fig. 1. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Waugh DS. Making the most of affinity tags. Trends Biotechnol. 2005;23:316–320. doi: 10.1016/j.tibtech.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 2.Esposito D, Chatterjee DK. Enhancement of soluble protein expression through the use of fusion tags. Curr Opin Biotechnol. 2006;17:353–358. doi: 10.1016/j.copbio.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 3.Kapust RB, Waugh DS. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 1999;8:1668–1674. doi: 10.1110/ps.8.8.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hammarström M, Hellgren N, Van den Berg S, Berglund H, Härd T. Rapid screening for improved solubility of small human proteins produced as fusion proteins in Escherichia coli. Protein Sci. 2002;11:313–321. doi: 10.1110/ps.22102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shih YP, Kung WM, Chen JC, Yeh CH, Wang AHJ, Wang TF. High-throughput screening of soluble recombinant proteins. Protein Sci. 2002;11:1714–1719. doi: 10.1110/ps.0205202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dyson MR, Shadbolt SP, Vincent KJ, Perera RL, McCafferty J. Production of soluble mammalian proteins in Escherichia coli: identification of protein features that correlate with successful expression. BMC Biotechnol. 2004;4:32. doi: 10.1186/1472-6750-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pryor KD, Leiting B. High-level expression of soluble protein in Escherichia coli using a His6-tag and maltose-binding protein double-affinity fusion system. Protein Expr Purif. 1997;10:309–319. doi: 10.1006/prep.1997.0759. [DOI] [PubMed] [Google Scholar]
- 8.Routzahn KM, Waugh DS. Differential effects of supplementary affinity tags on the solubility of MBP fusion proteins. J Struct Funct Genomics. 2002;2:83–92. doi: 10.1023/a:1020424023207. [DOI] [PubMed] [Google Scholar]
- 9.Telmer PG, Shilton BH. Insights into the conformational equilibria of maltose-binding protein by analysis of high affinity mutants. J Biol Chem. 2003;278:34555–34567. doi: 10.1074/jbc.M301004200. [DOI] [PubMed] [Google Scholar]
- 10.Marvin JS, Hellinga HW. Manipulation of ligand binding affinity by exploitation of conformational coupling. Nature Struct Biol. 2001;8:795–798. doi: 10.1038/nsb0901-795. [DOI] [PubMed] [Google Scholar]
- 11.Fox JD, Waugh DS. Maltose-binding protein as a solubility enhancer. Methods Mol Biol. 2003;205:99–117. doi: 10.1385/1-59259-301-1:99. [DOI] [PubMed] [Google Scholar]
- 12.Nallamsetty S, Austin BP, Penrose KJ, Waugh DS. Gateway vectors for the production of combinatorially-tagged His6-MBP fusion proteins in the cytoplasm and periplasm of Escherichia coli. Protein Sci. 2005;14:2964–2971. doi: 10.1110/ps.051718605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nallamsetty S, Waugh DS. Solubility-enhancing proteins MBP and NusA play a passive role in the folding of their fusion partners. Protein Expr Purif. 2006;45:175–182. doi: 10.1016/j.pep.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 14.Nallamsetty S, Kapust RB, Tozser J, Cherry S, Tropea JE, Copeland TD, Waugh DS. Efficient site-specific processing of fusion proteins by tobacco vein mottling virus protease in vivo and in vitro. Protein Expr Purif. 2004;38:108–115. doi: 10.1016/j.pep.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 15.Shirley BA. Urea and guanidine hydrochloride denaturation curves. Methods Mol Biol. 1995;40:177–90. doi: 10.1385/0-89603-301-5:177. [DOI] [PubMed] [Google Scholar]
- 16.Nallamsetty S, Dubey VK, Pande M, Ambasht PK, Jagannadham MV. Accumulation of partly folded states in the equilibrium unfolding of ervatamin A: Spectroscopic description of the native, intermediate, and unfolded states. Biochimie. 2007 doi: 10.1016/j.biochi. 2007.06.004.. in press. [DOI] [PubMed] [Google Scholar]
- 17.Sharff AJ, Rodseth LE, Spurlino JC, Quiocho FA. Crystallographic evidence of a large ligand-induced hinge-twist motion between the two domains of the maltodextrin binding protein involved in active transport and chemotaxis. Biochemistry. 1992;31:10657–10663. doi: 10.1021/bi00159a003. [DOI] [PubMed] [Google Scholar]
- 18.Fox JD, Routzahn KM, Bucher MH, Waugh DS. Maltodextrin-binding proteins from diverse bacteria and archaea are potent solubility enhancers. FEBS Lett. 2003;537:53–57. doi: 10.1016/s0014-5793(03)00070-x. [DOI] [PubMed] [Google Scholar]
- 19.Fox JD, Kapust RB, Waugh DS. Single amino acid substitutions on the surface of Escherichia coli maltose-binding protein can have a profound impact on the solubility of fusion proteins. Protein Sci. 2001;10:622–630. doi: 10.1110/ps.45201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nomine Y, Ristriani T, Laurent C, Lefevre JF, Weiss E, Trave G. Formation of soluble inclusion bodies by HPV E6 oncoprotein fused to maltose-binding protein. Protein Expr Purif. 2001;23:22–32. doi: 10.1006/prep.2001.1451. [DOI] [PubMed] [Google Scholar]
- 21.Douette P, Navet R, Gerkens P, Galleni M, Lévy D, Sluse FE. Escherichia coli fusion carrier proteins act as solubilizing agents for recombinant uncoupling protein 1 through interactions with GroEL. Biochem Biophys Res Comm. 2005;333:686–693. doi: 10.1016/j.bbrc.2005.05.164. [DOI] [PubMed] [Google Scholar]
- 22.Huang YS, Chuang DT. Mechanisms for GroEL/GroES-mediated Folding of a Large 86-kDa Fusion Polypeptide in Vitro. J Biol Chem. 1999;274:10405–10412. doi: 10.1074/jbc.274.15.10405. [DOI] [PubMed] [Google Scholar]
- 23.Zhang YB, Howitt J, McCorkle S, Lawrence P, Springer K, Freimuth P. Protein aggregation during overexpression limited by peptide extensions with large net negative charge. Protein Expr Purif. 2004;36:207–216. doi: 10.1016/j.pep.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 24.Su Y, Zou Z, Feng S, Zhou P, Cao L. The acidity of protein fusion partners predominantly determines the efficacy to improve the solubility of the target proteins expressed in Escherichia coli. J Biotech. 2007;129:373–382. doi: 10.1016/j.jbiotec.2007.01.015. [DOI] [PubMed] [Google Scholar]