

Abstract
Proteins that discriminate between cisplatin–DNA adducts and oxaliplatin–DNA adducts are thought to be responsible for the differences in tumor range, toxicity, and mutagenicity of these two important chemotherapeutic agents. However, the structural basis for differential protein recognition of these adducts has not been determined and could be important for the design of more effective platinum anticancer agents. We have determined high-resolution NMR structures for cisplatin–GG and undamaged DNA dodecamers in the AGGC sequence context and have compared these structures with the oxaliplatin–GG structure in the same sequence context determined previously in our laboratory. This structural study allows the first direct comparison of cisplatin–GG DNA and oxaliplatin–GG DNA solution structures referenced to undamaged DNA in the same sequence context. Non-hydrogen atom rmsds of 0.81 and 1.21 were determined for the 15 lowest-energy structures for cisplatin–GG DNA and undamaged DNA, respectively, indicating good structural convergence. The theoretical NOESY spectra obtained by back-calculation from the final average structures showed excellent agreement with the experimental data, indicating that the final structures are consistent with the NMR data. Several significant conformational differences were observed between the cisplatin–GG adduct and the oxaliplatin–GG adduct, including buckle at the 5′ G6•C19 base pair, opening at the 3′ G7•C18 base pair, twist at the A5G6•T20C19 base pair step, slide, twist, and roll at the G6G7•C19C18 base pair step, slide at the G7C8•C18G17 base pair step, G6G7 dihedral angle, and overall bend angle. We hypothesize that these conformational differences may be related to the ability of various DNA repair proteins, DNA binding proteins, and DNA polymerases to discriminate between cisplatin–GG and oxaliplatin–GG adducts.
Cisplatin [CP, cis-diamminedichloroplatinum(II)] and carboplatin [CBDCA, cis-diammine-1,1-cyclobutanedicarboxylatoplatinum(II)] are widely used for treatment of testicular cancer, ovarian cancer, head and neck tumors, and a variety of other solid tumors. However, many tumors are intrinsically resistant or develop acquired resistance to these chemotherapeutic agents, and tumors that are resistant to one of these platinum compounds are usually cross-resistant to the other. The mutagenicity of CP in vivo (1) is also of concern because secondary malignancies have been associated with CP chemotherapy (2). Considerable effort has been spent to develop third-generation platinum anticancer agents that would not share these limitations. Oxaliplatin [OX, trans-(R,R)-1,2-diaminocyclohexaneoxalatoplatinum(II)] is one such compound and has recently been approved for the treatment of colorectal cancer and tumors that are resistant to CP and CBDCA. While OX does exhibit some mutagenicity (3), it appears to be less mutagenic than CP (4). CP and CBDCA form Pt–DNA adducts that contain the cis-diammine carrier ligands, while OX forms Pt–DNA adducts that contain the trans-(R,R)-1,2-diaminocyclohexane carrier ligand. For the sake of simplicity, we will refer to these as CP–DNA and OX–DNA adducts, respectively, throughout this work. Other than the differences in carrier ligand, the adducts formed by CP, CBDCA, and OX appear to be identical in terms of the type of adduct formed (60–65% intrastrand GG, 25–30% intrastrand AG, 5–10% intrastrand GNG, and 1–3% interstrand) and the site of adduct formation (5-7).
Because of their abundance, the intrastrand GG adducts are thought to be major determinants of the cytotoxic response to platinum anticancer agents. The basis for the differences in tumor range and mutagenicity of OX compared to CP and CBDCA is not known but is thought to be determined by the ability of proteins involved in damage recognition, damage repair, and/or damage tolerance to discriminate between CP and OX adducts. For example, hMSH2 and MutS bind with greater affinity to CP–GG adducts than to OX–GG adducts (8, 9), and as might be expected from this difference in binding affinity, defects in mismatch repair result in resistance to CP and CBDCA, but not to OX (8, 10-13). Similarly, a number of damage recognition proteins and transcription factors, especially those with HMG domains, have been shown to discriminate between CP– and OX–GG adducts (14, 15). The mechanism(s) by which the binding of these proteins to Pt–DNA adducts influences the cytotoxic response is not known but has been postulated to involve shielding of the adducts from DNA repair and tolerance mechanisms (16-19), activation of signaling pathways leading to cell cycle arrest or apoptosis, and/or hijacking of transcription factors needed for DNA replication or cell division (15, 20). The binding specificity has been determined for only a few of these proteins, but where it has been studied, these proteins bind to CP–GG adducts with higher affinity than to OX–GG adducts (14, 15, 21). Finally, translesion DNA polymerases such as pol β and pol η have been shown to bypass OX–GG adducts with higher efficiency than CP–GG adducts (19, 22, 23), which might contribute to the differences in CP and OX mutagenicity.
The CP– and OX–GG adducts form in the major groove and have been shown to bend the DNA in the direction of the major groove (24-28). The proteins that discriminate between CP– and OX–GG adducts either bind to bent DNA or bend the DNA in the direction of the major groove after binding (29-32). Because these proteins primarily interact with the minor groove, we have hypothesized that the ability of the proteins to discriminate between CP– and OX–GG adducts probably results from subtle differences in conformation or conformational dynamics in the DNA containing the two adducts rather than from physical interaction of the proteins with the carrier ligands of the adducts in the major groove (26). A number of structures have been reported for CP–GG and OX–GG adducts (24, 25, 27, 33, 34). The overall conformation of DNA containing these adducts appears to be similar, but exact comparisons have been difficult to make because the structures have been determined by different techniques (crystallography vs NMR), in different sequence contexts, and with oligonucleotides that differ in length. Further, the NMR structures obtained to date have varied with respect to the number and resolution of NMR constraints obtained and the molecular mechanics simulations used to convert the NMR constraints to final structures (24-26). X-ray crystallographic structures have been reported for the CP–GG and OX–GG adducts in the TGGT sequence context (27, 28), but differences between the solution and X-ray structures (26) of the OX–GG DNA adduct suggest that the X-ray structures may have been at least partially constrained by crystal packing restraints (26). CP–GG adducts appear to be more mutagenic in the AGG sequence context than in the CGG, TGG, or GGG sequence context (35-38). In addition, our previous studies with translesion DNA polymerases have utilized DNA templates with oxaliplatin located in the AGGC sequence context (19, 22, 23, 39). Thus, we have recently obtained a high-resolution NMR solution structure of the OX–GG adduct in the mutagenic AGGC sequence context (the underlined bases indicate the position of the OX–GG adduct) (26).
In this paper, we report high-resolution NMR solution structures of the CP–GG adduct and undamaged DNA in the same sequence context. Moreover, the solution structures were determined using the same experimental approach employed for the OX–GG adduct. This is the first direct comparison of the CP–GG adduct, the OX–GG adduct, and undamaged DNA solution structures in the same sequence context. This study has allowed us to resolve the effects of sequence context and platinum carrier ligand on several previously reported chemical shift anomalies. For example, we find that several unusual chemical shifts, previously reported for the OX–GG adduct compared to earlier NMR solution structures of CP–GG adducts, were also seen for the CP–GG adduct in the AGGC sequence context. Thus, these chemical shift differences appear to reflect the effect of sequence context rather than the presence of the trans-(R,R)-1,2-diaminocyclohexane carrier ligand. The direct comparison of NOESY data from the CP–GG adduct, the OX–GG adduct, and undamaged DNA in the same sequence context has also allowed us to more accurately determine the effect of CP–GG and OX–GG adducts on NOE cross-peak volumes of nearby DNA residues. For example, we have been able to show that the unusually weak cross-peak between H2′ of the C complementary to the 5′ G and H6/ H8 of the base complementary to the 5′ flanking residue reported by Marzilli et al. (25) is a feature of CP–GG adducts, but not of OX–GG adducts. Finally, the direct comparison of CP–GG and OX–GG solution structures has allowed us to identify several conformational differences not seen in the crystallographic comparison of the two adducts in the TGGT sequence context. For example, significant differences were observed between the two adducts: buckle at the 5′ G6•C19 base pair, opening at the 3′ G7•C18 base pair, twist at the A5G6•T20C19 base pair step, slide, twist, and roll at the G6G7•C19C18 base pair step, slide at the G7C8•C18G17 base pair step, G6G7 dihedral angle, and overall bend angle. These subtle conformational differences may allow differential recognition of CP– and OX–GG adducts by critical DNA binding proteins that influence the cytotoxic response to these adducts.
MATERIALS AND METHODS
Preparation and HPLC Purification of the CP–GG Oligonucleotide
The GG strand (the strand containing the Pt–GG adduct) and complement (CC strand) were purchased from Operon Biotechnologies, Inc. The GG strand containing the cisplatin adduct was prepared using our previously described procedure (26). The DNA duplex (Figure 1) was prepared by adding the CC strand solution to the GG strand in NMR buffer [100 mM NaCl and 5 mM phosphate buffer (pH 7.0)] at a calculated 1:1 ratio. The DNA was annealed by cooling overnight from 70 °C to room temperature. The 1:1 ratio of CC strand to GG strand was verified by analysis of two-dimensional (2D) DQF-COSY NMR spectra as described previously (26).
FIGURE 1.
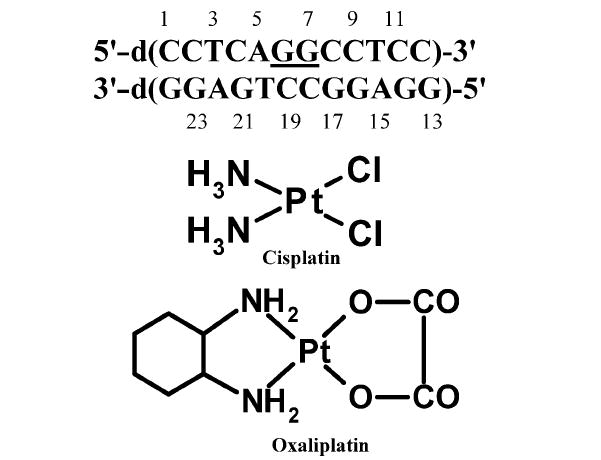
Duplex DNA sequence used in this study. The underlined bases show the position of the platinum adduct. Chemical structures of cisplatin and oxaliplatin.
Characterization of the CP–GG Adduct by Liquid Chromatography and Mass Spectroscopy (LC–MS)
LC–MS characterizations of the CP–GG adduct and undamaged DNA in the AGGC sequence context were performed following the procedure reported previously, with slight modifications (26). The platinated and undamaged 12-mer GG strand were first separated by HPLC. Following desalting and concentration, the purity of the CP-adducted 12-mer GG strand was confirmed by denaturing gel electrophoresis. No unplatinated 12-mer GG strand was detected by this method, indicating that the CP-adducted 12-mer GG strand was >95% pure (data not shown). The CP[d(GpG)] adduct and undamaged DNA digests were analyzed on a Surveyor LC system, connected to Surveyor photodiode array (PDA) and LCQDECA ion trap mass spectrometer (Thermo Finnigan, San Jose, CA). A Clarity 3 μm Oligo-RP column (100 mm × 4.60 mm) (Phenomenex, Torrance, CA) was operated with a linear gradient from 100% 10 mM ammonium acetate (pH 3.5) to 80% MeOH in 10 mM ammonium acetate for 20 min at a flow rate of 200 μL/min. UV spectra (λ = 200–400 nm) were recorded with a Finnigan Surveyor PDA detector, and positive and negative full scan (m/z 80–1000) mass spectra were acquired with Xcalibur (Thermo Finnigan).
NMR Experiments
Two NMR samples were prepared for both the undamaged DNA and CP–GG DNA adduct, one in 5% D2O/95% H2O buffer (H2O sample) for detection of exchangeable protons and the other in 100% D2O buffer (D2O sample) for detection of nonexchangeable protons. All NMR spectra were acquired on a Varian Inova 600 or 800 MHz spectrometer. The carrier frequency for protons was set on the H2O signal. One-dimensional (1D) proton spectra were recorded at temperatures ranging from 2 to 40 °C for detection of exchangeable protons. NOESY spectra were recorded at 800 MHz using a mixing time of 200 ms, 32 transients, and 320 or 400 complex FIDs corresponding to spectral widths of 20 and 9 ppm in both dimensions for the samples in H2O and D2O, respectively. WATERGATE was employed to suppress the water peak in water samples. Distance constraints were obtained from the 200 ms NOESY spectra in both H2O and D2O (40). The assignments were obtained initially from NOE connectivities and confirmed by analysis of 2D DQF-COSY (41) (2048 × 720 complex points, 9 ppm spectral width in both dimensions, 32 transients) and TOCSY data (2048 × 320 complex points, 9 ppm spectral width in both dimensions, 32 transients). 2D DQF-COSY data were also used to extract J coupling constraints for determination of sugar pucker. 2D 1H–31P HETCOR (42) (heteronuclear correlation spectroscopy) data were collected for the D2O samples (800 × 112 complex points, spectral width of 2040 × 900 Hz, 320 transients) at 600 MHz. The 1H and 31P chemical shifts were referenced relative to internal TSP and external 10% (w/v) trimethyl phosphate at 0.0 ppm, respectively. NMR data were processed with NMRPipe and analyzed with Felix (version 2000; Molecular Simulations, Inc., San Diego, CA).
Structure Calculations
The structure of the DNA duplexes was calculated using CNS (Crystallographic and NMR System) version 1.1 with a simulated annealing protocol (43). To construct the CP intrastrand platinum cross-link between G6 and G7, two new residues (XGU and YGU) were added to the topology and parameter database of CNS. Residue YGU was modified for G6 by adding a bond between the N7 atom of G6 and the Pt atom of NH3-Pt. XGU was modified for G7 by combining the Pt(NH3)2 moiety with N7 of the G7 residue. The new bonds, angles, dihedral angles, and force constants were obtained from ref 44. An initial extended structure was produced from the duplex sequence. Two types of constraints were used in the structure calculations: experimentally derived and halonomic distance constraints. A total of 551 and 920 NOE-derived distance restraints were employed in the structure calculations for CP–GG DNA and undamaged DNA, respectively. The NOE cross-peaks were standardized to the Cyt H5–H6 cross-peaks, with a H5–H6 distance of 2.5 Å. One additional angstrom was added for distance constraints involving methyl groups.
Planarity was also restrained for all base pairs (45) with the exception of the base pairs associated with the CP adduct, where the force constant was reduced by half. Although the sugar pucker conformation was estimated from analysis of NOE constraints and simulations of cross-peaks in DQF-COSY spectra to encompass both A and B DNA conformations, a broad range of torsion angle restraints were employed for the sugar–phosphate torsion angles, i.e., α (−70 ± 50°), β (180 ± 50°), γ (60 ± 35°), ζ (−85 ± 50°), χ (−120 ± 50°), δ (120 ± 50°), and ε (180 ± 50°) in the structure calculations. Finally, six distance restraints per base pair were employed to describe Watson–Crick base pairing (45). The lower and upper bounds for H-bond distance constraints were set to 7%. However, for base pairs associated with the CP adduct, the bounds were increased to 10%. Of 20 calculated structures, the 15 with the lowest energy were accepted as a family.
RESULTS AND DISCUSSION
Characterization of the Cisplatin–GG Intrastrand Cross–Link
The purity of the CP-adducted GG strand was >95% as determined by HPLC and denaturing gel electrophoresis (see Materials and Methods). Following hybridization with the complementary strand, the CP–GG 12-mer duplex was further characterized following the same procedure reported by Wu et al. (26) for the OX–GG 12-mer duplex. Both undamaged 12-mer duplex and 12-mer duplex containing the CP–GG intrastrand cross-link were digested to the deoxynucleoside and CP[d(GpG)] level, respectively, with a combination of deoxyribonuclease I, nuclease P1, and alkaline phosphatase as described by Eastman (46) and were characterized by HPLC–UV–MS. Using this procedure, the Pt–GG intrastrand cross-link is digested to Pt[d(GpG)], the Pt–AG intrastrand cross-link to Pt[d(ApG)], and the Pt–G monoadduct to Pt(dG). In addition, both the GNG intrastrand cross-link and the GG interstrand cross-link are digested to dG–Pt–dG (46). The HPLC–UV chromatogram of the digestion products derived from the undamaged 12-mer duplex exhibited four peaks (Figure 2, I). These peaks were identified as dC, dT, dG, and dA on the basis of their retention time, UV spectrum, and mass spectra (data not shown) that were identical to those of authentic standards (data not shown). The relative abundance of the individual deoxynucleosides was as expected for the 12-mer duplex. The 12-mer duplex containing the CP–GG intrastrand cross-link showed the four normal deoxynucleosides and one additional peak eluting just before dG (Figure 2, IIA). This additional digestion product had the same retention time and UV spectrum as the synthetic CP[d(GpG)] standard (data not shown). This additional peak was further identified as CP[d(GpG)] by the presence of the expected molecular ions in both the positive (m/z 824.89) (Figure 2, IIB) and negative (m/z 822.90) (data not shown) modes. In addition, the presence of a peak with a mass of 806.98 (positive m/z 808.01 and negative m/z 805.93) corresponds to the molecular weight of CP[d(GpG)] after dissociation of an ammonia molecule of cisplatin (Figure 2, IIB). Last, the MS spectra also showed the expected Pt isotopes, confirming the presence of a Pt compound. No digestion products were detected with the masses and isotopic pattern expected for the dG–CP–dG, CP[d(ApG)], or CP(dG) adducts (data not shown). These data demonstrate that the 12-mer duplex employed in this NMR study consisted exclusively of the CP–GG intrastrand cross-link. The lack of any detectable CP–AG intrastrand cross-link is consistent with previous reports that the Pt–AG diadduct comprises less than 3% of the total Pt intrastrand cross-links formed in the AGG sequence context (26, 47).
FIGURE 2.
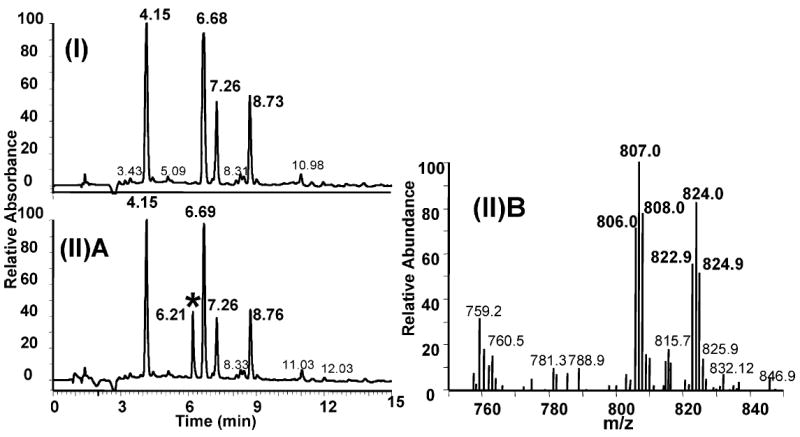
HPLC–UV–MS chromatogram of digestion products obtained from an undamaged 12-mer duplex (I) and from the same 12-mer duplex containing the CP–GG intrastrand cross-link. The spectra include (I) the HPLC–UV elution profile of the digestion products obtained from the undamaged 12-mer duplex, (II)A the HPLC–UV elution profile of the digestion products obtained from the 12-mer duplex containing the CP–GG adduct [the peak with the asterisk corresponds to the elution position of a CP–d(GpG) standard], and (II)B MS-positive mass spectrum of the peak identified with the asterisk in Figure 2(II)A.
Proton Assignments
Chemical shift assignments for both the CP and undamaged DNA samples were obtained as described previously for the OX–GG adduct in the same 12-mer duplex (26). Assignments of the nonexchangeable base and sugar protons were obtained by analysis of NOESY spectra recorded in D2O buffer at 25 °C. For example, the NOESY region for the CP–DNA duplex in Figure 3 shows NOE correlations between the base (purine H8/pyrimidine H6) and the H1′ sugar protons and between the base (purine H8/pyrimidine H6) and the H2′ and H2″ sugar protons. Sequential connectivities can be observed without interruptions from C1 to C12 in the GG strand and from G13 to G24 in the CC strand. Comparable data for the undamaged DNA are shown in Figure S1 of the Supporting Information. Once sequential assignments were obtained for H8/H6, H1′, H2′, and H2″, the assignments of the other (H3′, H4′, H5′, and H5″) sugar protons were extended on the basis of the NOESY, DQF-COSY, and TOCSY spectra, following standard procedures (41). Assignments of the exchangeable protons were obtained by analyzing distance connectivities between the imino and base/amino proton regions of NOESY spectra in H2O buffer at 2 °C. The chemical shift assignments for the CP–DNA duplex and undamaged DNA are listed in Tables S1 and S2 of the Supporting Information, respectively.
FIGURE 3.
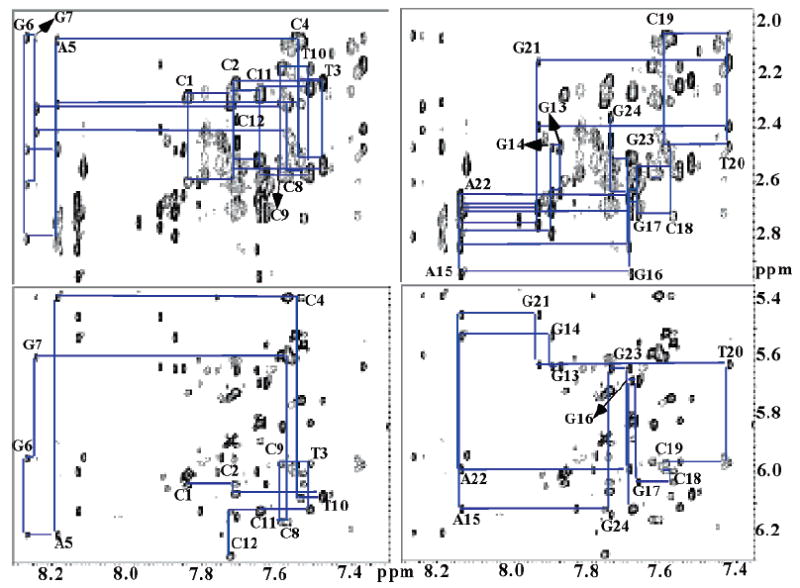
Expanded nonexchangeable proton regions taken from the 2D NOESY (200 ms) spectrum acquired in D2O for the CP–GG 12-mer duplex. The H6/H8–H2′/H2″ (top) and H6/H8–H1′ (bottom) regions are shown. The left and right regions correspond to the GG (the strand containing the Pt–GG adduct) and CC (complementary) strands, respectively.
Previous NMR studies of Pt–GG adducts have examined a single type of adduct (CP or OX) in a single sequence context and have compared both the proton chemical shifts and NOE cross-peak intensities with average values for undamaged B-DNA in a variety of sequence contexts (24, 25, 27, 33) (34). However, it is unclear whether some of the observed differences are due to differences in sequence context rather than between the platinated and unplatinated strand. We are in a unique position to directly determine the effect of Pt–GG adducts on both the proton chemical shifts and NOE cross-peak intensities of the DNA in the region of the Pt–GG adduct, as our NMR data were collected on OX–GG DNA (26), CP–GG DNA, and undamaged B-DNA in the same AGGC sequence context. Chemical shift differences among OX–GG DNA, CP–GG DNA, and undamaged B-DNA for the H6/H8, H1′, H2′, and H2″ protons are shown in Figure 4. It is clear from this comparison that the formation of Pt–GG adducts primarily affects chemical shifts of protons within the central 4 bp region. Moreover, a larger difference in chemical shifts is observed for protons on the strand containing the adduct relative to the complementary strand. Both the CP–GG and OX–GG adducts exhibited downfield chemical shifts for the H8 proton of both G6 and G7, the H6 proton of C8, and the H1′ protons of A5, G6, and C8. In contrast, upfield chemical shifts were observed for the H1′ proton of G7, the H2′ proton of G6 and G7, and the H2″ proton of G7.
FIGURE 4.
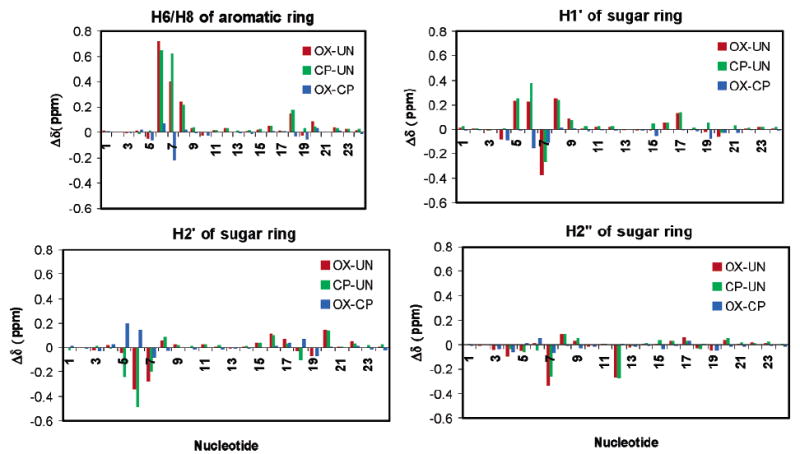
Effect of platination on chemical shifts of selected base and sugar proton chemical shifts. The values were determined by using experimental chemical shifts (Tables S1 and S2 and ref 26).
Previous studies have not made direct comparisons of platinated adducts with undamaged DNA in the same sequence context and thus were unable to determine the effect of Pt–DNA adducts on proton chemical shifts with this precision. However, downfield chemical shifts have been reported for the H8 protons of the 5′ and 3′ G residues for all Pt–GG adducts studied to date (24-26, 33), so this has been considered a universal feature of Pt–GG adducts. In their analysis of a CP–GG adduct, Marzilli et al. (25) reported a much greater downfield chemical shift for the H8 proton on the 5′ G residue than for the 3′ G residue and a significant upfield shift of the H2′ proton on the C complementary to the 5′ G residue, which they considered universal features of all CP–GG adducts. However, we do not observe either of these features in direct comparisons of the CP–GG DNA (Table S1) adducts with undamaged DNA (Table S2), so these chemical shifts do not appear to be features of all CP–GG adducts. Our adducts differ from all previous adducts in that they are in the AGGC sequence context, so the differences in proton chemical shifts between our Pt–GG adducts and the CP–GG adduct reported by Marzilli et al. (25) may reflect an effect of sequence context (AGGC vs CGGC). Experiments are currently underway to clarify the effect of sequence context on NMR data and conformation of the Pt–GG adducts.
With respect to NOE cross-peaks (Table S3 of the Supporting Information) for both CP–GG and OX–GG adducts, we observed stronger G6 H8–G7 H8 NOE cross-peaks and weaker A5 H1′–G6 H8 and G6 H1′–G7 H8 cross-peaks compared to those in undamaged DNA, consistent with data reported previously for other CP–GG adducts. We also see some significant difference in NOE cross-peak intensities between CP–GG and OX–GG adducts. Marzilli et al. (25) had previously suggested that an unusually weak NOE cross-peak between H2′ of the C complementary to the 5′ G residue and H6/H8 of the base complementary to the 5′ flanking base was a universal feature of all CP–GG adducts. Here, we find that the C19 H2′–T20 H6 NOE cross-peak for the CP–GG adduct in the AGGC sequence context is also weaker than that in undamaged DNA (Table S3). In contrast, we have previously reported (26) that the NOE cross-peak between C19 H2′ and T20 H6 for the OX–GG adduct was near normal. In addition, compared to the OX–DNA adduct, the CP–DNA adduct exhibited a stronger G6 H1′–G7 H8 NOE cross-peak and a weaker G6 H8–G7 H8 NOE cross-peak (Table S3). These differences in NOE cross-peak intensity are consistent with the differences in DNA helical parameters of CP– and OX–DNA adducts, such as the differences observed for the twist in the A5 T20•G6 C19 base pair step and twist and roll in the G6 C19•G7 C18 base pair step (see below).
The CP–GG and OX–GG adducts also differ significantly from each other in that the extent of the downfield chemical shift for the G7 H8 and G6 H1′ protons and the upfield chemical shift of the A5 and G6 H2′ protons was significantly greater for the CP–GG adduct than the OX–GG adduct (Figure 4). The difference in the chemical shift of the G7 H8 proton is particularly interesting because it could theoretically arise from a difference in the canting of the 3′ G residue in relation to the 5′ G residue for the OX–GG and CP–GG adducts. The degree of base canting for both the 5′ and 3′ G residues was determined by measuring the C5–N7–Pt–cis N and C8–N7–Pt–cis N dihedral angles (48) (Table S4 of the Supporting Information). However, no significant differences were observed in these dihedral angles for the OX–GG and CP–GG adducts. It is likely that the differences in G7 H8 chemical shifts between the OX–GG and CP–GG adducts result from the anisotropic effects of the (NH3)2Pt and 1,2-diaminocyclohexane-Pt moieties, since sequence context and buffer conditions were identical.
Temperature Dependence of Imino–Solvent Exchange
The temperature dependence of the imino proton resonances associated with the duplexes (Figure 5) was monitored by 1D 1H NMR in H2O buffer solution [100 mM NaCl and 5 mM Na2HPO4/NaH2PO4 (pH 7.0)] at temperatures ranging from 2 to 40 °C. For both CP–DNA and OX–DNA duplexes, the imino peaks of G6 and G7 totally disappear while other imino peaks survive at 40 °C, indicating that the binding of both CP and OX results in enhanced solvent accessibility of the G6 and G7 guanines. These data are consistent with previous reports for other Pt–GG intrastrand cross-links (24). Moreover, for both CP and OX, the imino peak for G6 shows a greater loss of intensity at 25 °C relative to the imino peak for G7, suggesting that the exchange rate for the G6 imino proton is faster than for the G7 imino proton. These observations are consistent with previous reports for other Pt–GG adducts (24, 25, 33) and suggest that both the CP and OX adducts may be more distorted and/or flexible on the 5′ side of the adduct than on the 3′ side.
FIGURE 5.
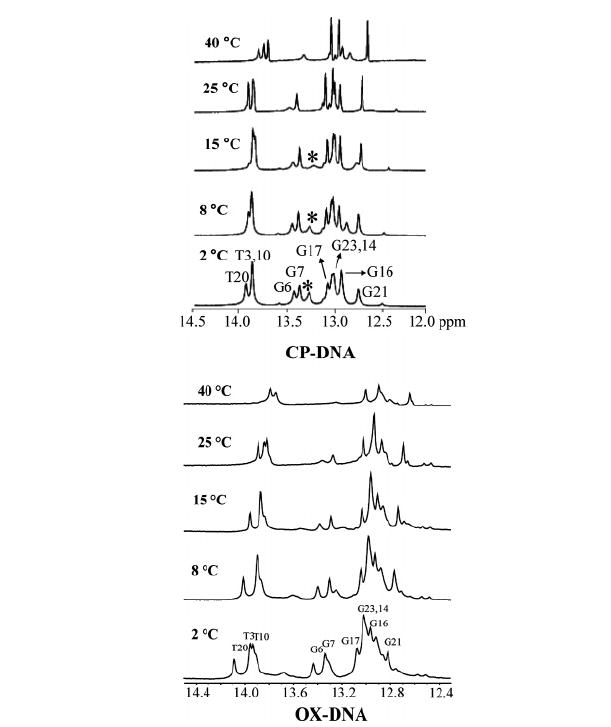
Expanded imino region from 1D 1H NMR spectra of the CP–DNA (top) and OX–DNA (bottom) (26) duplexes recorded in H2O buffer at various temperatures. The positions of the nucleotides in the 12-mer duplexes that give rise to the resonances are indicated. The asterisk in the 1D 1H NMR spectra of the CP–DNA adduct indicates the terminal guanine (G24). The experimental temperatures are shown at the left.
Assignment of31 P Resonances
Phosphorus chemical shifts are influenced strongly by backbone torsion angles. To investigate the effects of the CP–GG adduct on the phosphodiester backbone conformation, 2D 1H–31P HETCOR spectra were recorded for both the CP–DNA duplex and undamaged DNA (Figure S2A,B of the Supporting Information). Phosphorus signals were assigned from the H3′(i)–P(i) and P(i)–H4′(i+1) cross-peaks with the chemical shifts of the phosphorus atoms listed in Tables S1 and S2. All backbone phosphate resonances of the CP–GG 12-mer duplex were dispersed between −3.41 and −4.50 ppm over a range of 1.09 ppm. This is only slightly larger than the range (−3.80 to −4.40 ppm) observed for the phosphodiester backbone in undamaged B-DNA. Marzilli et al. (25) reported that for their CP–GG adduct the 31P signal for the phosphate group between the two G residues coordinated to platinum was significantly shifted downfield (to −3.08 ppm). However, we find almost no difference in the chemical shift of the 31P signal among the CP–DNA duplex (Table S1), the OX–DNA duplex (26), and undamaged DNA (Table S2). These data indicate that the formation of a CP– or OX–GG adduct in the AGGC sequence context does not significantly perturb the DNA backbone.
J Coupling Constants and Sugar Pucker Estimates
Figure 6 shows an expanded portion of the H1′–H2′ and H1′–H2″ region of the DQF–COSY spectra for the undamaged DNA and CP–DNA duplex at 25 °C in D2O with the A5, G6, and G7 cross-peaks indicated by the dotted lines. The relative intensity of the H1′–H2″ and H1′–H2′ DQF–COSY cross-peak reflects the relative size of the coupling between these sugar protons and can be used to provide information about sugar conformation (25, 49). Sugars with a C2′-endo (S) conformation give fairly weak H1′–H2″ and strong H1′–H2′ DQF–COSY cross-peaks, while sugars with near-C3′-endo (N) conformations give the opposite relative intensity pattern. To obtain sugar pucker for both the CP–DNA duplex and undamaged DNA, the procedure reported previously for the OX–DNA adduct was used (26). J coupling constants were obtained by simulating DQF-COSY cross-peaks using Chords version 2.0. Tables S5 and S6 of the Supporting Information list the J coupling constants obtained for all residues except terminal residues for the CP–DNA duplex and undamaged DNA, respectively. The sums of the H1′ J coupling constants, Σ1′, were used to calculate fractions of south (C2′-endo), fs, conformers for each sugar residue by assuming a two-state dynamic equilibrium and using the equation
FIGURE 6.
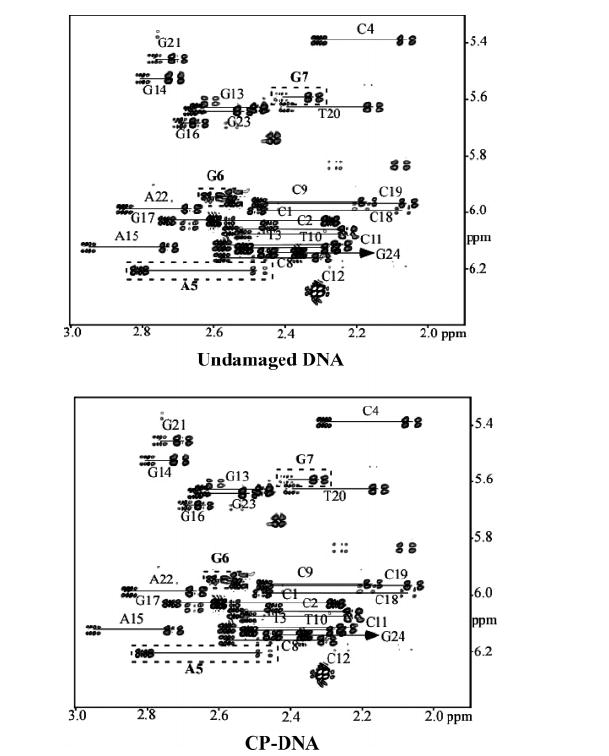
Expanded region of the DQF-COSY spectra showing the H1′–H2′ and H1′–H2″ cross-peaks of the undamaged DNA and CP–DNA duplex at 25 °C in H2O. Assigned peaks are labeled.
where Σ1′ = JH1′−H2′ + JH1′−H2″.
Table 1 summarizes the assigned sugar pucker (×c4s) for undamaged DNA, the CP–GG adduct, and the OX–GG adduct. For the undamaged DNA, all sugar puckers have the S conformation, including A5, G6, and G7. In contrast, for the OX–GG adduct, the G6 sugar pucker was found to be mostly N and the A5 sugar pucker primarily S, but with some N characteristics (26). For the CP–GG adduct, both the A5 and G6 sugar puckers are intermediate between S and N. The other residues have primarily the S sugar pucker, characteristic of B-DNA. These data are consistent with previous reports (24, 25) and indicate that compared to undamaged DNA, platinated DNA adducts exhibit distortion only in the sugar rings for the 5′ G residue and the base 5′ to the G.
Table 1.
Percent South Conformation (fs) Derived from Quantitative Simulation of DQF-COSY Data Obtained for Undamaged DNA and CP–DNA and OX–DNA Adducts in the AGGC Sequence Context
| residue | undamaged | CP–DNA | OX–DNA |
|---|---|---|---|
| C1 | 0.86 | 0.93 | |
| C2 | 0.93 | 0.89 | 0.91 |
| T3 | 0.97 | 0.92 | 0.98 |
| C4 | 0.88 | 0.86 | 0.93 |
| A5 | 0.85 | 0.62 | 0.72 |
| G6 | 0.83 | 0.63 | 0.32 |
| G7 | 0.86 | 0.95 | 0.88 |
| C8 | 0.66 | 0.71 | |
| C9 | 0.95 | 0.73 | 0.63 |
| T10 | 0.83 | 0.80 | 0.98 |
| C11 | 0.92 | 0.92 | |
| C12 | |||
| G13 | 0.81 | ||
| G14 | 0.83 | 0.81 | 0.93 |
| A15 | 0.90 | 0.83 | 0.88 |
| G16 | 0.95 | 0.86 | |
| G17 | 0.95 | 0.95 | 0.88 |
| C18 | 0.93 | 0.80 | 0.73 |
| C19 | 0.85 | 0.80 | 0.71 |
| T20 | 0.95 | 0.80 | 0.88 |
| G21 | 0.78 | 0.95 | 0.90 |
| A22 | 0.86 | 0.88 | 0.84 |
| G23 | |||
| G24 | 0.97 |
Structure Calculations
We obtained 551 and 920 experimental distance constraints for the CP–DNA duplex and undamaged DNA, respectively, from the 200 ms NOESY spectra in both H2O and D2O. These experimental distance constraints were then used as input for CNS to calculate the respective solution structures of the CP–DNA duplex and undamaged DNA. Additional constraints included base pair constraints between heavy atoms involved in hydrogen bonding and a planarity constraint to keep the base pairs in a plane (45). Force constants and charges for CP bound to the two G residues were obtained as described in Materials and Methods. The structural parameter statistics for the CP–DNA duplex and undamaged DNA are listed in Table 2. Of 20 calculated structures for each DNA, the 15 with the lowest energy were accepted as a family. The rmsd for the superimposition of the non-H atoms for all 15 final structures was 0.81 Å for the CP–DNA duplex and 1.21 for undamaged DNA, which indicates good convergence for both structures. The fact that there were no distance and dihedral angle restraint violations >0.5 Å and 5°, respectively, indicated that the final structures satisfied the imposed experimental restraints. The stereoviews of the average solution structures obtained for the CP–DNA duplex and undamaged DNA are shown in Figure 7.
Table 2.
Structural Statistics for CP–DNA and Undamaged DNA Structuresa
| structure-related information | CP–DNA | undamaged DNA |
|---|---|---|
| Number of Experimental Distance Constraints | ||
| intraresidue | 367 | 531 |
| inter-residue | 144 | 324 |
| cross-strand | 40 | 65 |
| total | 551 | 920 |
| Number of Empirical Constraints | ||
| hydrogen bonds | 72 | 72 |
| backbone dihedral angles | 168 | 168 |
| Structural Statistics | ||
| distance violation per structure (>0.5 Å) | 0 | 1 |
| dihedral angle violations per structure (>5°) | 0 | 0 |
| rmsd from ideal covalent geometry | ||
| bond lengths (Å) | 0.01 ± 0.00 | 0.01 ± 0.01 |
| bond angles (deg) | 0.66 ± 0.03 | 0.78 ± 0.03 |
| dihedral angles (deg) | 0.00 ± 0.00 | 0.00 ± 0.00 |
| Structure Quantity | ||
| rmsd from the mean structure | ||
| within the family (Å) | ||
| all atoms | 0.92 ± 0.30 | 1.37 ± 0.30 |
| non-H atoms | 0.81 ± 0.26 | 1.21 ± 0.30 |
Of 20 calculated structures, the 15 with the lowest energy are accepted as a family.
FIGURE 7.
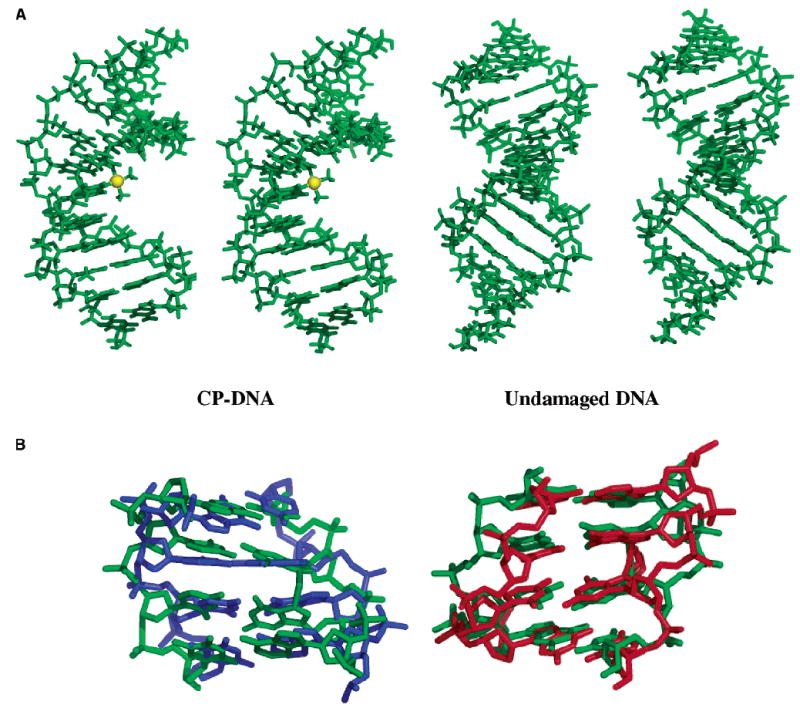
(A) Stereoviews of the average solution structures of the CP–DNA duplex (left) and undamaged DNA (right) in the AGGC sequence context. The platinum atom in the CP–DNA adduct is shown as a yellow sphere. (B) Comparison of the central 4 bp of the CP–DNA adduct (green) with those of the undamaged DNA (blue) and OX–DNA adduct (red) (26).
Accuracy of the Structures
To verify the accuracy of the structures, theoretical NOESY spectra were back-calculated from the average final structure and known chemical shifts using the Model module and the matrix-doubling capability (MSI) of Felix. The accuracy of the computed structure can be judged by comparing the back-calculated NOEs with the experimental 2D NOE data. The comparison of experimental and back-calculated NOEs for the entire molecule is shown in Figures S3 and S4 of the Supporting Information for undamaged DNA and the CP–DNA duplex, respectively. Experimental cross-peak volumes were compared to their theoretical counterparts to compute R-factors:
in which Rc is the crystallographic-equivalent R-factor, Rx is the sixth-root residual index, and Ie and It are the experimental and theoretical cross-peak volumes for the ith cross-peak, respectively. R-Factors were computed for the NOESY experiments (mixing time of 300 ms). For undamaged DNA, Rc and Rx values of the average structure were 0.21 and 0.04, respectively. For the CP–DNA duplex, Rc and Rx values of the average structure were 0.16 and 0.04, respectively. The obtained R-factors indicate that the structures are in good agreement with the measured NOEs.
Comparison with CP–DNA and OX–DNA Conformations
We are able to directly compare the solution structures of the CP–GG and OX–GG (26) adducts in the AGGC sequence context, since the DNA sequences are identical and the NMR data and structural calculations were performed in the same manner as our previous structural study of the OX–GG adduct (26). The platinum geometry is compared in Table S7 of the Supporting Information. However, as the location of the platinum atom is not determined by NMR constraints, the significance of these observations is unclear. An overlay of the central 4 bp of the CP–DNA duplex with undamaged DNA and with the OX–DNA duplex (26) is shown in Figure 7, and selected DNA helical parameters are compared in Table 3. The CP–GG and OX–GG adducts differ significantly in terms of twist at the A5-G6 base pair step, slide, twist, and roll at the G6-G7 base pair step, slide at the G7-C8 base pair step, buckle for the G6•C19 base pair, opening for the G7•C18 base pair, G6-G 7 dihedral angle, and overall bend. One or more of these conformational differences may be important for the differential recognition of CP–GG and OX–GG adducts by some mismatch repair proteins, DNA binding proteins (including certain transcription factors), and DNA polymerases. For example, the HMG domain of HMG-B1 has been shown to bind with higher efficiency to CP–GG adducts than to OX–GG adducts (14), and a crystal structure of the HMG–CP–DNA complex has been reported (31). The DNA helical parameters for the HMG–CP–DNA complex have been included in Table 3 for comparison. Many of the DNA helical parameters for the DNA–adduct complex differ from those of the solution structures of both the CP–GG and OX–GG adducts, which probably reflects the severe DNA distortion imposed by binding of the HMG protein. However, the twist at the A5-G6 and G6-G7 base pair steps and slide at the G7-C8 base pair steps for the HMG–CP–GG DNA complex are much closer to those of the CP–GG solution structure than the OX–GG solution structure. If any of these conformations are characteristic of the protein–DNA conformation in the initial recognition step, this may facilitate recognition of the CP–GG adduct by the HMG domain. The conformational difference at the G7-C8 base pair step might be particularly critical because both the affinity of the HMG domain for the CP–GG adduct (14, 50, 51) and the ability of the HMG domain to discriminate between CP–GG and OX–GG adducts (14) are highly dependent on the base to the 3′ side of the adduct. There are, of course, a number of important limitations inherent in this type of comparison. First, we are comparing a crystal structure of the HMG–CP–GG DNA complex with the solution structures of the CP–GG and OX–GG DNA adducts. Second, it is likely that the final, stable protein–DNA complex has a conformation different from that of the initial protein–DNA recognition complex. Third, the sequence context of the CP–GG adduct in the HMG–CP–GG complex differs from that in the CP–GG and OX–GG solution structures. Hence, experiments are underway to determine solution NMR structures of CP–GG and OX–GG adducts in complex with an HMG domain from one or more proteins that discriminate between these adducts, in the same AGGC sequence context, to better characterize conformational differences that are important for differential protein recognition of the adducts.
Table 3.
Comparison of Selected 3-DNA and MADBEND Parameters for Undamaged DNA, CP–GG DNA, OX–GG DNA (26), and DNA from the HMG–CP–GG DNA Complex (31)
| undamaged | CP–DNA | OX–DNA | HMG–CP–DNA | |
|---|---|---|---|---|
| A5•T20/G6•C19 | ||||
| shift (Å) | 0.44 ± 0.05 | −0.04 ± 0.10 | 0.17 ± 0.04 | 1.17 |
| slide (Å) | 0.09 ± 0.08 | −0.65 ± 0.10 | −0.89 ± 0.10 | 1.32 |
| twist (deg) | 27.81 ± 0.58 | 35.3 ± 1.40 | 22.1 ± 1.30 | 31.7 |
| G6•C19/G7•C18 | ||||
| shift (Å) | 0.24 ± 0.05 | 0.99 ± 0.08 | 0.84 ± 0.14 | −0.78 |
| slide (Å) | −1.55 ± 0.08 | −0.55 ± 0.10 | −1.36 ± 0.30 | −1.66 |
| twist (deg) | 30.29 ± 0.58 | 18.40 ± 1.40 | 25.20 ± 2.00 | 19.20 |
| roll (deg) | 1.12 ± 0.74 | 36.30 ± 3.20 | 28.30 ± 3.20 | 57.20 |
| G7•C18/C8•G17 | ||||
| shift (Å) | 0.24 ± 0.05 | −0.99 ± 0.17 | −1.24 ± 0.1 | −0.45 |
| slide (Å) | −1.55 ± 0.08 | −0.33 ± 0.13 | 0.93 ± 0.10 | 0.08 |
| G6•C19 | ||||
| buckle (deg) | 10.39 ± 0.31 | 4.20 ± 2.20 | 12.60 ± 2.40 | 29.00 |
| G7•C18 | ||||
| buckle (deg) | −0.90 ± 0.31 | −4.58 ± 1.67 | −3.18 ± 1.70 | −11.80 |
| opening (deg) | 0.83 ± 0.24 | −1.24 ± 0.93 | 5.73 ± 0.53 | −5.20 |
| G6G7 dihedral angle | 42.70 ± 3.10 | 35.60 ± 2.80 | ||
| overall bend (deg) (MADBEND) | 22 ± 10 | 31 ± 10 |
Supplementary Material
Tables listing 1H chemical shifts of the 12-mer CP–GG adduct (Table S1) and those of the undamaged B-DNA (Table S2), a table listing a comparison of cross-peak ratios of select internucleotide NOEs associated with undamaged DNA, the CP–DNA duplex, and the OX–DNA duplex in the AGGC sequence context (Table S3), a table listing canting measurements (degrees) for the G bases of CP– and OX–DNA adducts in the AGGC seuqnece context (Table S4), tables listing deoxyribose J coupling constants (hertz) and percent south conformation (fs) derived from quantitative simulation of DQF-COSY cross-peaks for undamaged DNA (Table S5) and the CP–DNA adduct (Table S6) in the AGGC sequence context, a table listing a summary of the platinum geometry for the CP–GG and OX–GG adducts (Table S7), expanded regions of a 1H–1H 2D NOESY spectrum of the undamaged DNA 12-mer duplex (Figure S1), the 1H–31P HETCOR NMR spectrum of the 12-mer CP–DNA duplex and undamaged DNA duplex (Figure S2), and comparisons of the experimental and back-calculated 2D NOESY spectra of the aromatic and sugar region of the undamaged DNA (Figure S3) and those of the CP–DNA adduct (Figure S4). This material is available free of charge via the Internet at http://pubs.acs.org.
Footnotes
This work was supported by NIH Grant CA8440 (to S.G.C.) and NIEHS Grant P30ES10126 (to J.A.S.).
Coordinates are available from the Protein Data Bank (PDB): 2NPW for the averaged structure of the CP–GG adduct and 2NQ0 for the family of the 15 best structures, 2NQ1 for the averaged structure of undamaged DNA in the AGGC sequence context, and 2NQ4 for the family of the 15 best structures.
References
- 1.Greene MH. Is Cisplatin a Human Carcinogen. J Natl Cancer Inst. 1992;84:306–312. doi: 10.1093/jnci/84.5.306. [DOI] [PubMed] [Google Scholar]
- 2.Travis LB, Curtis RE, Storm H, Hall P, Holowaty E, Van Leeuwen FE, Kohler BA, Pukkala E, Lynch CF, Andersson M, Bergfeldt K, Clarke EA, Wiklund T, Stoter G, Gospodarowicz M, Sturgeon J, Fraumeni JF, Jr, Boice JD., Jr Risk of second malignant neoplasms among long-term survivors of testicular cancer. J Natl Cancer Inst. 1997;89:1429–1439. doi: 10.1093/jnci/89.19.1429. [DOI] [PubMed] [Google Scholar]
- 3.Silva MJ, Costa P, Dias A, Valente M, Louro H, Boavida MG. Comparative analysis of the mutagenic activity of oxaliplatin and cisplatin in the Hprt gene of CHO cells. Environ Mol Mutagen. 2005;46:104–115. doi: 10.1002/em.20138. [DOI] [PubMed] [Google Scholar]
- 4.Bassett E, King NM, Bryant MF, Hector S, Pendyala L, Chaney SG, Cordeiro-Stone M. The role of DNA polymerase η in translesion synthesis past platinum-DNA adducts in human fibroblasts. Cancer Res. 2004;64:6469–6475. doi: 10.1158/0008-5472.CAN-04-1328. [DOI] [PubMed] [Google Scholar]
- 5.Page JD, Husain I, Sancar A, Chaney SG. Effect of the diaminocyclohexane carrier ligand on platinum adduct formation, repair, and lethality. Biochemistry. 1990;29:1016–1024. doi: 10.1021/bi00456a026. [DOI] [PubMed] [Google Scholar]
- 6.Jennerwein MM, Eastman A, Khokhar AR. Characterization of adducts produced in DNA by isomeric 1, 2-diaminocyclohexaneplatinum(II) complexes. Chem -Biol Interact. 1989;70:39–49. doi: 10.1016/0009-2797(89)90061-6. [DOI] [PubMed] [Google Scholar]
- 7.Woynarowski JM, Chapman WG, Napier C, Herzig MCS, Juniewicz P. Sequence- and region-specificity of oxaliplatin adducts in naked and cellular DNA. Mol Pharmacol. 1998;54:770–777. doi: 10.1124/mol.54.5.770. [DOI] [PubMed] [Google Scholar]
- 8.Fink D, Nebel S, Aebi S, Zheng H, Cenni B, Nehme A, Christen RD, Howell SB. The role of mismatch repair in platinum drug resistance. Cancer Res. 1996;56:4881–4886. [PubMed] [Google Scholar]
- 9.Zdraveski ZZ, Mello JA, Farinelli CK, Essigmann JM, Marinus MG. MutS preferentially recognizes cisplatin- over oxaliplatin-modified DNA. J Biol Chem. 2002;277:1255–1260. doi: 10.1074/jbc.M105382200. [DOI] [PubMed] [Google Scholar]
- 10.Aebi S, Kurdihaidar B, Gordon R, Cenni B, Zheng H, Fink D, Christen RD, Boland CR, Koi M, Fishel R, Howell SB. Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res. 1996;56:3087–3090. [PubMed] [Google Scholar]
- 11.Fink D, Zheng H, Nebel S, Norris PS, Aebi S, Lin T-P, Nehme A, Christen RD, Haas M, MacLeod CL, Howell SB. In vitro and in vivo resistance to cisplatin in cells that have lost DNA mismatch repair. Cancer Res. 1997;57:1841–1845. [PubMed] [Google Scholar]
- 12.Vaisman A, Varchenko M, Umar A, Kunkel TA, Risinger JI, Barrett JC, Hamilton TC, Chaney SG. The role of hMLH1, hMSH3, and hMSH6 defects in cisplatin and oxaliplatin resistance: Correlation with replicative bypass of platinum-DNA adducts. Cancer Res. 1998;58:3579–3585. [PubMed] [Google Scholar]
- 13.Brown R, Hirst GL, Gallagher WM, Mcilwrath AJ, Margison GP, Vanderzee AGJ, Anthoney DA. hMLH1 expression and cellular responses of ovarian tumour cells to treatment with cytotoxic anticancer agents. Oncogene. 1997;15:45–52. doi: 10.1038/sj.onc.1201167. [DOI] [PubMed] [Google Scholar]
- 14.Wei M, Cohen SM, Silverman AP, Lippard SJ. Effects of spectator ligands on the specific recognition of intrastrand platinum-DNA cross-links by high mobility group box and TATA-binding proteins. J Biol Chem. 2001;276:38774–38780. doi: 10.1074/jbc.M106374200. [DOI] [PubMed] [Google Scholar]
- 15.Zhai X, Beckmann H, Jantzen H-M, Essigmann JM. Cisplatin-DNA adducts inhibit ribosomal RNA synthesis by hijacking the transcription factor human upstream binding factor. Biochemistry. 1998;37:16307–16315. doi: 10.1021/bi981708h. [DOI] [PubMed] [Google Scholar]
- 16.Huang JC, Zamble DB, Reardon JT, Lippard SJ, Sancar A. HMG-domain proteins specifically inhibit the repair of the major DNA adduct of the anticancer drug cisplatin by human excision nuclease. Proc Natl Acad Sci U S A. 1994;91:10394–10398. doi: 10.1073/pnas.91.22.10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mcanulty MM, Lippard SJ. The HMG-domain protein Ixr1 blocks excision repair of cisplatin-DNA adducts in yeast. Mutat Res. 1996;362:75–86. doi: 10.1016/0921-8777(95)00037-2. [DOI] [PubMed] [Google Scholar]
- 18.Hoffmann JS, Locker D, Viliani G, Leng M. HMG1 protein inhibits the translesion synthesis of the major DNA cisplatin adduct by cell extracts. J Mol Biol. 1997;270:539–543. doi: 10.1006/jmbi.1997.1143. [DOI] [PubMed] [Google Scholar]
- 19.Vaisman A, Lim SE, Patrick SM, Copeland WC, Hinkle DC, Turchi JJ, Chaney SG. Effect of DNA Polymerases and High Mobility Group Protein 1 on the Carrier Ligand Specificity for Translesion Synthesis past Platinum-DNA Adducts. Biochemistry. 1999;38:11026–11039. doi: 10.1021/bi9909187. [DOI] [PubMed] [Google Scholar]
- 20.Treiber DK, Zhai XQ, Jantzen HM, Essigmann JM. Cisplatin-DNA adducts are molecular decoys for the ribosomal RNA transcription factor hUBF (human upstream binding factor) Proc Natl Acad Sci U S A. 1994;91:5672–5676. doi: 10.1073/pnas.91.12.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coin F, Frit P, Viollet B, Salles B, Egly JM. TATA binding protein discriminates between different lesions on DNA, resulting in a transcription decrease. Mol Cell Biol. 1998;18:3907–3914. doi: 10.1128/mcb.18.7.3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vaisman A, Masutani C, Hanaoka F, Chaney SG. Efficient translesion replication past oxaliplatin and cisplatin GpG adducts by human DNA polymerase η. Biochemistry. 2000;39:4575–4580. doi: 10.1021/bi000130k. [DOI] [PubMed] [Google Scholar]
- 23.Vaisman A, Chaney SG. The efficiency and fidelity of translesion synthesis past cisplatin and oxaliplatin GpG adducts by human DNA polymerase β. J Biol Chem. 2000;275:13017–13025. doi: 10.1074/jbc.275.17.13017. [DOI] [PubMed] [Google Scholar]
- 24.Gelasco A, Lippard SJ. NMR solution structure of a DNA dodecamer duplex containing a cis-diammineplatinum-(II) dGpG intrastrand cross-link, the major adduct of the anticancer drug cisplatin. Biochemistry. 1998;37:9230–9239. doi: 10.1021/bi973176v. [DOI] [PubMed] [Google Scholar]
- 25.Marzilli LG, Saad JS, Kuklenyik Z, Keating KA, Xu Y. Relationship of solution and protein-bound structures of DNA duplexes with the major intrastrand cross-link lesions formed on cisplatin binding to DNA. J Am Chem Soc. 2001;123:2764–2770. doi: 10.1021/ja0007915. [DOI] [PubMed] [Google Scholar]
- 26.Wu Y, Pradhan P, Havener J, Boysen G, Swenberg JA, Campbell SL, Chaney SG. NMR solution structure of an oxaliplatin 1, 2-d(GG) intrastrand cross-link in a DNA dodecamer duplex. J Mol Biol. 2004;341:1251–1269. doi: 10.1016/j.jmb.2004.06.066. [DOI] [PubMed] [Google Scholar]
- 27.Takahara PM, Frederick CA, Lippard SJ. Crystal structure of the anticancer drug cisplatin bound to duplex DNA. J Am Chem Soc. 1996;118:12309–12321. [Google Scholar]
- 28.Spingler B, Whittington DA, Lippard SJ. 2.4 Å crystal structure of an oxaliplatin 1,2-d(GpG) intrastrand cross-link in a DNA dodecamer duplex. Inorg Chem. 2001;40:5596–5602. doi: 10.1021/ic010790t. [DOI] [PubMed] [Google Scholar]
- 29.Lamers MH, Perrakis A, Enzlin JH, Winterwerp HHK, de Wind N, Sixma TK. The crystal structure of DNA mismatch repair protein MutS binding to a G center dot T mismatch. Nature. 2000;407:711–717. doi: 10.1038/35037523. [DOI] [PubMed] [Google Scholar]
- 30.Obmolova G, Ban C, Hsieh P, Yang W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature. 2000;407:703–710. doi: 10.1038/35037509. [DOI] [PubMed] [Google Scholar]
- 31.Ohndorf UM, Rould MA, He Q, Pabo CO, Lippard SJ. Basis for recognition of cisplatin-modified DNA by high-mobility-group proteins. Nature. 1999;399:708–712. doi: 10.1038/21460. [DOI] [PubMed] [Google Scholar]
- 32.Sawaya MR, Prasad R, Wilson SH, Kraut J, Pelletier H. Crystal structures of human DNA polymerase β complexed with gapped and nicked DNA: Evidence for an induced fit mechanism. Biochemistry. 1997;36:11205–11215. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- 33.Yang D, van Bloom SSGE, Reedijk J, van Bloom JH, Wang AHJ. Structure and isomerization of an intrastrand cisplatin-cross-linked octamer DNA duplex by NMR analysis. Biochemistry. 1995;34:12912–12920. doi: 10.1021/bi00039a054. [DOI] [PubMed] [Google Scholar]
- 34.Herman F, Kozelka J, Stoven V, Guittet E, Girault JP, Huynh-Dinh T, Igolen J, Lallemand JY, Chottard JC. A d(GpG)-platinated decanucleotide duplex is kinked. An extended NMR and molecular mechanics study. Eur J Biochem. 1990;194:119–133. doi: 10.1111/j.1432-1033.1990.tb19435.x. [DOI] [PubMed] [Google Scholar]
- 35.Bubley GJ, Ashburner BP, Teicher BA. Spectrum of cis-diamminedichloroplatinum(II)-induced mutations in a shuttle vector propagated in human cells. Mol Carcinog. 1991;4:397–406. doi: 10.1002/mc.2940040512. [DOI] [PubMed] [Google Scholar]
- 36.de Boer JG, Glickman BW. Sequence specificity of mutations induced by the antitumor drug cisplatin in the CHO aprt gene. Carcinogenesis. 1989;10:1363–1367. doi: 10.1093/carcin/10.8.1363. [DOI] [PubMed] [Google Scholar]
- 37.de Boer JG, Glickman BW. Mutations Recovered in the Chinese Hamster aprt Gene After Exposure to Carboplatin: A Comparison with Cisplatin. Carcinogenesis. 1992;13:15–17. doi: 10.1093/carcin/13.1.15. [DOI] [PubMed] [Google Scholar]
- 38.Pillaire MJ, Margot A, Villani G, Sarasin A, Defais M, Gentil A. Mutagenesis in monkey cells of a vector containing a single d(GPG) cis-diamminedichloroplatinum(II) adduct placed on codon 13 of the human H-ras proto-oncogene. Nucleic Acids Res. 1994;22:2519–2524. doi: 10.1093/nar/22.13.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bassett E, Vaisman A, Havener JM, Masutani C, Hanaoka F, Chaney SG. Efficiency of extension of mismatched primer termini across from cisplatin and oxaliplatin adducts by human DNA polymerases β and η in vitro. Biochemistry. 2003;42:14197–14206. doi: 10.1021/bi035359p. [DOI] [PubMed] [Google Scholar]
- 40.Gruschus JM, Ferretti JA. 15N-edited three-dimensional NOESY–HMQC with water flipback: Enhancement of weak labile 1H resonances of protein side chains contacting DNA. J Magn Reson. 1998;135:87–92. doi: 10.1006/jmre.1998.1532. [DOI] [PubMed] [Google Scholar]
- 41.Wuthrich K. NMR of proteins and nucleic acids. Wiley; New York: 1986. [Google Scholar]
- 42.Sklenar V, Miyashiro H, Zon G, Miles HT, Bax A. Assignment of the 31P and 1H resonances in oligonucleotides by two-dimensional NMR spectroscopy. FEBS Lett. 1986;208:94–98. doi: 10.1016/0014-5793(86)81539-3. [DOI] [PubMed] [Google Scholar]
- 43.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54(Part 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 44.Yao S, Plastaras JP, Marzilli LG. A molecular mechanics AMBER-type force field for modeling platinum complexes of guanine derivatives. Inorg Chem. 1994;33:6061–6077. [Google Scholar]
- 45.Kuszewski J, Schwieters C, Clore GM. Improving the accuracy of NMR structures of DNA by means of a database potential of mean force describing base-base positional interactions. J Am Chem Soc. 2001;123:3903–3918. doi: 10.1021/ja010033u. [DOI] [PubMed] [Google Scholar]
- 46.Eastman A. Reevaluation of the interactions of cis-diamminedichloro(ethylenediamine)platinum(II) with DNA. Biochemistry. 1986;25:3912–3915. doi: 10.1021/bi00361a026. [DOI] [PubMed] [Google Scholar]
- 47.Comess KM, Burstyn JN, Essigmann JM, Lippard SJ. Replication Inhibition and Translesion Synthesis on Templates Containing Site-Specifically Placed cis-Diamminedichloroplatinum(II) DNA Adducts. Biochemistry. 1992;31:3975–3990. doi: 10.1021/bi00131a013. [DOI] [PubMed] [Google Scholar]
- 48.Bhattacharyya D, Marzilli PA, Marzilli LG. Exploring the universality of unusual conformations of the 17-membered Pt(dG*pG*)) macrochelate ring. Dependence of conformer formation on a change in bidentate carrier ligand from an sp3 to an sp2 nitrogen donor. Inorg Chem. 2005;44:7644–7651. doi: 10.1021/ic050925l. [DOI] [PubMed] [Google Scholar]
- 49.Chen X, McDowell JA, Kierzek R, Krugh TR, Turner DH. Nuclear magnetic resonance spectroscopy and molecular modeling reveal that different hydrogen bonding patterns are possible for GU pairs: One hydrogen bond for each G•U pair in r(GGCGUGCC)2 and two for each G•U pair in r(GAGUGCUC)2. Biochemistry. 2000;39:8970–8982. [PubMed] [Google Scholar]
- 50.Cohen SM, Mikata Y, He Q, Lippard SJ. HMG-Domain protein recognition of cisplatin 1,2-intrastrand d(GpG) cross-links in purine-rich sequence contexts. Biochemistry. 2000;39:11771–11776. doi: 10.1021/bi001352l. [DOI] [PubMed] [Google Scholar]
- 51.Dunham SU, Lippard SJ. DNA sequence context and protein composition modulate HMG-domain protein recognition of ciplatin-modified DNA. Biochemistry. 1997;36:11428–11436. doi: 10.1021/bi9709452. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables listing 1H chemical shifts of the 12-mer CP–GG adduct (Table S1) and those of the undamaged B-DNA (Table S2), a table listing a comparison of cross-peak ratios of select internucleotide NOEs associated with undamaged DNA, the CP–DNA duplex, and the OX–DNA duplex in the AGGC sequence context (Table S3), a table listing canting measurements (degrees) for the G bases of CP– and OX–DNA adducts in the AGGC seuqnece context (Table S4), tables listing deoxyribose J coupling constants (hertz) and percent south conformation (fs) derived from quantitative simulation of DQF-COSY cross-peaks for undamaged DNA (Table S5) and the CP–DNA adduct (Table S6) in the AGGC sequence context, a table listing a summary of the platinum geometry for the CP–GG and OX–GG adducts (Table S7), expanded regions of a 1H–1H 2D NOESY spectrum of the undamaged DNA 12-mer duplex (Figure S1), the 1H–31P HETCOR NMR spectrum of the 12-mer CP–DNA duplex and undamaged DNA duplex (Figure S2), and comparisons of the experimental and back-calculated 2D NOESY spectra of the aromatic and sugar region of the undamaged DNA (Figure S3) and those of the CP–DNA adduct (Figure S4). This material is available free of charge via the Internet at http://pubs.acs.org.