

Abstract
Caspase-activated DNase (CAD) cleaves chromosomal DNA during apoptosis. Here, we report isolation of two classes of human CAD cDNAs from a human KT-3 leukemic cell cDNA library. One class of cDNA encoded a protein comprising 338 amino acids, which showed a marked similarity to its murine counterpart. In vitro transcription and translation of this cDNA resulted in a functional CAD protein when the protein was synthesized in the presence of its inhibitor (inhibitor of CAD). The other cDNA class contained many deletions, insertions, and point mutations in the sequence corresponding to the coding region, suggesting that it is derived from a pseudogene. The functional CAD gene was localized to human chromosome 1p36.3 by fluorescent in situ hybridization. The CAD mRNA was expressed in a limited number of human tissues, including pancreas, spleen, prostate, and ovary. The expression of the CAD mRNA in human cell lines correlated with their ability to show DNA fragmentation during apoptosis. Overexpression of CAD potentiated DNA fragmentation by apoptotic stimuli in these cell lines, indicating that CAD is responsible for the apoptotic DNA degradation.
Apoptosis is a cell death process that removes toxic and/or useless cells during mammalian development (1). Apoptosis can be induced by a variety of stimuli, including the death factors of the tumor necrosis factor family, γ or UV irradiation, anticancer drugs, and survival factor deprivation (2, 3). The apoptotic process is accompanied by shrinkage and fragmentation of the cells and nuclei and degradation of the chromosomal DNA into nucleosomal units (4). Recently, the molecular mechanism underlying the signal transduction of the death factor/receptor-induced apoptosis has been elucidated (2, 5). Binding of the Fas ligand, a member of the tumor necrosis factor family, to its receptor Fas (also called APO-1 or CD95), causes trimerization of the receptor. The trimerized receptor recruits caspase 8 through the adaptor molecule FADD/MORT1 and activates it at the plasma membrane by forming the death-inducing signaling complex (6). The activated caspase 8 sequentially activates other members of the caspase family, such as caspases 3 and 6 which act downstream in the caspase cascade (7). The activated caspases then cleave various cellular substrates such as poly(ADP ribose) polymerase, lamin, actin, rho–GDI, and fodrin to cause the morphological cellular changes observed during apoptosis (8, 9). The involvement of caspases in other apoptotic systems such as the apoptosis induced by anticancer drugs (10) and factor deprivation (11) has also been demonstrated.
One of the hallmarks of apoptosis is the degradation of chromosomal DNA into nucleosomal units (12). We have recently found a DNase, designated as caspase-activated DNase (CAD), that can be activated by caspase 3. Purification and molecular cloning of CAD from mouse lymphoid cells indicated that murine CAD is a protein comprised of 344 amino acids (13). It exists as a complex with its inhibitor, inhibitor of CAD (ICAD) in the growing cells. Caspase, which is activated during the apoptotic process, cleaves ICAD, and the CAD released from ICAD seems to enter the nuclei to cleave the chromosomal DNA (13, 14).
Recently, cancer therapy involving the induction of apoptosis in tumor cells has been proposed (15, 16). Although some tumor cells are sensitive to proapoptotic stimuli such as death factors and anticancer drugs and undergo DNA degradation, others are resistant to these reagents and do not show DNA fragmentation (17, 18). To clarify the mechanisms behind these phenomena, it is important to examine the structure and expression of the molecules that are involved in the apoptotic process in these tumor cells.
In this report, we describe the molecular cloning and characterization of human CAD cDNA. The human CAD cDNA was found to be expressed in cells that easily undergo to DNA fragmentation in response to apoptotic stimuli, but not in those that do not show apoptotic DNA fragmentation. Introduction of the CAD expression vector resulted in enhanced DNA fragmentation. These results indicated that the expression of CAD, at least in part, is responsible for DNA degradation during apoptosis.
MATERIALS AND METHODS
Materials, Cell Lines, and Transformation.
Mouse liver nuclei were prepared as described (19). The recombinant mouse ICAD-L [glutathione S-transferase–-mouse ICAD-L) was produced as a fusion protein with Escherichia coli glutathione S-transferase, and purified by glutathione affinity column as described (14). The His-tagged human recombinant caspase 3 was produced in E. coli and purified as described (20).
Human squamous carcinoma CHU-2 (21), cervix adenocarcinoma HeLa S3 (ATCC CCL 2.2), hepato adenocarcinoma SK-HEP-1 (ATCC HTB 52), bladder carcinoma 5637 (ATCC HTB-9), and glioblastoma U-87 MG (ATCC HTB-14) were cultured in DMEM containing 5% fetal calf serum. Human acute T cell leukemia Jurkat (ATCC TIB-152) and histiocytic lymphoma U937 (ATCC CRL-1593) were maintained in RPMI 1640 medium supplemented with 10% fetal calf serum. HeLa cells were cotransfected by electroporation with mouse CAD expression plasmid and pBSpocΔP carrying the puromycin-resistant gene. From among puromycin-resistant clones, the clones expressing mouse CAD were identified by Western blotting using a rabbit anti-mouse CAD antibody (M.E. and S.N., manuscript in preparation).
Isolation of Human CAD cDNA Clones and Nucleotide Sequence Analysis.
A human CHU-2 cDNA library in a λgt10 vector (21) and a KT-3 cDNA library in a pCEV4 vector (22) have been described previously. The CHU-2 cDNA library was screened by plaque hybridization with a 1.0-kb fragment carrying the full-length coding sequence of mouse CAD cDNA (13). Hybridization was carried out as described (23), except that the hybridization temperature was lowered to 35°C and the filter was washed at 37°C in 15 mM NaCl/1.5 mM sodium citrate (pH 7.2)/1 mM NaH2PO4/0.1 mM EDTA/0.1% SDS. Three positive clones were obtained from 4 × 105 screened, of which one carried an insert of about 800 bp. A DNA fragment from this clone that contained about 200 bp from its most 5′ end was used to screen the KT-3 cDNA library under stringent hybridization conditions. Screening of 3.0 × 105 clones by colony hybridization yielded five positive clones. These clones were characterized by restriction enzyme analysis and DNA sequencing. The DNA sequence was determined using a DNA sequencer (Alfred; Pharmacia, Uppsala, Sweden) and a thermo sequenase fluorescent-labeled primer cycle sequencing kit from Amersham.
In Vitro Transcription and Translation.
To express human CAD in a cell-free system, a 1.0-kb fragment carrying the full-length coding sequence of human CAD was prepared by PCR, placed under the T7 promoter of the pBluescript SK+ vector (Invitrogen), and designated pBS-hCAD. Coupled transcription and translation of CAD was carried out using a TNT in vitro transcription/translation kit (Promega) according to the manufacturer’s instructions. In brief, pBS-hCAD DNA (1 μg) was incubated at 30°C for 2 h with 40 μl of the TNT reagents and 40 μCi of [35S]methionine (Amersham). An aliquot of 8 μl was heated at 90°C for 5 min in Laemmli’s sample buffer, fractionated by electrophoresis on a 10–20% gradient polyacrylamide gel, and exposed to an X-Omat x-ray film (Kodak) with an intensifying screen.
CAD Assay and DNA Fragmentation in Cells.
The CAD activity was determined using mouse liver nuclei or plasmid DNA as described (13). In brief, nuclei (2 × 105) were incubated at 30°C for 2 h with samples in 20 μl of the reaction mixture consisting of 10 mM Hepes–KOH (pH 7.2), 50 mM NaCl, 20% (vol/vol) glycerol, 2 mM MgCl2, 5 mM EGTA, 5 mM DTT, and 1 mg/ml BSA, and 90 ng of caspase 3. After the reaction, DNA was extracted from the nuclei and analyzed by electrophoresis on a 1.5% agarose gel. For the DNase assay, the nuclei in the above reaction mixture were replaced with 1 μg of plasmid DNA.
To determine the amount of DNA fragmentation in cells, cells were incubated for various periods of time at 37°C with 10 μM staurosporine or 1 μg/ml anti-human Fas antibody (CH-11, MBL) in the presence of 0.05 μg/ml actinomycin D. All cells were collected (attached or detached from the plate) and then incubated at 37°C overnight with 0.2 mg/ml of proteinase K in 500 μl of 100 mM Tris⋅HCl (pH 8.5), 5 mM EDTA, 200 mM NaCl, and 0.2% SDS. DNA was precipitated with isopropanol, treated with 0.1 mg/ml RNase A, and analyzed on an agarose gel.
Northern Blot Hybridization and In Situ Hybridization to Human Chromosomes.
For Northern blot hybridization, total RNA was prepared from various cell lines using the acid/phenol extraction method according to Chomczynsky and Sacci (24). RNA was denatured at 65°C for 5 min in 50% formamide and 2.2 M formaldehyde, fractionated by electrophoresis through a 1.5% agarose gel containing 6.6% formaldehyde, and transferred to a nylon membrane (Amersham). Nylon membranes blotted with poly(A) RNA from various human tissues were purchased from CLONTECH. The probe was a 1.0-kb DNA fragment carrying the coding sequence of human CAD cDNA and was labeled with 32P using a random primer labeling kit (Boehringer Mannheim). A 32P-labeled 1.8-kb BamHI fragment of human EF-1α cDNA (25) was used as a control. Hybridization was carried out as described (23), under high stringency.
To assign the human CAD gene location on human chromosomes, a cosmid clone carrying the human CAD chromosomal gene was isolated and used as a probe. Fluorescent in situ hybridization was carried out as described (26). The cosmid DNA was labeled with biotin–16-dUTP by nick-translation and hybridized to the denatured chromosomes at a final concentration of 25 ng/ml in 50% formamide, 10% dextran sulfate, 2× SSC, 0.2 mg/ml Cot-1 DNA (GIBCO/BRL), 2 mg/ml salmon sperm DNA, and 2 mg/ml E. coli tRNA. The hybridized signals were detected with fluorescein isothiocyanate–avidin (Boehringer Mannheim). Metaphase cells were counterstained with 4′,6-diamidino-2-phenylindole, and the slides were examined through a Nikon epigluorescent microscope equipped with a CCD camera (Photometrics, Tucson, AZ). Images were captured with Quips (Vysis) software and processed with Adobe Photoshop 3.0 software.
RESULTS
Isolation and Characterization of Human CAD cDNAs.
To isolate cDNA clones containing the sequence for human CAD, a CHU-2 cDNA library was screened as described in Materials and Methods and three positive clones were identified. These clones carried a sequence highly homologous to that of mouse CAD, but none of them contained the full-length coding sequence. The KT-3 cDNA library was therefore screened with the 5′ portion of the longest cDNA from the CHU-2 cDNA library. The screening of 3 × 105 clones yielded five positive ones, which could be divided into two classes. Most of them (four clones) belonged to the first class, represented by pKT-hCAD, which carried a cDNA of 4.2 kb, and contained a long reading frame that encoded a protein consisting of 338 amino acids with a calculated Mr of 39,109 (Fig. 1A). As found in mouse CAD, human CAD was rich in cysteine residues (11 residues) and had a basic character (pI = 9.3). Human CAD also contained a stretch of basic amino acids at the C terminus, which may act as a nuclear localization signal. The overall identity of human CAD to mouse CAD was 75.9% at the amino acid sequence level. Of particular note, the positions of 10 cysteine residues were conserved between human and mouse CAD proteins (Fig. 1B).
Figure 1.

Human CAD cDNA. (A) Nucleotide sequences of human CAD cDNA and the CAD pseudogene. The nucleotide sequence of human CAD cDNA (upper line) is shown with that of the pseudogene (lower line). The dashed line indicates identity between the two sequences. The dots indicate positions where the corresponding nucleotides are deleted. The amino acid sequence of human CAD was deduced from the cDNA nucleotide sequence and is numbered starting from Met-1. (B) Alignment of the amino acid sequences of human CAD with mouse CAD. The identical amino acids are shown in bold. The conserved cysteine residues and the putative nuclear localization signal are indicated by double and single underlines, respectively. The amino acid sequence of murine CAD is from DNA Data Base in Japan/GenBank/European Molecular Biology Laboratory (accession no. AB009377).
The fifth clone carried a cDNA of 4.2 kb and contained a sequence that is highly homologous (86.26%) to that in pKT-hCAD cDNA (Fig. 1A). However, the sequence had many point mutations, deletions, and insertions in the coding region, suggesting that this cDNA is derived from a pseudogene of human CAD.
Expression of the Functional Human CAD.
To confirm that the cDNA borne by pKT-hCAD coded for the functional CAD protein, the protein was expressed in an in vitro transcription and translation-coupled system. Specifically, human CAD mRNA was synthesized with T7 polymerase using pBS-hCAD as the template and translated in reticulocyte lysates. Previously, we noted that functional mouse CAD can be produced only in the presence of ICAD (13). Therefore, the synthesis of human CAD in reticulocyte lysates was carried out in the presence or absence of the recombinant ICAD. In both cases, human CAD was synthesized as a protein with the expected Mr of 40,000, which is slightly larger than that of mouse CAD (Fig. 2A). The human CAD synthesized in the absence of ICAD showed little CAD activity, whereas the CAD protein synthesized in the presence of ICAD efficiently cleaved chromosomal DNA in nuclei and digested plasmid DNA, when the products were treated with caspase 3 (Fig. 2B). These results indicated that ICAD is required to generate the functional human CAD, as found with mouse CAD.
Figure 2.
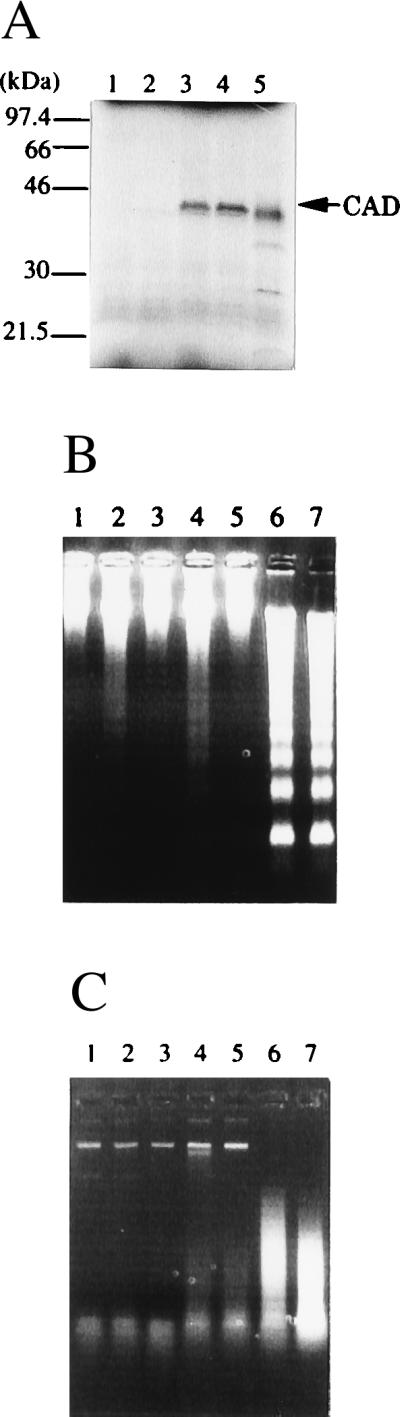
Expression of human CAD in a cell-free system. (A) Synthesis of human CAD protein in reticulocyte lysates. The pBS-hCAD carrying human CAD cDNA under the T7 promoter (lanes 3 and 4), pcDNA-CAD carrying mouse CAD under the T7 promoter (lane 5), or empty vector (lanes 1 and 2) was subjected to in vitro transcription and translation in the presence of [35S]Met in a final volume of 50 μl. The reaction mixtures in lanes 2, 4, and 5 included 160 ng of glutathione S-transferase–mouse ICAD-L. After the reaction, 8-μl aliquots were analyzed by electrophoresis on a 10–20% gradient polyacrylamide gel, followed by autoradiography. Molecular masses of the standard proteins are shown in kDa. (B and C) The activity of the human CAD synthesized in reticulocyte lysates. Human CAD (lanes 3–6) or mouse CAD (lane 7) was synthesized in the in vitro transcription and translation system in a final volume of 50 μl in the absence (lanes 3 and 4) or presence (lanes 5–7) of 160 ng of glutathione S-transferase–mouse ICAD-L. Using 3-μl aliquots, the CAD activity with nuclei (B) or plasmid DNA (C) was determined in the absence (lanes 3 and 5) or presence (lanes 4, 6, and 7) of 90 ng of caspase 3. The empty vector (pBluescript) (lanes 1 and 2) was subjected to a similar procedure in the absence (lane 1) or presence (lane 2) of caspase 3.
Expression of CAD in Human Cell Lines and Tissues.
Total RNA from various human cell lines was analyzed by Northern blot hybridization. As shown in Fig. 3A, the RNAs from human Jurkat and U937 cells showed a strong band of 3.4 kb hybridizing with the human CAD cDNA probe. Weak expression of CAD mRNA was observed in SK-Hep-1, 5637, CHU-2, and HeLa cells. However, no hybridizing band was observed in RNA from the U-87 MG cell line. Northern blot hybridization analysis of mRNAs from human tissues also indicated limited expression of the CAD mRNA in some tissues. As shown in Fig. 3B, the heart, placenta, kidney, pancreas, spleen, prostate, ovary, colon, and peripheral blood leukocytes expressed a significant level of CAD mRNA. In contrast, almost no expression of the CAD mRNA was observed in the brain, lung, liver, skeletal muscle, thymus, testis, and small intestine.
Figure 3.

Expression of CAD mRNA in human cell lines and tissues. (A) Expression of CAD mRNA in human cell lines. Total RNA was prepared from the indicated human cell lines, and 10 μg of RNA each was subjected to electrophoresis and analyzed by Northern blot hybridization. For probes, human CAD cDNA (Upper) or human EF-1α cDNA (Lower) was labeled with 32P. The positions of the 28S and 18S rRNAs are indicated on the right. (B) Expression of CAD mRNA in human tissues. Human RNA Master Blots (CLONTECH) were analyzed by Northern blot hybridization using 32P-labeled human CAD cDNA as the probe. The positions of the size markers are indicated on the right.
To examine the correlation of CAD expression with DNA degradation during apoptosis, Jurkat, CHU-2, and HeLa cells were treated for various periods of time with 10 μM staurosporine, and DNA fragmentation was analyzed. As shown in Fig. 4A, staurosporine caused rapid DNA fragmentation in Jurkat cells, which express a high level of CAD mRNA. Most of the chromosomal DNA in Jurkat cells was fragmented within 2 h of staurosporine treatment. On the other hand, CHU-2 and HeLa cells, which expressed only a marginal level of CAD mRNA, showed little DNA fragmentation after 4 h.
Figure 4.
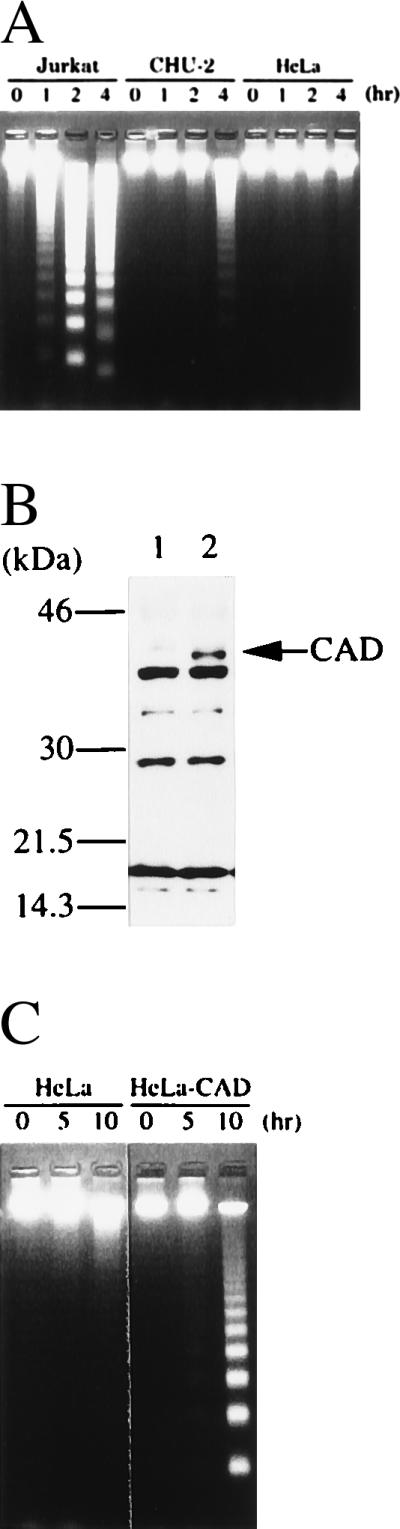
Enhancement of DNA fragmentation during apoptosis by the overexpression of CAD. (A) DNA fragmentation in human cell lines during apoptosis. Five × 105 human Jurkat cells or 2 × 105 CHU-2 or HeLa cells were treated at 37°C for the indicated periods of time with 10 μM staurosporine, and the chromosomal DNA was analyzed by electrophoresis on an agarose gel. (B) Overexpression of CAD in HeLa cells transformed with the CAD expression vector. The total cell lysates were prepared from the parental HeLa cells (lane 1) and the CAD-transformed clones (lane 2) and analyzed by Western blotting using rabbit antibody against mouse CAD. (C) Enhanced DNA fragmentation in HeLa cells overexpressing CAD. Parental HeLa cells or the transformants overexpressing CAD were incubated at 37°C with 1.0 μg/ml of the agonistic anti-human Fas antibody (CH-11) in the presence of 0.05 μg/ml actinomycin D. At the indicated time, chromosomal DNA was prepared. Analysis was by electrophoresis on an agarose gel.
To explore further the role of CAD in apoptotic DNA degradation, the expression plasmid for mouse CAD was introduced into HeLa cells. One of the stable transformant clones abundantly expressed CAD, as revealed by Western blotting analysis with rabbit antibody against mouse CAD (Fig. 4B). The parental HeLa cells and the transformant clones overexpressing mouse CAD were then treated with an agonistic anti-human Fas antibody. As shown in Fig. 4C, no DNA fragmentation was observed in the parental HeLa cells within 15 h after treatment with the anti-Fas antibody. On the other hand, the transformant overexpressing CAD showed DNA fragmentation within 10 h after treatment with the anti-Fas antibody. These results indicated that CAD expression levels can control the occurrence of DNA fragmentation during apoptosis.
Chromosomal Localization of Human CAD Gene.
The chromosomal region of the human CAD gene was determined by fluorescent in situ hybridization as described in Materials and Methods. A total of 20 metaphase cells were examined and a specific signal appearing as two symmetrical spots on both chromatids was detected on the distal part of the short arm of chromosomes 1 in 18 cells. No specific signal was observed on any other regions of the chromosomes. By comparison with the 4′,6-diamidino-2-phenylindole banding pattern, the CAD gene was assigned to the band 1p36.3 (Fig. 5).
Figure 5.

Assignment of the human CAD gene at chromosome 1p36.3. Arrows indicate the twin-spot signals specific for the CAD cosmid. The colors of fluorescein isothiocyanate signals and 4′,6-diamidino-2-phenylindole bands of chromosomes were electronically changed as the red and the black-and-white, respectively.
DISCUSSION
In this report, we describe the isolation of cDNAs for intact and pseudo genes for human CAD. Southern blot hybridization analysis of human chromosomal DNA showed different specific bands for the intact and pseudo genes (N.M. and S.N., unpublished results). These results suggest that human chromosomes carry one functional gene and one pseudogene for CAD per haploid genome. The functional CAD gene was assigned to human chromosome 1p36.3, which is syntenic to the region of mouse chromosome 4 where the mouse CAD gene is located (M.E., S.N., N. Jenkins, and N. Copeland, unpublished results). This distal region of human chromosome 1p is often deleted and translocated in human tumors, particularly in neuroblastoma, suggesting the existence of a tumor suppressor gene in this locus (27–29). Because CAD is a DNase that is involved in apoptotic DNA fragmentation, its loss-of-function mutation may cause cellular transformation because the damaged DNA cannot be destroyed. Thus, it would be interesting to examine possible mutations of the CAD gene in human neuroblastoma and other tumors.
A search by BLAST and FASTA of all sequences in the databases of National Biomedical Research Foundation and SwissProt for sequences homologous to human CAD revealed that an N-terminal domain of human CAD of about 80 amino acids is homologous not only with that of murine CAD, but also with the corresponding region of the human DNA fragmentation factor 45-kDa subunit (DFF-45) (30) and murine ICAD (13) (Fig. 6). These molecules are identical at 24 of 83 positions, with a similarity of 43.4% if conservative amino acid substitutions are allowed. In contrast, there was no similarity between CAD and ICAD in the rest of the region. ICAD/DFF-45 inhibits the DNase activity of CAD by binding to it (14). It may also work as a chaperon to help the correct folding of the CAD protein (13). Recently, several proteins that mediate the apoptotic signal transduction have been identified. These include FADD/MORT1, caspases, and Bcl-2 family members (2, 8, 31). These mediators carry domains such as the death domain, death-effector domain, and Bcl-2 homology domain. These domains have the ability to interact homophilically. In this regard, it is possible that CAD and ICAD also interact homophilically through their homologous N-terminal regions. Several other molecules, including the mouse FSP-27 protein whose function is unknown (32), carry a domain homologous to the N-terminal domain of CAD/ICAD. These proteins may also interact with ICAD and/or CAD.
Figure 6.

An N-terminal domain homologous between CAD and ICAD. The N-terminal amino acid sequences of human CAD (hCAD), mouse CAD (mCAD), human DFF-45/ICAD (hDFF-45), and mouse ICAD (mICAD) are aligned to give maximum homology by introducing several gaps (−). The residues identical in at least three of four members are shown in bold, and residues regarded as favored substitutions are underlined. Favored amino acid substitutions are defined as pairs of residues belonging to one of the following groups: S, T, P, A, and G; N, D, E, and Q; H, R, and K; M, I, L, and V; F, Y, and W.
The expression of CAD was found in cell lines that easily undergo DNA fragmentation by an apoptotic stimulus. The exogenous overexpression of CAD in cells that normally express only marginal levels of CAD mRNA enhanced the DNA fragmentation, indicating that the CAD expression level can determine the ability of cells to undergo DNA fragmentation. In contrast, overexpression of ICAD inhibits the CAD-mediated DNA fragmentation (14). ICAD should be cleaved by caspase to activate CAD (14), suggesting that the balance of CAD and ICAD and the ability of the apoptotic stimuli to activate caspase in cells would also control the apoptotic DNA fragmentation. In this regard, it will be interesting to examine the expression of CAD and ICAD in tumors that do not show DNA fragmentation in response to apoptotic stimuli. CAD mRNA was detected in only a few human tissues. This is in contrast to the expression of mouse CAD mRNA which could be found in most tissues except for the heart, liver, and kidney (M.E. and S.N., unpublished observation). Whether the CAD-negative tissues do not undergo DNA fragmentation during apoptosis, or whether they have a CAD-independent mechanism for DNA fragmentation, remains to be studied.
Acknowledgments
We thank Dr. R. Fukunaga for valuable comments and discussion and Ms. S. Kumagai for secretarial assistance. This work was supported in part by Grants-in-Aid from the Ministry of Education, Science, Sports, and Culture in Japan.
ABBREVIATION
- CAD
caspase-activated DNase
- ICAD
inhibitor of CAD
Note Added in Proof
After submission of this manuscript, we learned that Halenbeck et al. (33) also isolated a human cDNA for CAD (called CPAN).
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the DNA Data Base in Japan/European Molecular Biology Laboratory/GenBank databases (accession no. AB013918, human CAD cDNA).
References
- 1.Jacobson M D, Weil M, Raff M C. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- 2.Nagata S. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 3.Raff M C, Barres B A, Burne J F, Coles H S, Ishizaki Y, Jacobson M D. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- 4.Wyllie A H, Kerr J F R, Currie A R. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 5.Baker S, Reddy E. Oncogene. 1996;12:1–9. [PubMed] [Google Scholar]
- 6.Medema J P, Scaffidi C, Kischkel F C, Shevchenko A, Mann M, Krammer P H, Peter M E. EMBO J. 1997;16:2794–2804. doi: 10.1093/emboj/16.10.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirata H, Takahashi A, Kobayashi S, Yonehara S, Sawai H, Okazaki T, Yamamoto K, Sasada M. J Exp Med. 1998;187:587–600. doi: 10.1084/jem.187.4.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicholson D, Thornberry N. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- 9.Cohen G. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mashima T, Naito M, Kataoka S, Kawai H, Tsuruo T. Biochem Biophys Res Commun. 1995;209:907–915. doi: 10.1006/bbrc.1995.1584. [DOI] [PubMed] [Google Scholar]
- 11.Iversen P O, Hercus T R, Zacharakis B, Woodcock J M, Stomski F C, Kumar S, Nelson B H, Miyajima A, Lopez A F. J Biol Chem. 1997;272:9877–9883. doi: 10.1074/jbc.272.15.9877. [DOI] [PubMed] [Google Scholar]
- 12.Wyllie A H. Nature (London) 1980;284:555–556. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]
- 13.Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. Nature (London) 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 14.Sakahira H, Enari M, Nagata S. Nature (London) 1998;391:96–99. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- 15.Hickman J A. Cancer Metastasis Rev. 1992;11:121–139. doi: 10.1007/BF00048059. [DOI] [PubMed] [Google Scholar]
- 16.Hickman J A. Eur J Cancer. 1996;32A:921–926. doi: 10.1016/0959-8049(96)00080-9. [DOI] [PubMed] [Google Scholar]
- 17.Friesen C, Fulda S, Debatin K M. Leukemia. 1997;11:1833–1841. doi: 10.1038/sj.leu.2400827. [DOI] [PubMed] [Google Scholar]
- 18.Chapman R S, Chresta C M, Herberg A A, Beere H M, Heer S, Whetton A D, Hickman J A, Dive C. Cytometry. 1995;20:245–256. doi: 10.1002/cyto.990200308. [DOI] [PubMed] [Google Scholar]
- 19.Newmeyer D D, Wilson K L. Methods Cell Biol. 1991;36:607–634. doi: 10.1016/s0091-679x(08)60299-x. [DOI] [PubMed] [Google Scholar]
- 20.Kamens J, Paskind M, Hugunin M, Talanian R, Allen H, Banach D, Bump N, Hackett M, Johnston C, Li P, Mankovich J, et al. J Biol Chem. 1995;270:15250–15256. doi: 10.1074/jbc.270.25.15250. [DOI] [PubMed] [Google Scholar]
- 21.Nagata S, Tsuchiya M, Asano S, Kaziro Y, Yamazaki T, Yamamoto O, Hirata Y, Kubota N, Oheda M, Nomura H, Ono M. Nature (London) 1986;319:415–418. doi: 10.1038/319415a0. [DOI] [PubMed] [Google Scholar]
- 22.Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y, Nagata S. Cell. 1991;66:233–243. doi: 10.1016/0092-8674(91)90614-5. [DOI] [PubMed] [Google Scholar]
- 23.Sambrook J, Fretsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 24.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 25.Uetsuki T, Naito A, Nagata S, Kaziro Y. J Biol Chem. 1989;264:5791–5798. [PubMed] [Google Scholar]
- 26.Inazawa J, Saito H, Ariyama T, Abe T, Nakamura Y. Genomics. 1993;17:153–162. doi: 10.1006/geno.1993.1297. [DOI] [PubMed] [Google Scholar]
- 27.Schleiermacher G, Peter M, Michon J, Hugot J P, Vielh P, Zucker J M, Magdelénat H, Thomas G, Delattre O. Genes Chromosomes Cancer. 1994;10:275–281. doi: 10.1002/gcc.2870100409. [DOI] [PubMed] [Google Scholar]
- 28.Hunt J D, Tereba A. Genes Chromosomes Cancer. 1990;2:137–146. doi: 10.1002/gcc.2870020210. [DOI] [PubMed] [Google Scholar]
- 29.Barker P E, Savelyeva L, Schwab M. Oncogene. 1993;8:3353–3358. [PubMed] [Google Scholar]
- 30.Liu X, Zou H, Slaughter C, Wang X. Cell. 1997;89:175–184. doi: 10.1016/s0092-8674(00)80197-x. [DOI] [PubMed] [Google Scholar]
- 31.Reed J. Nature (London) 1997;387:773–776. doi: 10.1038/42867. [DOI] [PubMed] [Google Scholar]
- 32.Danesch U, Hoeck W, Ringold G. J Biol Chem. 1992;267:7185–7193. [PubMed] [Google Scholar]
- 33.Halenbeck R, McDonald H, Rouston A, Chen T T, Conroy L, Williams L T. Curr Biol. 1998;8:537–540. doi: 10.1016/s0960-9822(98)79298-x. [DOI] [PubMed] [Google Scholar]