

Abstract
HIV-1 Vif regulates viral infectivity by inhibiting the encapsidation of APOBEC3G (APO3G) through proteasomal degradation of the protein. Here we compared various Vif proteins for their ability to induce APO3G degradation and rescue viral infectivity. We found that Vif expressed from proviral vectors caused relatively inefficient degradation of APO3G in HeLa cells yet was very effective in inhibiting APO3G’s antiviral activity. On the other hand, Vif expressed autonomously from a codon-optimized vector caused very efficient APO3G degradation and also effectively inhibited APO3G’s antiviral effects. In contrast, a Vif chimera containing an N-terminal fluorescent tag efficiently induced APO3G degradation but was unable to restore viral infectivity. The lack of a direct correlation between APO3G degradation and rescue of viral infectivity suggests that these two properties of Vif are functionally separable. Our data imply that intracellular degradation of APO3G may not be the sole activity of Vif required for the production of infectious virions from APO3G-expressing cells.
Keywords: Vif, APOBEC3G, protein degradation, virus-host interactions
Introduction
Replication of HIV-1 in most primary cells and some immortalized T cell lines is dependent on the expression of a functional Vif protein. In the absence of Vif, virus replication is restricted by several host deaminases, most notably APOBEC3G (APO3G) (Sheehy et al., 2002). In the absence of Vif, APO3G is efficiently incorporated into virus particles where it causes extensive hypermutation of the viral cDNA during reverse transcription (Lecossier et al., 2003; Mangeat et al., 2003; Mariani et al., 2003; Zhang et al., 2003). The conversion of cytidine to deoxyuridine on the minus-strand cDNA results in guanine to adenine changes on the viral plus-strand cDNA to yield highly mutated viral genomes. Virus replication may be inhibited through accumulation of mutations in the viral genome or through degradation of the deaminated viral cDNA via a cellular DNA repair mechanism (reviewed in (Beard et al., 2003; Kavli et al., 2002)). Alternatively, APO3G may inhibit virus replication through deamination-independent mechanisms (Bishop et al., 2006; Chiu et al., 2005; Guo et al., 2006; Newman et al., 2005).
Vif is a 23-kDa basic protein that is expressed late during infection. Previous immunocytochemical analyses revealed a largely cytoplasmic localization of Vif (Goncalves et al., 1994; Karczewski and Strebel, 1996; Simon et al., 1997). Vif is efficiently incorporated into HIV virions during productive infection through an interaction with viral genomic RNA and associates with viral nucleoprotein complexes (Kao et al., 2003a; Karczewski and Strebel, 1996; Khan et al., 2001; Liu et al., 1995). In the presence of Vif, the steady-state levels of cell-associated APO3G are reduced by 2–10 fold when compared to Vif-negative controls (Conticello et al., 2003; Kao et al., 2003b; Mariani et al., 2003; Marin et al., 2003; Mehle et al., 2004b; Sheehy et al., 2003; Stopak et al., 2003; Yu et al., 2003). Such Vif-induced reduction of intracellular APO3G has been attributed to proteasome-dependent degradation of the protein and requires a physical interaction of Vif and APO3G (Marin et al., 2003; Sheehy et al., 2003; Stopak et al., 2003; Yu et al., 2003). Other studies, however, suggest that intracellular degradation of APO3G may not be the sole mechanism by which Vif neutralizes its antiviral activity (Kao et al., 2003b; Kao et al., 2004; Mariani et al., 2003; Mehle et al., 2004a).
The current study aimed at a more detailed characterization of the effects of Vif on the stability of APO3G in transiently transfected HeLa and 293T cells and its correlation with the production of infectious virions. As part of this project we analyzed three different types of HIV-1 Vif, including (i) Vif, expressed from a proviral vector (A1-Vif); (ii) Vif expressed from a codon-optimized vector in the absence of other viral proteins (hVif); and (iii) Vif fused at its N-terminus to yellow fluorescent protein (YFP-Vif). The YFP-Vif chimera was previously found to efficiently induce proteasome degradation of APO3G (Wichroski et al., 2005). Our own results are consistent with these observations. In fact, in all of our experiments, YFP-Vif and hVif were more effective in inducing APO3G degradation than A1-Vif protein. Interestingly, despite their different effects on APO3G stability, A1-Vif and hVif both efficiently counteracted APO3G’s antiviral activity. Surprisingly, however, YFP-Vif was severely impaired in its ability to direct the production of infectious HIV-1 particles from APO3G-expressing cells. The lack of correlation between APO3G degradation and infectivity of virus particles suggests that these two functions of Vif can be separated. The efficiency with which Vif induced APO3G degradation appeared to be dependent on the expression system and was most pronounced when Vif was expressed from autonomous (i.e. Tat- and Rev-independent) expression vectors, e.g. hVif and YFP-Vif. The inefficient degradation of APO3G by A1-Vif in HeLa cells was not due to isolate-specific differences or the co-expression of additional pNL-A1-encoded viral proteins. However, there were cell type-specific differences as APO3G degradation by A1-Vif was somewhat more efficient in 293T cells than in HeLa cells. Taken together our data demonstrate that the ability of HIV-1 Vif to counteract the anti-viral effects of APO3G is not directly linked to the efficiency of Vif-induced APO3G degradation by cellular proteasomes. Our results also demonstrate that the functional properties of Vif vary depending on the expression system used. Importantly, our results suggest that intracellular degradation of APO3G may not be the sole function of Vif required for the production of infectious virions from APO3G-expressing cells. These results are consistent with our recent identification of a degradation resistant APO3G variant whose virus encapsidation and antiviral activity was nonetheless efficiently controlled by Vif (Opi et al., 2007).
Results
Variability in the efficiency of Vif-dependent APO3G degradation
In the course of productive virus replication, Vif is expressed from a partially spliced transcript and requires the activity of Rev. Most of our previous studies on Vif involved the proviral vector pNL-A1 that expresses Vif from an authentic, partially spliced Vif mRNA (Strebel et al., 1987). In this vector, Vif expression is Rev dependent. Of note, pNL-A1 also expresses other viral proteins including Tat, Rev, Nef, Vpr, Vpu, and Env. To avoid the complexity of multigenic proviral vectors, expression vectors have been developed that allow expression of Vif in the absence of additional viral proteins (Goncalves et al., 1994; Nguyen et al., 2004; Wichroski et al., 2005). In this study, we employed two such vectors; (i) pcDNA-hVif expressing Vif from a codon-optimized gene (Nguyen et al., 2004) and (ii) pYFP-Vif expressing Vif fused at its N-terminus to yellow fluorescent protein (Wichroski et al., 2005). Both of these vectors allow for the Rev-independent expression of Vif and do not encode additional viral proteins. The various Vif expression vectors used in this study are schematically shown in Fig. 1A.
Fig. 1.
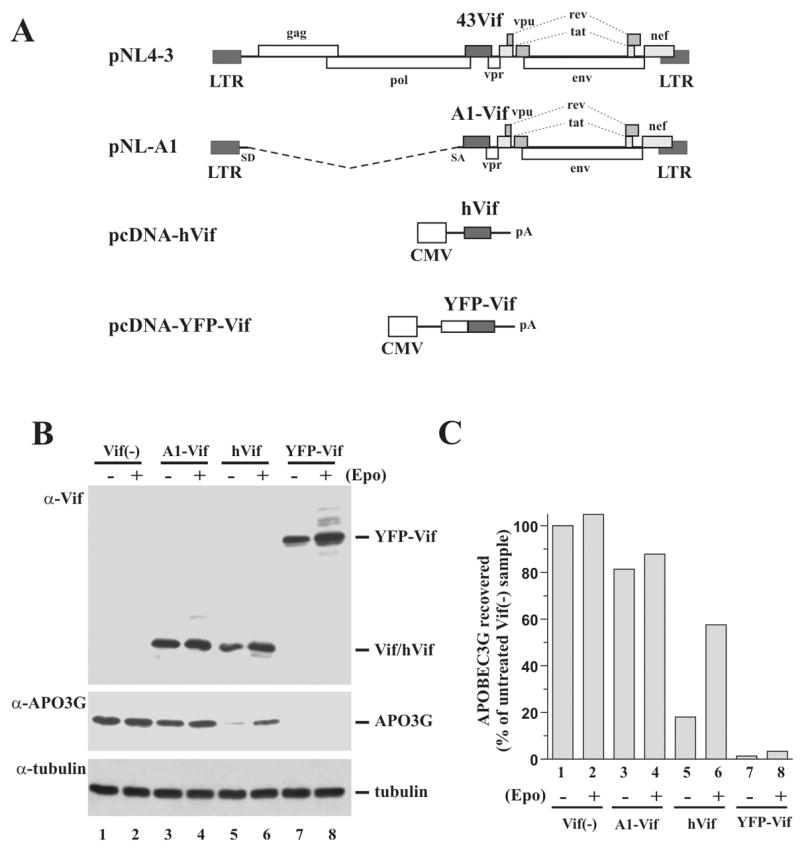
Variability in the efficiency of Vif-induced APO3G degradation. (A) Vectors used for the expression of Vif in this study are schematically shown. The full-length NL4-3 molecular clone, which is shown at the top for reference is a chimeric virus. Its 5′half, including Vif, is derived from the NY5 HIV-1 isolate while the 3′half is derived from LAV (Adachi et al., 1986). The pNL-A1 vector was derived from pNL4-3 by removing its gag/pol/vif region and replacing it with a Vif cDNA derived from the HIV-1 HXB2 isolate (Strebel et al., 1987). Aside from HXB2 vif, pNL-A1 encodes NL4-3 derived vpr, tat, rev, vpu, env, and nef genes but lacks gag and pol genes. To express NL4-3 Vif in the context of pNL-A1, vif sequences in pNL-A1 were replaced via a PCR-based strategy by NL4-3 vif resulting in pNL-A1/43Vif. Plasmid pcDNA-hVif (Nguyen et al., 2004) encodes a codon-optimized HIV-1 Vif (NL4-3 isolate). Expression of Vif from this vector is Tat- and Rev-independent and under the control of a CMV immediate early promoter. No other viral proteins are encoded by this vector. Plasmid pYFP-Vif encodes HIV-1 Vif (HXB2 isolate) fused to the C-terminus of yellow fluorescent protein (Wichroski et al., 2005). Protein expression is under the control of a CMV immediate early promoter and is Tat- and Rev-independent. (B) HeLa cells were transfected with pcDNA-Apo3G (0.5 μg) and 2.5 μg of either pNL-A1vif(−) (lanes 1–2), pNL-A1 (lanes 3–4), pcDNA-hVif (lanes 5–6), or pYFP-Vif (lanes 7–8). One day later, cells were treated for 10 h with either DMSO (EPO−) or the proteasome inhibitor epoxomicin (5μM; (EPO+)). Whole cell lysates were analyzed by immunoblotting using antibodies to Vif (α-Vif) or APO3G (α-APO3G). The APO3G blot was subsequently re-probed with an α-tubulin-specific monoclonal antibody (α-tubulin). (C) APO3G-specific bands in panel B were quantified by densitometric scanning and plotted as percentage of the untreated vif(−) control, which was defined as 100% (lane 1). Lane numbers correspond to those in panel B. Epo = epoxomicin.
We first compared the relative effect of Vif expressed from the various expression vectors on APO3G stability (Fig. 1B). HeLa cells were transfected with pcDNA-Apo3G and either pNL-A1vif(−) (Fig. 1B, Vif(−)), pNL-A1 (A1-Vif), pcDNA-hVif (hVif), or pYFP-Vif (YFP-Vif) plasmid DNAs. Vif-expressing vectors were provided in 5-fold excess over APO3G-expressing vector (2.5 μg and 0.5 μg, respectively) to maximize the impact of Vif on APO3G stability. To verify proteasome-dependent degradation of APO3G, the proteasome inhibitor epoxomicin (5 μM) was added to one set of each sample 24 h after transfection. The second set remained untreated. Cells were incubated for an additional 10 h prior to cell lysis. Whole cell lysates were then analyzed by immunoblotting using a Vif-specific monoclonal antibody (Fig. 1B, α-Vif) or an APO3G-specific polyclonal antibody (Fig. 1B, α-APO3G). The APO3G blot was subsequently reprobed with an antibody to α-tubulin to control for experimental variations (Fig. 1B, α-tubulin). APO3G steady-state levels were quantified by densitometric scanning of APO3G-specific bands and expressed relative to the amount of APO3G observed in the absence of Vif and inhibitor (Fig. 1C). As expected, the absence of Vif resulted in stable expression of APO3G (Fig. 1B/C, lanes 1). Epoxomicin-treatment resulted in a small (5%) increase in APO3G (Fig. 1B/C, lanes 2) presumably reflecting the inhibition of the normal turnover of APO3G in transfected HeLa cells. Expression of Vif from pNL-A1 resulted in a moderate 20% reduction of the APO3G level (Fig. 1B/C, lanes 3) that was partially reversed by addition of epoxomicin (Fig. 1B/C, lanes 4). In contrast, expression of Vif from the codon-optimized vector (hVif) or in fusion with yellow fluorescent protein (YFP-Vif) dramatically reduced APO3G expression in the untreated samples (Fig. 1B/C, lanes 5 & 7). The presence of epoxomicin significantly stabilized APO3G levels in the hVif expressing sample (Fig. 1B/C, lanes 6) verifying the involvement of proteasomes in the cellular depletion of APO3G by hVif. Interestingly, epoxomicin had very little effect on APO3G stability in YFP-Vif expressing cells even though YFP-Vif itself was stabilized by the drug (Fig. 1B/C, lanes 8). This result was unexpected and could be explained by the incomplete inhibition of proteasome-associated trypsin- and caspase-like proteolytic activities by epoxomicin. Similar results were reported by Wichroski et al. who found that at high levels of Vif treatment with the proteasome inhibitor ALLN did not restore APO3G expression in transfected 293T cells (Wichroski et al., 2005). At lower concentrations of YFP-Vif, proteasome inhibitor treatment resulted in partial recovery of APO3G (data not shown). Thus, when Vif expression vectors were provided at 5-fold excess over the APO3G vector, hVif and YFP-Vif caused efficient degradation of APO3G while A1-Vif had only a modest effect on APO3G stability. The observation that epoxomicin treatment also increased the levels of Vif proteins in this experiment was expected since Vif itself is degraded by proteasomes (Fujita et al., 2004).
APO3G degradation by hVif and YFP-Vif is dose dependent and occurs at low levels of Vif
In the experiments shown in figure 1, Vif DNA was provided at 5-fold excess over APO3G DNA. Next, the dose dependence of Vif-induced APO3G degradation was analyzed (Fig. 2). For that purpose, HeLa cells were transfected with a constant amount of pcDNA-Apo3G (1 μg) and increasing amounts (0.1, 0.5, or 1.5 μg) of plasmid DNA expressing A1-Vif (Fig. 2A, lanes 3–5) or YFP-Vif (lanes 10–12). The ratios of APO3G:Vif vectors were 10:1 (lanes 3 & 10), 2:1 (lanes 4 & 11), and 1:1.5 (lanes 5 & 12). Amounts of transfected pcDNA-hVif DNA (lanes 7–9) were 2-fold higher than for A1-Vif and YFP-Vif (i.e. 0.2, 1, and 3 μg, respectively) to compensate for the lower overall expression of hVif from the codon-optimized vector when compared to A1-Vif or YFP-Vif. Vif-negative vectors pNL-A1vif(−) (lane 2) or pcDNA3.1 (lane 6) were included as controls. Lane 1 is a mock-transfected control. Total amounts of transfected DNA were adjusted to 5 μg by addition of appropriate amounts of Vif-negative pNL-A1vif(−) (Fig. 2A, lanes 2–5) or empty pcDNA3.1 vector DNA (Fig. 2A, lanes 6-12). Whole cell lysates were prepared 24 h after transfection and analyzed by immunoblotting for the expression of Vif (Fig. 2A, top panel) or APO3G (middle panel). The APO3G blot was subsequently re-probed with an antibody to α-tubulin to control for loading errors (bottom panel). APO3G signals were quantified as described for figure 1C and the results are shown in figure 2B. All three Vif proteins were expressed in a dose-dependent manner and at comparable levels (Fig. 2A, α-Vif). As before, A1-Vif had very little effect on APO3G expression at all ratios tested (Fig. 2B, solid circles). In contrast, hVif (Fig. 2B, open circles) and YFP-Vif (triangles) reduced APO3G expression in a dose-dependent manner and with comparable efficiency. Even when APO3G vector was used at 5- or 10-fold excess relative to hVif and YFP-Vif vectors, respectively, APO3G expression was effectively reduced (Fig. 2B, 10:1).
Fig. 2.
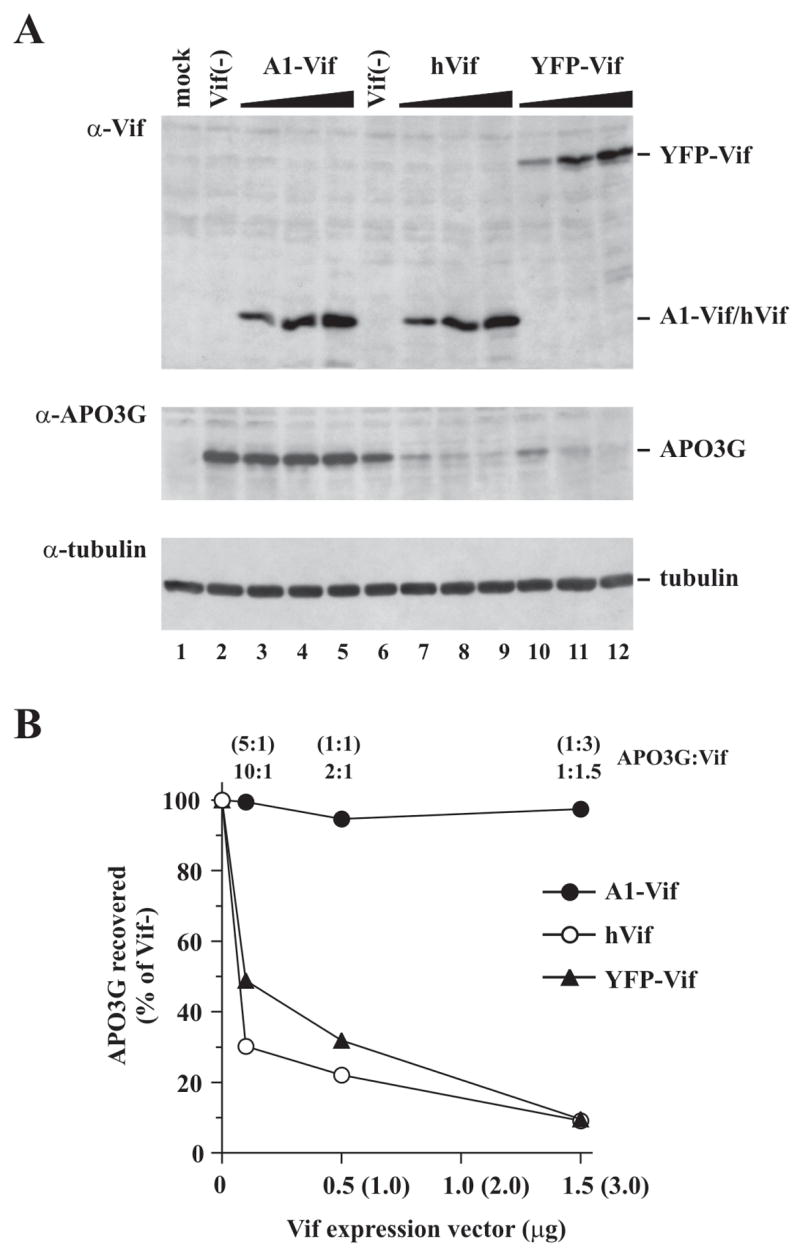
Dose-dependent effect of Vif on cellular APO3G expression. (A) HeLa cells were transfected with 1 μg of pcDNA-Apo3G (lanes 2–12) together with increasing amounts (0.1, 0.5, 1.5 μg) of pNL-A1 (lanes 3–5), or pYFP-Vif (lanes 10–12). Amounts of transfected pcDNA-hVif were 0.2, 1, and 3 μg (lanes 7–9) to compensate for the lower expression of hVif from the codon-optimized vector and to achieve comparable expression of the three Vif types in this experiment. Vif-negative controls are shown in lanes 2 & 6. Total transfected DNA in all samples was adjusted to 5 μg using pNL-A1vif(−) (lanes 2–5) or pcDNA3.1 vector DNA (lanes 6–12). The Vif(−) controls shown in lanes 2 and 6 each contained 1 μg of pcDNA-Apo3G; however the samples differ in that lane 2 contained 4μg of pNL-A1vif(−) while lane 6 contained 4μg of pcDNA3.1 vector DNA as filler DNA. These two controls were included because of the different vector backbones used for Vif expression in this experiment. Lane 1 shows a mock-transfected sample (no APO3G DNA). Cells were harvested 24 h after transfection and whole cell lysates were analyzed by immunoblotting with antibodies to Vif (α-Vif) or APO3G (α-APO3G). The APO3G blot was subsequently re-probed with an antibody to tubulin to control for loading errors (α-tubulin). (B) APO3G-specific bands from panel A were quantified. Results are expressed as percentage of the respective vif-negative controls and were plotted as a function of Vif levels. APO3G:Vif vector ratios are listed at the top and x-axis values show the absolute amounts of transfected Vif vectors. Numbers in parentheses refer to pcDNA-hVif DNA.
Viral proteins encoded by pNL-A1 do not interfere with Vif-induced APO3G degradation
Unlike pcDNA-hVif and pYFP-Vif, which encode only Vif, the pNL-A1 vector encodes additional viral proteins including Tat, Rev, Nef, Env, Vpu, and Vpr (Strebel et al., 1987). Interestingly, cotransfection of APO3G and pNL-A1vif(−) plasmid DNAs in figure 2 resulted in slightly (< 2 fold) increased levels of APO3G (Fig. 2A, lane 2, α-APO3G) when compared to APO3G co-transfected with pcDNA3.1 vector DNA (Fig. 2A, lane 6, α-APO3G). It is therefore conceivable that pNL-A1-encoded proteins affect APO3G expression or modulate the ability of A1-Vif to induce APO3G degradation.
To assess the possible effect of Tat or other viral proteins encoded by pNL-A1 on APO3G expression or stability, constant levels of APO3G and hVif were expressed in the presence of increasing amounts of pNL-A1vif(−) or pNL-A1. For that purpose, HeLa cells were transfected with 0.5 μg of pcDNA-Apo3G (Fig. 3, lanes 2–12) together with 0.5 μg of pcDNA-hVif (lanes 3–12). In addition, increasing amounts (0.5, 1.5, 2.5, 3.5 μg, respectively) of pNL-A1vif(−) (Fig. 3, lanes 4–7) or pNL-A1 (lanes 9–12) plasmid DNA were co-transfected. Total transfected DNA in all samples was adjusted to 5 μg using pcDNA3.1 vector DNA (lanes 2–12). Lanes 3 & 8 are replicate samples and show the effect of hVif on APO3G stability in the absence of co-transfected pNL-A1 vectors. Expression of APO3G in the absence of hVif or A1-Vif is shown in lane 2. Lane 1 is a mock-transfected control. Cells were harvested 24 h after transfection and cell lysates were subjected to immunoblot analysis using a Vif-specific monoclonal antibody (Fig. 3A, α-Vif) or polyclonal antibodies to gp120 (α-Env) or APO3G (α-APO3G). The APO3G blot was subsequently re-probed with a monoclonal antibody to α-tubulin to control for loading errors (α-tubulin). APO3G signals were quantified by densitometric scanning of APO3G-specific bands and corrected for variations in tubulin signals. The results were plotted as percentage of APO3G recovered relative to the vif-negative control (lane 2), which was defined as 100% (Fig. 3B).
Fig. 3.
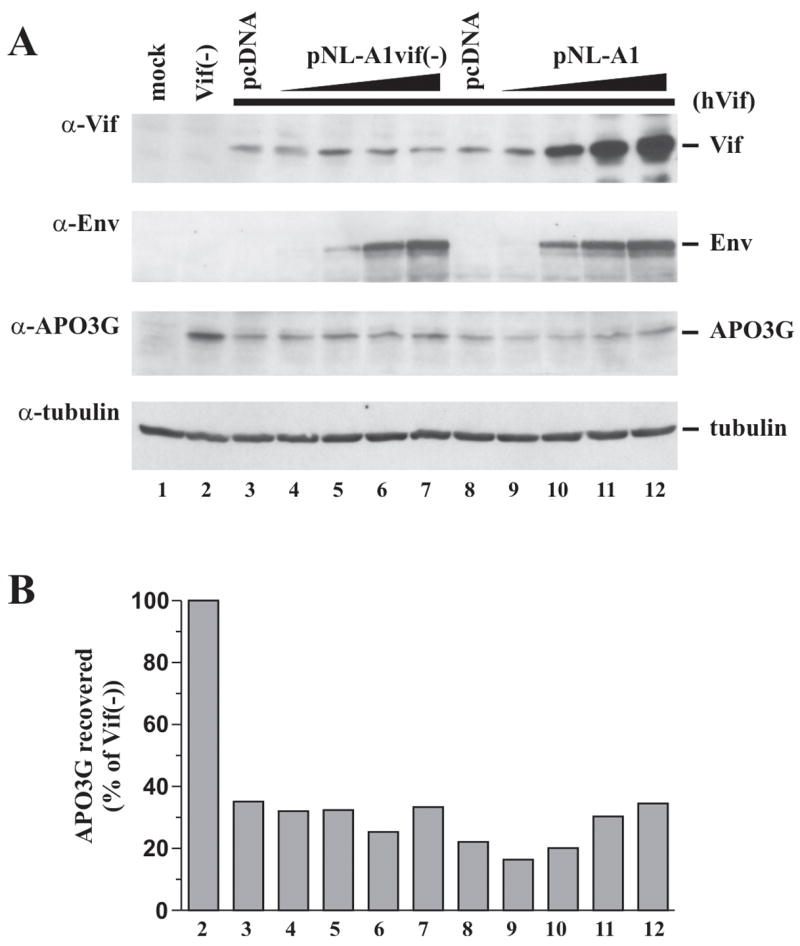
Viral proteins encoded by pNL-A1 do not interfere with hVif-induced APO3G degradation. (A) HeLa cells were either mock transfected (no DNA, lane 1) or transfected with constant amounts of pcDNA-Apo3G (0.5 μg; lanes 2–12) and pcDNA-hVif (0.5 μg, lanes 3–12) together with increasing amounts of pNL-A1vif(−) (lanes 4–7) or pNL-A1 DNA (lanes 9–12) as described in the text. Cells were harvested 24 h post transfection and whole cell lysates were subjected to immunoblot analysis using antibodies to Vif (α-Vif), Env (α-Env), or APO3G (α-APO3G). The APO3G blot was subsequently re-probed with antibodies to tubulin to control for loading errors (α-tubulin). (B) APO3G expression was quantified by densitometric scanning of the APO3G-specific bands in panel A. Results were corrected for variations in tubulin levels and plotted as percentage of APO3G recovered from the Vif-negative sample (lane 2), which was defined as 100%. Lane numbers correspond to those in panel A.
As expected, expression of hVif in the absence of pNL-A1 vectors led to a marked reduction of APO3G expression relative to the vif-negative control (Fig. 3A/B, compare lane 2 with lanes 3 & 8). The slight variation observed for APO3G levels in the replicate samples shown in lanes 3 & 8 reflects experimental variation. Importantly, increasing amounts of pNL-A1vif(−) (lanes 4–7) or pNL-A1 (lanes 9–12) had no detectable effect on the ability of hVif to induce degradation of APO3G (Fig. 3B). Expression of pNL-A1vif(−) and pNL-A1 vectors was monitored by measuring Env expressed from these vectors using an Env-specific polyclonal antibody (Fig. 3A, α-Env). As expected, Env expression from pNL-A1vif(−) and pNL-A1 was comparable and increased in a dose-dependent manner (Fig. 3A, α-Env, compare lanes 4–7 & 9–12). Of note, expression of hVif, which is under the control of a CMV immediate early promoter, was largely unaffected by increasing amounts of pNL-A1vif(−) (Fig. 3A, α-Vif, lanes 4–7) suggesting that neither Tat nor any of the other pNL-A1-encoded proteins significantly affected CMV promoter activity in these experiments. As expected, co-transfection of pNL-A1 led to a dose-dependent increase in Vif expression (Fig. 3A, α-Vif, lanes 9–12). Interestingly, despite the dramatic increase in Vif expression, APO3G levels were not further reduced confirming that A1-Vif, even at 7-fold excess of pNL-A1 DNA over pcDNA-hVif DNA, was unable to exacerbate the hVif-induced degradation of APO3G (Fig. 3B, compare lane 8 with lanes 9–12). The finding that the presence of increasing amounts of pNL-A1vif(−) or pNL-A1 had no effect on the ability of hVif to induce APO3G degradation implies that none of the pNL-A1 encoded proteins modulates Vif function to a significant extent and suggests that the inability of A1-Vif to efficiently induce degradation of APO3G in figures 1 & 2 is a cis-effect and not due to the expression of extraneous viral proteins such as Tat or Nef.
Comparative analysis of A1-Vif induced APO3G degradation in HeLa and 293T cells
Previous studies have reported more pronounced effects of A1-Vif on APO3G degradation than observed in our hands (Mehle et al., 2004b; Stopak et al., 2003; Wichroski et al., 2005). The reason for this discrepancy is unclear but could be due to (i) cell type-specific differences (most labs use 293T cells as opposed to HeLa cells used in our study); (ii) differences in the relative ratio of Vif:APO3G vectors; or (iii) differences in the APO3G vector itself since the APO3G construct used in our study has two amino acid differences (S162N and D370Y) relative to the published sequence for APO3G (Kao et al., 2003b). To address these issues, we performed a side-by-side analysis of our APO3G clone (referred to here as APO3G-NY) with a variant (referred to here as APO3G wt), in which the two residues were changed to the published sequence. Parallel experiments were performed in HeLa and 293T cells. HeLa cells (Fig. 4A/B) or 293T cells (Fig. 4C/D) were cotransfected with pNL-A1 (Vif+) or pNL-A1vif(−) (Vif−) and either pcDNA-APO3Gwt-myc (APO3G wt) or pcDNA-APO3G-NY-myc (APO3G-NY) using various ratios of Vif:APO3G vectors as indicated in figure 4. Amounts of transfected DNA are listed in the legend to figure 4. Cells were harvested 36 h following transfection and cell lysates were analyzed by immunoblotting with antibodies to Vif (Fig. 4A/C, top panels) or APO3G (Fig. 4A/C, middle panels). The APO3G-specific blots were subsequently reprobed with a tubulin-specific antibody to control for loading errors (Fig. 4A/C, lower panels). APO3G-specific bands were quantified by densitometric scanning and signals for APO3G from Vif-positive samples were expressed as percentage of the signal observed for the corresponding Vif-negative sample (Figs. 4B & D). In HeLa cells, the effects of A1-Vif on APO3G wt or APO3G-NY stability were modest but dose dependent. At 10-fold excess of transfected Vif vector over APO3G vector we noticed a ~30–40% reduction of APO3G wt and APO3G-NY levels while at 1:1 transfection ratios, the effect of A1-Vif on APO3G wt or APO3G-NY stability was virtually undetectable (Fig. 4B). Interestingly, the effects of A1-Vif on APO3G stability were somewhat more pronounced in 293T cells. For instance, transfection of Vif and APO3G vectors at a 10:1 ratio reduced APO3G levels ~4-5-fold (Fig. 4D). There was no significant difference, however, between APO3G wt and APO3G-NY with respect to protein expression and sensitivity to A1-Vif in HeLa or 293T cells. Thus, while 293T cells appeared to be more permissive overall to Vif-induced APO3G degradation, the two amino acid difference between APO3G wt and APO3G-NY did not alter their sensitivity to Vif.
Fig. 4.
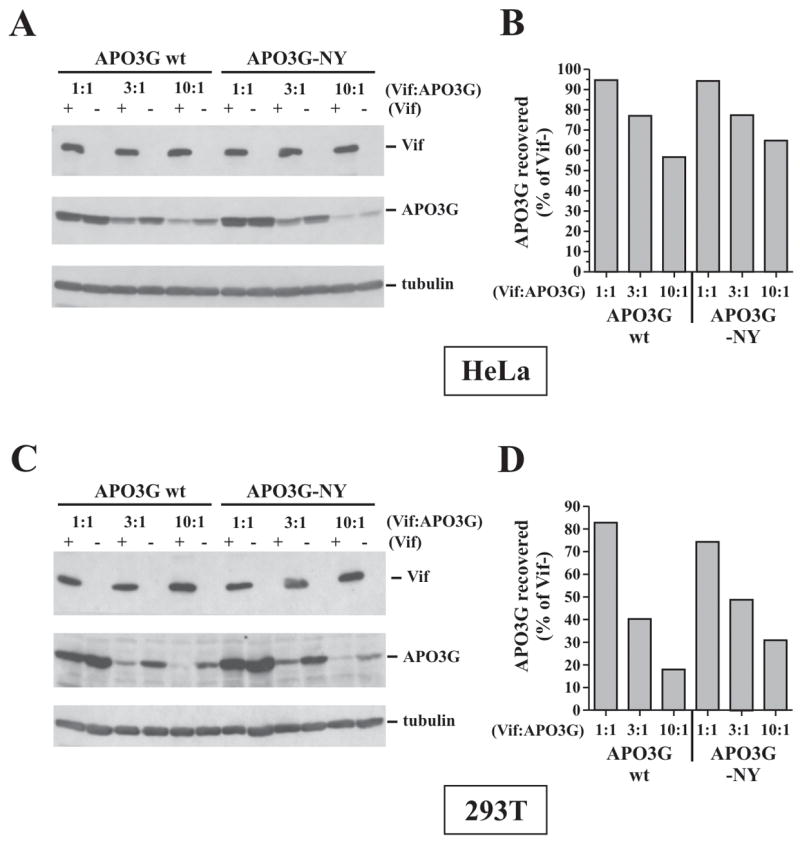
Comparative analysis of A1-Vif-induced APO3G degradation in HeLa and 293T cells. Hela cells (A & B) or 293T cells (C & D) were transfected with 2.5 μg of either pNL-A1 (Vif+) or pNL-A1vif(−) vectors (Vif−) together with decreasing amounts (2.5, 0.83, 0.25 μg) of vectors encoding APO3G wt or APO3G-NY to achieve the plasmid ratios as indicated in the figure. Transfected DNA amounts in all samples were adjusted with empty pcDNA3.1 vector to 5 μg total DNA. (A & C) Cells were harvested 36 h post transfection and cell lysates were subjected to immunoblot analysis using antibodies to Vif and APO3G. The APO3G blot was subsequently re-probed with an antibody to α-tubulin. Proteins are identified on the right. (B & D) APO3G-specific bands were quantified by densitometric scanning and the amounts of APO3G recovered from Vif-expressing samples were plotted as percentage of the signals obtained for the corresponding Vif-negative samples.
A1-Vif and hVif but not YFP-Vif proteins inhibit the antiviral activity of APO3G
The ability of hVif and YFP-Vif to efficiently reduce cellular APO3G levels in the experiment shown in figure 2 predicted that hVif and YFP-Vif would be highly effective and presumably more efficient than A1-Vif in inhibiting the antiviral activity of APO3G. To test this hypothesis, vif-defective NL4-3 virus was produced in the presence of constant amounts of APO3G and increasing amounts of A1-Vif, hVif, or YFP-Vif as detailed in the legend to figure 5. The ratios of Vif to APO3G vectors tested here were 1:10 (Fig. 5, lanes 3, 7, 10), 1:2 (lanes 4, 8, 11), and 1.5:1 (lanes 5, 9, 12). As a control, vif-defective NL4-3 virus was produced without exogenous Vif in the absence (lane 1) or presence (lane 2) of APO3G. Virus-containing supernatants from transfected cultures were collected 48 h after transfection, normalized for reverse transcriptase (RT) activity and used for the infection of LuSIV indicator cells (Fig. 5). Relative infectivity of the viruses was determined by measuring the virus-induced expression of luciferase in the LuSIV cells 24 h later. The infectivity of vif-defective virus produced in the absence of APO3G was defined as 100% (Fig. 5, lane 1). As expected, the infectivity of vif-defective viruses produced in the presence of APO3G was near background levels (Fig. 5, lanes 2 & 6). In contrast, expression of A1-Vif (lanes 3–5) and hVif (lanes 7–9) increased viral infectivity in a dose-dependent manner. The drop in infectivity at high levels of A1-Vif (Fig. 5, lane 5) is consistent with our previous observation that high level expression of Vif can inhibit viral infectivity (Akari et al., 2004). Surprisingly, YFP-Vif did not restore viral infectivity to a significant extent despite its potent effect on APO3G stability (see Figs. 1, 2 & 6A). The results shown in figure 5 were derived from multiple independent experiments. In particular, experiments involving YFP-Vif were performed 19 times because of the unexpected result.
Fig. 5.
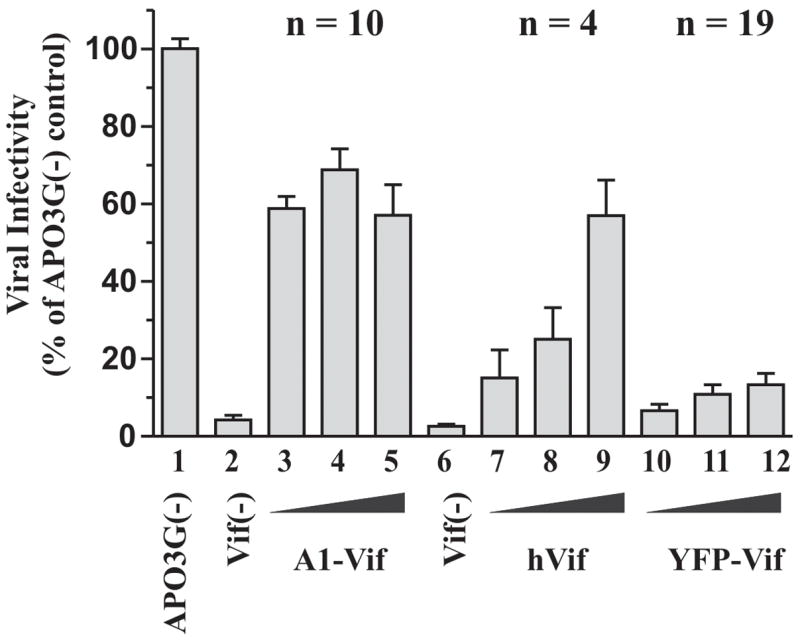
A1-Vif and hVif efficiently inhibit the APO3G antiviral activity. (A) HeLa cells were transfected with constant amounts of vif-defective pNL4-3 (3 μg) and pcDNA-Apo3G DNA (1μg) together with increasing amounts of A1-Vif, hVif, or YFP-Vif (0.1μg: lanes 3, 7, 10; 0.5μg: lanes 4, 8, 11; 1.5μg: lanes 5, 9, 12). Cell-free, virus-containing supernatants were harvested 48 h post-transfection, normalized for reverse transcriptase activity and used to infect LuSIV indicator cells. Vif-defective viruses produced in the absence (lane 1) and presence (lane 2 & 6) of APO3G were included as controls. Virus-induced activation of luciferase was determined 24 h later in a standard luciferase assay. Results were calculated relative to the signal obtained with vif-defective virus produced in the absence of APO3G (lane 1), which was defined as 100%. Error bars reflect standard error calculated from multiple independent experiments (n = number of independent analyses).
Fig. 6.
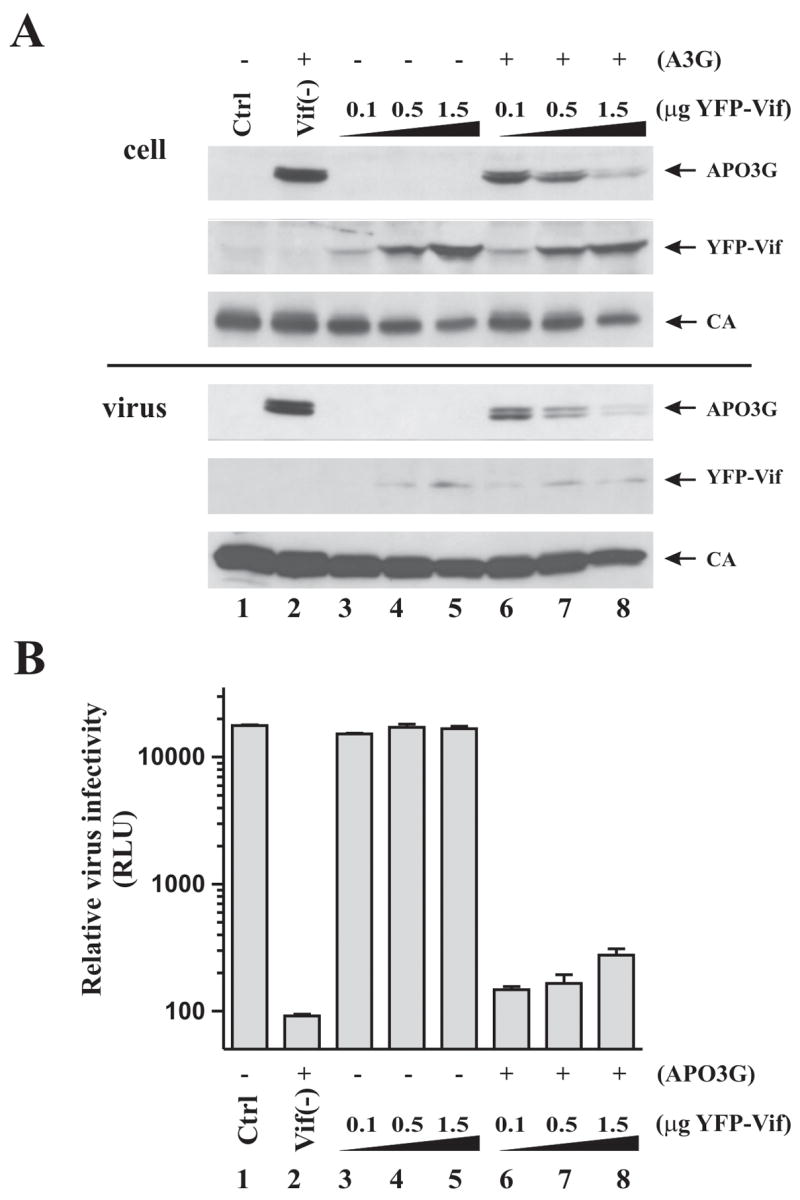
The inability of YFP-Vif to restore viral infectivity is not due to a non-specific inhibitory effect of YFP-Vif. Vif-defective viruses were produced in HeLa cells by transfection with pNL4-3vif(−) (3 μg) in the presence of increasing amounts of YFP-Vif (0.1, 0.5, 1.5 μg) and the absence (lanes 3–5) or presence (lanes 6–8) of 1 μg APO3G vector. Note that the ratio of Vif to APO3G vectors in lanes 6–8 was the same as in figure 5 (lanes 10–12). Vif-defective viruses produced in the absence (lane 1) and presence (lane 2) of APO3G were included as controls. (A) Cells and virus-containing supernatants were harvested 48 h after transfection and processed for immunoblotting as described in Materials and Methods. Filters were probed with antibodies to APO3G or Vif, or were reacted with an HIV-positive patient serum for the detection of capsid protein (CA). Proteins are identified on the right. (B) Virus-containing supernatants were normalized for reverse transcriptase activity and used for the infection of LuSIV indicator cells as in figure 5. Relative luciferase units (RLU) are shown. A log scale was chosen to visualize the small increase in viral infectivity by YFP-Vif. Error bars reflect the standard error from duplicate infections.
To control for possible toxic effects of YFP-Vif that could affect viral infectivity independent of APO3G, HeLa cells were transfected with vif-defective NL4-3 together with increasing amounts of YFP-Vif either in the absence (lanes 3–5) or presence of APO3G (Fig. 6A, lanes 6–8). Vif-defective NL4-3 produced in the absence (lane 1) or presence of APO3G (lane 2) but without YFP-Vif were included as controls. Intracellular protein expression and virus production were monitored by immunoblotting (Fig. 6A). As predicted, increasing levels of YFP-Vif correlated with decreasing amounts of intracellular APO3G and was paralleled by decreasing levels of virus-associated APO3G. Importantly, however, small amounts of virus-associated APO3G were detectable even at the highest level of YFP-Vif tested. Also, YFP-Vif had a small but identifiable effect on expression of viral protein and release of viral particles (Fig. 6A, CA; compare lanes 3–5 and 6–8). This effect was independent of APO3G and the Vif portion of the YFP-Vif chimera and was seen with other YFP- or GFP-tagged expression constructs ((Khan et al., 2005) and data not shown). Viral infectivity was determined as described for figure 5. Consistent with the results from figure 5, expression of increasing amounts of YFP-Vif in the presence of APO3G only modestly increased viral infectivity (Fig. 6B, lanes 6–8). Importantly, however, YFP-Vif did not significantly affect infectivity of viral particles in the absence of APO3G (Fig. 6B, lanes 3–5). These results rule out that the lack of infectivity of virus produced in the presence of APO3G and YFP-Vif was caused by a non-specific inhibitory or toxic effect of YFP-Vif.
Discussion
Since the identification of APO3G as a potent Vif-sensitive antiviral host factor (Sheehy et al., 2002), a tremendous effort has been made to understand how Vif counteracts the inhibitory effect of APO3G and other cellular cytidine deaminases. Two key observations have aided in this effort: (i) Vif was found to inhibit the packaging of APO3G into viral particles (Kao et al., 2003b; Mariani et al., 2003; Marin et al., 2003; Mehle et al., 2004b; Sheehy et al., 2003; Stopak et al., 2003) and (ii) Vif reduced the cellular steady-state levels of APO3G through proteasomal degradation (Conticello et al., 2003; Marin et al., 2003; Mehle et al., 2004b; Sheehy et al., 2003; Stopak et al., 2003; Yu et al., 2003). Since packaging of APO3G was found to be directly correlated with cellular expression levels, it follows that the Vif-induced reduction of cellular APO3G contributes directly to the exclusion of APO3G from HIV-1 particles. On the other hand, several reports noted that the inhibition of APO3G encapsidation by Vif can be more pronounced than the reduction of intracellular steady-state levels (Kao et al., 2003b; Mariani et al., 2003) suggesting additional degradation-independent mechanism(s) of interference with APO3G packaging. Indeed, we previously reported that intracellular depletion of APO3G by Vif is not a prerequisite for the rescue of viral infectivity (Kao et al., 2004). The results from the current study support these observations by demonstrating that a moderate reduction of intracellular APO3G levels by A1-Vif was sufficient to overcome the inhibitory effects of APO3G with similar efficiency as hVif, which efficiently depleted cellular APO3G. These results lead us to conclude that intracellular depletion of APO3G and rescue of viral infectivity are not necessarily linked but may in fact be separable functions of Vif. These conclusions are supported by our recent identification and characterization of an APO3G mutant that was resistant to Vif-induced degradation but whose antiviral activity and viral encapsidation were nonetheless efficiently controlled by Vif (Opi et al., 2007). Our conclusions are further supported by the recent finding that mutation of a serine residue at position 144 in Vif (S144A) did not impair its effect on APO3G expression yet severely impaired its ability to govern the production of infectious viruses from APO3G-expressing cells (Mehle et al., 2004a).
It is unclear why YFP-Vif despite substantial degradation of cellular APO3G did not restore viral infectivity. We can rule out non-specific cytotoxicity of YFP-Vif (Fig. 6). It is important to note, however, that despite extensive degradation in YFP-Vif expressing cells small amounts of APO3G remained detectable in cell-free virus preparations even at the highest level of YFP-Vif (Fig. 6A). It is conceivable that such residual amounts of virus-associated APO3G are sufficient to inhibit viral infectivity. Indeed, a recent study suggested that only a few copies of APO3G per virion were required to inhibit HIV-1 replication (Xu et al., 2007). It should be pointed out, however, that even viruses produced in the presence of A1-Vif or hVif invariably contained small amounts of virus-associated APO3G (data not shown). Whether such levels were below a critical threshold is unclear. Alternatively, it is possible that A1-Vif and hVif proteins have functional properties required to control residual virus-associated APO3G that were lost in YFP-Vif. These questions will be addressed in future studies.
It also remains to be investigated why YFP-tagged Vif and hVif are more effective in inducing APO3G degradation than A1-Vif. The difference in APO3G degradation cannot be explained by relative Vif protein levels since in the experiment shown in figure 2 all Vif proteins were expressed at very similar levels. Also, we can rule out isolate-specific differences since NL4-3 Vif and HXB2 Vif were both inefficient in inducing APO3G degradation when expressed in the backbone of pNL-A1 (data not shown) but were equally efficient when expressed from autonomous vector systems (Fig. 2, cf. hVif and YFP-Vif). Finally, we ruled out possible modulating effects on APO3G degradation by pNL-A1-encoded viral proteins (e.g. Tat, Rev, Nef, Env, Vpr, or Vpu; Fig. 3). One important difference between A1-Vif and hVif or YFP-Vif vectors is the structure of the Vif-encoding mRNA. A1-Vif are expressed from a Rev-dependent mRNA that requires the cellular Crm-1 pathway for nuclear export (for review see (Strebel and Bour, 1999)). In contrast, hVif and YFP-Vif mRNAs are Rev-independent and are exported from the nucleus via a Crm-1 independent pathway. It is possible that differences in mRNA export affect the cellular targeting of Vif. The idea that differences in mRNA targeting may translate into different functional properties of the encoded protein is not unprecedented as it was previously reported that assembly and release of HIV capsids from rodent cells was influenced by differences in the mode of nuclear mRNA export (Swanson et al., 2004).
Our finding that A1-Vif appeared to be more effective in targeting APO3G into a proteasomal degradation pathway in 293T cells when compared to HeLa cells (Fig. 4) is interesting and could explain some of the existing differences between our observations regarding A1-Vif and APO3G in HeLa cells and those reported by others using 293T cells (Mehle et al., 2004b; Wichroski et al., 2005; Xu et al., 2004). It is unclear why 293T cells seem more susceptible to APO3G degradation by A1-Vif. However, 293T cells are generally believed to be more susceptible to transfection. Since the Vif effect is dose-dependent, a simple explanation would be that Vif is more efficiently expressed on a per cell level in 293T cells. Alternatively, it is possible that HeLa and 293T cells exhibit differences in the intracellular targeting of A1-Vif. In analogy to our discussion on the differential effects of A1-Vif and hVif in HeLa cells, one could speculate that A1-Vif expressed in 293T cells is more efficiently targeted to intracellular compartment(s) relevant to APO3G degradation. The functional relevance of the more efficient APO3G degradation by A1-Vif in 293T cells versus HeLa cells remains unclear, however, especially since viruses produced in APO3G-expressing HeLa cells in the presence of Vif are fully infectious.
The question of how Vif might prevent the packaging of APO3G into viral particles under conditions of little or no APO3G degradation remains under active investigation. We have reported that both Vif and APO3G are packaged through specific interactions with viral genomic RNA (Khan et al., 2001; Khan et al., 2007; Khan et al., 2005). It is therefore conceivable that Vif inhibits the packaging of APO3G through a competitive mechanism by binding to a common packaging signal on the viral RNA. Alternatively, intracellular Vif:APO3G interactions could lead to sequestration of the proteins in a cellular compartment that prohibits their packaging into viral particles without prior degradation. In such a scenario, the fate of APO3G (i.e. degradation vs sequestration) could depend on where in the cell or when after synthesis the Vif-APO3G interaction occurs. APO3G has the propensity to assemble into high molecular mass ribonucleoprotein complexes (Chiu et al., 2005; Gallois-Montbrun et al., 2007; Kozak et al., 2006). It is conceivable that Vif accelerates this process and experiments are ongoing to study the relative contribution of Vif to the assembly of APO3G into high molecular mass complexes.
Materials and Methods
Cell culture and transfections
HeLa cells were propagated in Dulbecco’s modified Eagles medium (DMEM) containing 10% fetal bovine serum. LuSIV cells are derived from CEMx174 cells and contain a luciferase indicator gene under the control of the SIVmac239 LTR. These cells were obtained through the NIH AIDS Research and Reference Reagent Program and were maintained in complete RPMI 1640 medium supplemented with 10% FBS and hygromycin B (300 μg/ml). For transfection of HeLa cells, cells were grown in 25 cm2 flasks to about 80% confluency. Cells were transfected using LipofectAMINE PLUS™ (Invitrogen Corp, Carlsbad CA) following the manufacturer’s recommendations. A total of 5 μg of plasmid DNA per 25 cm2 flask was generally used. Where appropriate, empty vector DNA (pcDNA3.1(−)MycHis (Invitrogen)) or vif-defective vector DNA (pNL-A1vif(−)) was used to adjust total DNA amounts. Cells were harvested 24 to 48 h post-transfection.
Antibodies
A peptide antibody to human APO3G was prepared by immunizing rabbits with KLH-coupled peptides corresponding to residues 367 to 384 of human APO3G. A monoclonal antibody to Vif (MAb #319) was used for all immunoblot analyses and was obtained from Michael Malim through the NIH AIDS Research and Reference Reagent Program (Cat No: 6459). A monoclonal antibody to alpha-tubulin (T-9026; Sigma-Aldrich, Inc., St. Louis MO) was used as a loading control. An HIV-positive patient serum was used for the identification of HIV-1 capsid (CA) protein. An HIV-1 Env-specific antibody was produced by immunizing rabbits with purified recombinant gp120.
Plasmids
Construction of the vif-defective pNL4-3 vif(−) vector was described previously (Karczewski and Strebel, 1996). Expression vectors pNL-A1 for the expression of HXB2 Vif in a proviral backbone and its vif-defective variant, pNL-A1vif(−), have been described previously (Khan et al., 2001; Strebel et al., 1987). Both vectors express additional HIV-1 proteins including Vpr, Tat, Rev, Vpu, Env, and Nef. The construction of pcDNA-hVif for the expression of NL4-3 Vif from a codon-optimized gene under the transcriptional control of a CMV promoter has been described elsewhere (Nguyen et al., 2004). The pYFP-Vif vector for the expression of Yellow Fluorescent Protein (YFP)-tagged HXB2 Vif under the control of a CMV promoter was a gift of Tariq Rana (Wichroski et al., 2005). The vector for the expression of APO3G carrying a C-terminal myc epitope tag, pcDNA-Apo3Gmyc, has been described previously (Kao et al., 2003a). A variant, pcDNA-Apo3G, expressing untagged APO3G was used for all experiments described in this study except figure 5 and was constructed by insertion of a stop codon at the end of the APO3G gene in pcDNA-Apo3Gmyc (Opi et al., 2006). Plasmid pcDNA-APO3Gwt-myc is a derivative of pcDNA-Apo3Gmyc and was created by site-specific mutagenesis to introduce N162S and Y370D changes that make it identical to the published sequence for CEM15 ((Sheehy et al., 2002); Genbank NM_021822 & AF182420).
Immunoblotting
For immunoblot analysis of intracellular proteins, whole cell lysates were prepared as follows: Cells were washed once with PBS, suspended in PBS and mixed with an equal volume of sample buffer (4% sodium dodecyl sulfate, 125 mM Tris-HCl, pH 6.8, 10% 2-mercaptoethanol, 10% glycerol, and 0.002% bromphenol blue). To analyze virus-associated proteins, cell-free filtered supernatants from transfected HeLa cells (5–7 ml) were pelleted (75 min, 35,000 rpm) through a 20% sucrose cushion (4 ml) in an SW41 rotor. The concentrated virus pellet was suspended in PBS and mixed with an equal volume of sample buffer. Proteins were solubilized by heating 10 to 15 min at 95°C. Cell and virus lysates were subjected to SDS-PAGE; proteins were transferred to PVDF membranes and reacted with appropriate antibodies as described in the text. Membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies (Amersham Biosciences, Piscataway NJ) and proteins were visualized by enhanced chemiluminescence (ECL, Amersham Biosciences).
Virus preparation
Virus stocks were prepared by transfection of HeLa cells with appropriate plasmid DNAs. Virus-containing supernatants were harvested 24 to 48 h after transfection. Cellular debris was removed by centrifugation (5 min, 1500 rpm) and clarified supernatants were filtered (0.45 μM) to remove residual cellular contaminations. Filtered virus stocks were further purified by pelleting through a 20% sucrose cushion (75 min, 4°C at 35,000 rpm in an SW41 rotor).
Viral infectivity assay
To determine viral infectivity, virus stocks were normalized for equal reverse transcriptase activity and used to infect 5 × 105 LuSIV cells (Roos et al., 2000) in a 24-well plate in a total volume of 1.2 to 1.4 ml. Infection was allowed for 24 h at 37°C. Cells were then harvested and lysed in 150 μl of Promega 1x reporter lysis buffer (Promega Corp., Madison WI). To determine the luciferase activity in the lysates, 50 μl of each lysate were combined with luciferase substrate (Promega Corp., Madison WI) by automatic injection and light emission was measured for 10 seconds at room temperature in a luminometer (Optocomp II, MGM Instruments, Hamden CT).
Acknowledgments
We are grateful to Judith Levin and Yasumasa Iwatani for the pcDNA-APO3Gwt-myc vector. We thank Michael Malim for the Vif monoclonal antibody (MAb #319) and Jason Roos and Janice Clements for the LuSIV indicator cell line. The latter reagents were obtained through the NIH AIDS Research and Reference Reagent Program. This work was supported by a Grant from the NIH Intramural AIDS Targeted Antiviral Program to K.S. and by the Intramural Research Program of the NIH, NIAID.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akari H, Fujita M, Kao S, Khan MA, Shehu-Xhilaga M, Adachi A, Strebel K. High level expression of human immunodeficiency virus type-1 Vif inhibits viral infectivity by modulating proteolytic processing of the Gag precursor at the p2/nucleocapsid processing site. J Biol Chem. 2004;279:12355–12362. doi: 10.1074/jbc.M312426200. [DOI] [PubMed] [Google Scholar]
- Beard BC, Wilson SH, Smerdon MJ. Suppressed catalytic activity of base excision repair enzymes on rotationally positioned uracil in nucleosomes. Proc Natl Acad Sci U S A. 2003;100:7465–7470. doi: 10.1073/pnas.1330328100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop KN, Holmes RK, Malim MH. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J Virol. 2006;80:8450–8458. doi: 10.1128/JVI.00839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YL, Soros VB, Kreisberg JF, Stopak K, Yonemoto W, Greene WC. Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature. 2005;435:108–114. doi: 10.1038/nature03493. [DOI] [PubMed] [Google Scholar]
- Conticello SG, Harris RS, Neuberger MS. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr Biol. 2003;13:2009–2013. doi: 10.1016/j.cub.2003.10.034. [DOI] [PubMed] [Google Scholar]
- Fujita M, Akari H, Sakurai A, Yoshida A, Chiba T, Tanaka K, Strebel K, Adachi A. Expression of HIV-1 accessory protein Vif is controlled uniquely to be low and optimal by proteasome degradation. Microbes Infect. 2004;6:791–798. doi: 10.1016/j.micinf.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Gallois-Montbrun S, Kramer B, Swanson CM, Byers H, Lynham S, Ward M, Malim MH. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J Virol. 2007;81:2165–2178. doi: 10.1128/JVI.02287-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves J, Jallepalli P, Gabuzda DH. Subcellular localization of the Vif protein of human immunodeficiency virus type 1. J Virol. 1994;68:704–712. doi: 10.1128/jvi.68.2.704-712.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Cen S, Niu M, Saadatmand J, Kleiman L. Inhibition of tRNA3Lys-primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J Virol. 2006;80:11710–11722. doi: 10.1128/JVI.01038-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao S, Akari H, Khan MA, Dettenhofer M, Yu XF, Strebel K. Human immunodeficiency virus type 1 Vif is efficiently packaged into virions during productive but not chronic infection. J Virol. 2003a;77:1131–1140. doi: 10.1128/JVI.77.2.1131-1140.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao S, Khan MA, Miyagi E, Plishka R, Buckler-White A, Strebel K. The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity. J Virol. 2003b;77:11398–11407. doi: 10.1128/JVI.77.21.11398-11407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao S, Miyagi E, Khan MA, Takeuchi H, Opi S, Goila-Gaur R, Strebel K. Production of infectious human immunodeficiency virus type 1 does not require depletion of APOBEC3G from virus-producing cells. Retrovirology. 2004;1:27. doi: 10.1186/1742-4690-1-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski MK, Strebel K. Cytoskeleton association and virion incorporation of the human immunodeficiency virus type 1 Vif protein. J Virol. 1996;70:494–507. doi: 10.1128/jvi.70.1.494-507.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavli B, Sundheim O, Akbari M, Otterlei M, Nilsen H, Skorpen F, Aas PA, Hagen L, Krokan HE, Slupphaug G. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J Biol Chem. 2002;277:39926–39936. doi: 10.1074/jbc.M207107200. [DOI] [PubMed] [Google Scholar]
- Khan MA, Aberham C, Kao S, Akari H, Gorelick R, Bour S, Strebel K. Human immunodeficiency virus type 1 Vif protein is packaged into the nucleoprotein complex through an interaction with viral genomic RNA. J Virol. 2001;75:7252–7265. doi: 10.1128/JVI.75.16.7252-7265.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA, Goila-Gaur R, Opi S, Miyagi E, Takeuchi H, Kao S, Strebel K. Analysis of the contribution of cellular and viral RNA to the packaging of APOBEC3G into HIV-1 virions. Retrovirology. 2007;4:48. doi: 10.1186/1742-4690-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA, Kao S, Miyagi E, Takeuchi H, Goila-Gaur R, Opi S, Gipson CL, Parslow TG, Ly H, Strebel K. Viral RNA is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J Virol. 2005;79:5870–5874. doi: 10.1128/JVI.79.9.5870-5874.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak SL, Marin M, Rose KM, Bystrom C, Kabat D. The anti-HIV-1 editing enzyme APOBEC3G binds HIV-1 RNA and messenger RNAs that shuttle between polysomes and stress granules. J Biol Chem. 2006;281:29105–29119. doi: 10.1074/jbc.M601901200. [DOI] [PubMed] [Google Scholar]
- Lecossier D, Bouchonnet F, Clavel F, Hance AJ. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science. 2003;300:1112. doi: 10.1126/science.1083338. [DOI] [PubMed] [Google Scholar]
- Liu H, Wu X, Newman M, Shaw GM, Hahn BH, Kappes JC. The Vif protein of human and simian immunodeficiency viruses is packaged into virions and associates with viral core structures. J Virol. 1995;69:7630–7638. doi: 10.1128/jvi.69.12.7630-7638.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 2003;114:21–31. doi: 10.1016/s0092-8674(03)00515-4. [DOI] [PubMed] [Google Scholar]
- Marin M, Rose KM, Kozak SL, Kabat D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med. 2003;9:1398–1403. doi: 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- Mehle A, Goncalves J, Santa-Marta M, McPike M, Gabuzda D. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 2004a;18:2861–2866. doi: 10.1101/gad.1249904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehle A, Strack B, Ancuta P, Zhang C, McPike M, Gabuzda D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J Biol Chem. 2004b;279:7792–7798. doi: 10.1074/jbc.M313093200. [DOI] [PubMed] [Google Scholar]
- Newman EN, Holmes RK, Craig HM, Klein KC, Lingappa JR, Malim MH, Sheehy AM. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr Biol. 2005;15:166–170. doi: 10.1016/j.cub.2004.12.068. [DOI] [PubMed] [Google Scholar]
- Nguyen KL, llano M, Akari H, Miyagi E, Poeschla EM, Strebel K, Bour S. Codon optimization of the HIV-1 vpu and vif genes stabilizes their mRNA and allows for highly efficient Rev-independent expression. Virology. 2004;319:163–175. doi: 10.1016/j.virol.2003.11.021. [DOI] [PubMed] [Google Scholar]
- Opi S, Kao S, Goila-Gaur R, Khan MA, Miyagi E, Takeuchi H, Strebel K. Human immunodeficiency virus type 1 Vif inhibits packaging and antiviral activity of a degradation-resistant APOBEC3G variant. J Virol. 2007;81:8236–8246. doi: 10.1128/JVI.02694-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opi S, Takeuchi H, Kao S, Khan MA, Miyagi E, Goila-Gaur R, Iwatani Y, Levin JG, Strebel K. Monomeric APOBEC3G is catalytically active and has antiviral activity. J Virol. 2006;80:4673–4682. doi: 10.1128/JVI.80.10.4673-4682.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos JW, Maughan MF, Liao Z, Hildreth JE, Clements JE. LuSIV cells: a reporter cell line for the detection and quantitation of a single cycle of HIV and SIV replication. Virology. 2000;273:307–315. doi: 10.1006/viro.2000.0431. [DOI] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med. 2003;9:1404–1407. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- Simon JH, Fouchier RA, Southerling TE, Guerra CB, Grant CK, Malim MH. The Vif and Gag proteins of human immunodeficiency virus type 1 colocalize in infected human T cells. J Virol. 1997;71:5259–5267. doi: 10.1128/jvi.71.7.5259-5267.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopak K, de Noronha C, Yonemoto W, Greene WC. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell. 2003;12:591–601. doi: 10.1016/s1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- Strebel K, Bour S. Molecular interactionsof HIV with host factors. AIDS. 1999;13(Suppl A):S13–S24. [PubMed] [Google Scholar]
- Strebel K, Daugherty D, Clouse K, Cohen D, Folks T, Martin MA. The HIV ‘A’ (sor) gene product is essential for virus infectivity. Nature. 1987;328:728–730. doi: 10.1038/328728a0. [DOI] [PubMed] [Google Scholar]
- Swanson CM, Puffer BA, Ahmad KM, Doms RW, Malim MH. Retroviral mRNA nuclear export elements regulate protein function and virion assembly. EMBO J. 2004;23:2632–2640. doi: 10.1038/sj.emboj.7600270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichroski MJ, Ichiyama K, Rana TM. Analysis of HIV-1 viral infectivity factor-mediated proteasome-dependent depletion of APOBEC3G: correlating function and subcellular localization. J Biol Chem. 2005;280:8387–8396. doi: 10.1074/jbc.M408048200. [DOI] [PubMed] [Google Scholar]
- Xu H, Chertova E, Chen J, Ott DE, Roser JD, Hu WS, Pathak VK. Stoichiometry of the antiviral protein APOBEC3G in HIV-1 virions. Virology. 2007;360:247–256. doi: 10.1016/j.virol.2006.10.036. [DOI] [PubMed] [Google Scholar]
- Xu H, Svarovskaia ES, Barr R, Zhang Y, Khan MA, Strebel K, Pathak VK. A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc Natl Acad Sci U S A. 2004;101:5652–5657. doi: 10.1073/pnas.0400830101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003;424:94–98. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]