

Abstract
HIV-1 integrase is an essential enzyme in the life cycle of the virus, responsible for catalyzing the insertion of the viral genome into the host cell chromosome; it provides an attractive target for antiviral drug design. The previously reported crystal structure of the HIV-1 integrase core domain revealed that this domain belongs to the superfamily of polynucleotidyltransferases. However, the position of the conserved catalytic carboxylic acids differed from those observed in other enzymes of the class, and attempts to crystallize in the presence of the cofactor, Mg2+, were unsuccessful. We report here three additional crystal structures of the core domain of HIV-1 integrase mutants, crystallized in the presence and absence of cacodylate, as well as complexed with Mg2+. These three crystal forms, containing between them seven independent core domain structures, demonstrate the unambiguous extension of the previously disordered helix α4 toward the amino terminus from residue M154 and show that the catalytic E152 points in the general direction of the two catalytic aspartates, D64 and D116. In the vicinity of the active site, the structure of the protein in the absence of cacodylate exhibits significant deviations from the previously reported structures. These differences can be attributed to the modification of C65 and C130 by cacodylate, which was an essential component of the original crystallization mixture. We also demonstrate that in the absence of cacodylate this protein will bind to Mg2+, and could provide a satisfactory platform for binding of inhibitors.
A necessary step in the retroviral replication cycle is the integration of viral DNA into the host cell chromosome. In the human immunodeficiency virus type 1 (HIV-1) this function is carried out by an integrase, a 32-kDa enzyme, in a reaction composed of two steps (for reviews, see refs. 1–4). First, the integrase removes two nucleotides from each of the 3′ ends of the viral DNA adjacent to a conserved CA sequence (a reaction termed “3′ processing”). In the second step, these processed viral ends are inserted into opposite strands of chromosomal DNA in a direct transesterification reaction. For HIV-1 integrase, the insertion sites on opposite chromosomal strands are five base pairs apart. Because integrase has no human counterpart, it forms an attractive target for drug design.
In the presence of divalent metal ions such as Mg2+ or Mn2+, recombinant HIV-1 integrase produced in an Escherichia coli expression system will carry out both 3′ processing and strand transfer in vitro when a synthetic double-stranded oligonucleotide substrate mimicking a single viral end is used. Recombinant integrase will also carry out the apparent reversal of the strand transfer step if presented with a Y-shaped oligonucleotide (5); this “disintegration” reaction also requires either Mg2+ or Mn2+.
The entire 32-kDa protein (residues 1–288) is required for 3′ processing and strand transfer, although smaller fragments of the molecule can carry out the disintegration reaction if they contain its central core domain, residues 50–212, indicating that this domain contains the enzyme active site (6). Further evidence supporting this conclusion was obtained from site-directed mutagenesis experiments in which it was demonstrated that even the most conservative substitutions of any of the three absolutely conserved carboxylate residues, D64, D116, and E152 (the so-called D,D-35-E motif), abolished catalytic activity (7–9). The conservation of these three amino acids extends beyond retroviral integrases, as retrotransposons and some prokaryotic transposases contain the same arrangement of catalytically essential carboxylates (8, 10).
We have previously presented the crystal structure of the central core domain of HIV-1 integrase (containing the F185K solubilizing mutation (11)) at 2.5-Å resolution (12). The protein crystallized in a trigonal space group with one core domain molecule per crystallographic asymmetric unit. On the basis of this crystal structure, we demonstrated that the integrase core domain is a member of a polynucleotidyltransferase superfamily whose members include RNase H (13), the bacteriophage Mu transposase (14), and the E. coli Holliday junction resolving enzyme, RuvC (15). Furthermore, on the basis of solvent-excluded surface calculations, we proposed that the dimer we observed in the crystal is most likely the authentic dimer, identical to that which forms in solution (16, 17). This interpretation was later confirmed by the crystal structure of the core domain of integrase from the avian sarcoma virus (ASV), which, despite different crystallization conditions, space group, and crystal packing interactions, showed an essentially identical dimer (18).
In our original structure determination, parts of the molecule displayed a significant degree of disorder, which was serious enough that one region of the polypeptide chain, residues 140–153, remained crystallographically invisible. This loop region has been observed to be flexible in other proteins of this superfamily (13, 14). However, in a recently reported crystal structure of the core domain of HIV-1 integrase F185H mutant (19) the complete active site loop was traced and appeared to be in an extended conformation with E152 pointing away from the other two catalytic carboxylates. Given the proposed role of these three residues in binding metal ions, the authors conclude that the conformation of the active site loop observed in these studies does not correspond to that adopted during catalysis.
Another discrepancy is observed when the conformations of the two catalytic aspartates (D64 and D116) of HIV are compared with those of their counterparts from ASV (D64 and D121). While the β-strands containing D64 superimpose quite well, the main chains surrounding D116 (D121) follow different paths. Moreover, the carboxylate of D64 of HIV-1 integrase makes a hydrogen bond to a main-chain nitrogen of D116, and it cannot participate in binding of a metal ion unless it undergoes a conformational change. Knowledge of the authentic active site structure is crucial for the understanding of the interactions between the enzyme and known inhibitors. It is also important to understand whether these differences reflect true conformational differences between integrases of different viruses, or simply correspond to differences in crystallization conditions.
In trying to account for these differences, we focused on the role of sodium cacodylate used in the original crystallization because cacodylate can react with free sulfhydryl groups. We have produced crystals in the absence of cacodylate and we present here three refined crystal structures of the core domain of HIV-1 integrase mutants, containing seven crystallographically independent monomer structures, crystallized in the presence and in absence of cacodylate, as well as complexed with the Mg2+ cofactor.
MATERIALS AND METHODS
Choice of Mutation Sites.
In an attempt to identify alternative crystallization conditions for the core domain, we examined the available structures of the HIV-1 integrase core domain for possible mutation sites. Crystal formation demonstrated an absolute requirement for DTT, implying that one or more cysteine residues plays an important role in crystallization. The structure of HIV-1 core domain showed that two cysteines, C65 and C130, were buried in a hydrophobic cavity, whereas C56 was disordered and, presumably, surface exposed, making it a good candidate for mutation. Another approach was to modify the protein surface to allow alternative crystal contacts. W131 was chosen for the following reasons: it is a completely surface-exposed large hydrophobic residue, it is located far from both the active site and the dimeric interface, and it is not involved in crystal contacts in the existing structures. New crystal forms were obtained for both mutants, indicating that such a structure-based approach is fruitful and potentially generalizable.
Protein Purification and Crystallization.
The soluble HIV-1 core domain, +HT IN50–212(F185K), was engineered to contain additional site-specific mutations, either C56S or W131K, as previously described (11). It was purified essentially as previously described for +HT IN213–288 (20). E. coli cells (4 liters) induced for expression were harvested, resuspended in 25 mM Hepes, pH 7.5/0.1 mM EDTA, and frozen in liquid nitrogen. The cells were allowed to thaw on ice, and were subsequently lysed by incubation with 0.2 mg/ml lysozyme, followed by sonication. The protein solution was centrifuged at 45,000 × g for 60 min, and the supernatant containing soluble protein was loaded onto a 10-ml Ni-affinity column (Pharmacia) equilibrated in 20 mM Hepes, pH 7.5/0.5 M NaCl/5 mM imidazole (Im)/2 mM 2-mercaptoethanol. The column was washed extensively with the equilibration buffer and then with 20–30 column volumes of equilibration buffer containing 60 mM Im. The protein was eluted with a gradient from 0.06 to 0.8 M Im (100 ml total); it eluted at ≈0.27 M Im.
The purified +HT proteins were cleaved with thrombin to remove the His6 tag and were characterized by analytical gel filtration as previously described (11). The proteins were readily concentrated to >10 mg/ml by ultrafiltration. When protein at 7.5 mg/ml was loaded onto a Sephadex 200 gel filtration column, ≈3% of the protein was present as high molecular weight material eluting in the void volume, with the remainder eluting at a position consistent with that of a dimer. Assays indicated that C56S was fully active in disintegration, and a full-length construct containing the W131E mutation was active in 3′-strand processing (data not shown). Crystallization conditions are listed in Table 1. In all cases the protein solution contained 20 mM Tris⋅HCl, pH 7.5/0.5 M NaCl/1 mM EDTA/5 mM DTT to which precipitant was added in a 1:1 ratio.
Table 1.
Crystallization conditions, data collection, and refinement statistics for three additional crystal forms of HIV-1 integrase core domain
| Form I | Form II | Form III | |
|---|---|---|---|
| Mutant | C56S | W131E | W131E |
| Cofactor | None | None | Mg2+ |
| Crystallization method | Sitting drop | Hanging drop | Hanging drop |
| Protein concentration, mg/ml | 8 | 4 | 2.5 |
| Precipitant solution | 15% PEG 8000/0.16 M ammonium sulfate/100 mM sodium cacodylate, pH 6.5/5 mM DTT* | 30% PEG 4000, 100 mM Hepes, pH 7.0/5 mM DTT | 30% PEG 4000/100 mM Hepes, pH 7.0/5 mM MgCl2/5 mM DTT |
| Space group | P212121 | P1 | C2 |
| Unit cell, a, b, and c in Å; α, β, and γ in ° | a = 50.84 b = 71.34 c = 91.88 | a = 45.09 b = 45.08 c = 49.43 α = 68.82 β = 64.74 γ = 62.63 | a = 76.22 b = 46.91 c = 140.27 β = 105.13 |
| Resolution, Å | 2.0 | 1.95 | 2.5 |
| Rmerge, % | 5.4 (23.7)† | 2.9 (10.6)† | 2.8 (8.3)† |
| Completeness, % | 92.3 (88.4)† | 93.8 (83.8)† | 96.0 (75.6)† |
| Monomers in asymmetric unit | 2 | 2 | 3 |
| R factor, % | 21.2 | 20.4 | 19.8 |
| Free R factor, % | 25.3 | 25.6 | 26.5 |
| rmsd bonds, Å | 0.028 | 0.009 | 0.008 |
| rmsd angles, ° | 2.490 | 1.695 | 1.874 |
Crystals of the usual trigonal morphology were formed initially. However, several weeks after the initial sitting drops were set up, one drop contained C56S crystals in two crystal forms: the original trigonal crystals and an orthorhombic form.
Figures in parentheses indicate the values for the outer shell of the data.
Data Collection and Structure Determination.
For data collection, crystals were gradually transferred to a cryoprotectant solution containing 20% (vol/vol) glycerol in the corresponding mother liquor. Data were collected at 95K on a Raxis IIC image plate detector mounted on a Rigaku RU200 rotating anode source operated at 50 kV and 100 mA with double-mirror focused CuKα radiation. All diffraction data were integrated and scaled with the HKL suite (21). The structures were solved by molecular replacement with amore (22) and refined with x-plor 3.1, using bulk solvent correction (23).
RESULTS AND DISCUSSION
Through the introduction of two additional point mutations into the catalytic core domain of HIV-1 integrase, three new crystal forms were obtained. Data collection and refinement statistics are presented in Table 1.
Overall Structure.
The first half of the protein up to residue F139 at the end of strand β5 in the new crystal forms is topologically very similar to the earlier crystal forms. In all cases, this half of the catalytic domain follows the α-β meander (24) or RNase H-like fold (see Fig. 1). The second half of the molecule (beyond F139) is mostly α-helical, with the last two helices forming the bulk of the solvent-excluded dimer interface. However, there is a new short two-stranded antiparallel β-sheet (strands β6 and β7) not present in the earlier structure determinations. In the original trigonal form, this region of the protein (residues 188–193) was fairly disordered and highly mobile, with some disconnected electron density, which was interpreted as a loop with no secondary structure. In one of the monomers of crystal form I, as well as in both monomers of crystal form II and in two monomers of crystal form III of the W131E mutant, crystal packing interactions not present in the trigonal form stabilized I191, the central residue in the type II β turn that separates strands β6 and β7. As a result of this interaction, the strands are well localized, with strong and continuous electron density. It is interesting to note that in the three-dimensional structure of the catalytic portion of the bacteriophage MuA transposase (the only other member of the polynucleotidyltransferase superfamily that also contains helices α5 and α6 in a very similar orientation relative to the rest of the core domain), the corresponding polypeptide segment also forms a very similar short two-stranded antiparallel β-sheet (14).
Figure 1.

Ribbon diagram of HIV-1 integrase core domain from crystal form II. Two short β-strands not present in previous structures are designated β6 and β7. Essential catalytic residues are shown as ball-and-stick models.
Structure of the Active Site and Effect of Cacodylate.
In the present crystal forms, there is an extension of helix α4 toward the amino terminus. In the original trigonal form, the lack of interpretable electron density for 14 residues between the end of strand β5 at F139 and M154 resulted in the helix starting at M154. The missing region contains E152, one of the catalytically essential acidic residues. In a second previously reported structure of the F185H mutant, the active site loop including E152 was traced (19). However, it appeared to possess a very extended conformation with the side chain of E152 pointing away from D64 and D116. The conformation of this loop was stabilized by crystal contacts with a symmetry-related molecule that is not part of the dimer, and very high temperature factors and poor electron density indicate that this region is extremely flexible.
In all three additional crystal forms reported here, continuous difference electron density typical for an α-helix is seen upstream of M154. The extent of this density varies in different crystal forms. However, it was possible to trace the main chain unambiguously to G149 in crystal form III, to S147 in crystal form I, and I151 in one of the monomers of crystal form II. In the second monomer of crystal form II helix α4 also starts at S147, but the whole loop connecting F139 and S147 can be traced (Fig. 2A). The temperature factors in the loop region are high, indicating its high flexibility. In all these structures the active site residue, E152, is located on helix α4, with its side chain facing in the general direction of D64 and D116 in all the described cases. The location of this residue is quite similar to that of the corresponding essential carboxylate of the ASV integrase (18). The observed position of E152 is likely to be relevant to the catalytic activity for the following reasons: it is consistent in seven independently refined monomers in three different crystal forms; it is similar to the conformation of E157 in ASV integrase; and there are no crystal contacts in the vicinity of E152 that could affect its conformation.
Figure 2.

(A) Difference omit map in the region of helix α4 and the preceding loop, contoured at 1.2 σ level (crystal form II). (B) Superposition of the active site loops of HIV-1 integrase crystal form II (blue), F185H mutant of HIV-1 integrase (19) (red), and ASV integrase (18) (green). Numbering in parentheses corresponds to ASV integrase. The positions of E152 are consistent in all the new structures of HIV-1 integrase and are close to that of the corresponding E157 of ASV integrase.
The crystal form II of the HIV-1 integrase core domain was obtained in absence of cacodylate. Superposition of the active site regions of crystal forms I and II and of ASV integrase is shown in Fig. 2B. The Cα atoms of the catalytically essential D116 in the two HIV-1 structures are shifted apart by 3.5 Å, whereas the distance between the corresponding carboxylate ends of the side chains is as long as 7 Å. In crystal form I, as well as in other cacodylate-containing structures, another essential residue, D64, forms a hydrogen bond to the main chain nitrogen of the displaced D116, thus distorting the geometry of the active site. The cacodylate-free structure is much closer to that of the ASV integrase core domain than to the original HIV-1 integrase structure. The root mean square deviation (rmsd) of Cα positions between the ASV core domain and the HIV-1 core from the crystal form I is 1.7 Å, which may be compared with 1.25 Å between the ASV core and crystal form II of HIV-1. The resemblance is especially strong in the active site region, where all three carboxylates superimpose well on their ASV counterparts.
Comparison of crystal forms I and II of HIV-1 integrase allows us to observe directly the effect of cacodylate modification on the conformation of the adjacent protein regions. The structure refinement of the orthorhombic crystal form I as well as of the original trigonal form revealed peaks of additional density approximately 2.1 Å from the sulfur atoms of C65 and C130. The density corresponded to the highest features in difference maps, suggesting that the cysteine residues had been covalently modified. Anomalous scattering data at 1.542- and 0.9879-Å wavelengths, corresponding to an energy just below the selenium K absorption edge, are consistent with the presence of an arsenic atom covalently bound to the cysteine sulfurs. We propose that the cysteine residues have been modified as follows (25):
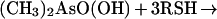 |
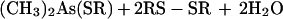 |
Two of the required thiol groups are supplied by DTT, while the third corresponds to the side chain of a cysteine. The modifying group (CH3)2As- fits well into the observed electron density. The side chains of both modified cysteines are pointing into a hydrophobic pocket formed by the α-helices α1, α2, and α3 and strands β1 and β4. To accommodate these bulky moieties, the loop that connects strand β4 to helix α2 together with this helix have been reoriented (Fig. 3). Because cacodylate and reducing agents are widely used in protein crystallization, it is necessary to keep in mind while interpreting structures obtained in their presence that protein conformation may be seriously affected by modification of cysteines by cacodylate. It should be noted that such a modification has been previously reported in the crystal structure determination of transducin (26).
Figure 3.

Effect of covalent modification of cysteines by cacodylate on the conformation of adjacent protein regions. Ribbon diagram in green depicts crystal form I, the one in red, crystal form II. Positions of modified cysteines and two of the catalytic residues, D64 and D116, are shown.
Binding of Magnesium.
The structure of the cacodylate-free core domain in crystal form II showed that the metal-binding site ligands are positioned such that no major rearrangement appears necessary to bind metals. Nevertheless, when these crystals were soaked in solutions containing metal ions, they ceased to diffract. In contrast, crystals could be readily obtained when grown in the presence of Mg2+ and belonged to a different space group (see Table 1, crystal form III). The protein conformation is very similar to that of crystal form II with rmsd of Cα positions of 0.3 Å. The only observed difference is in the region of residues 187–193 in one of the monomers, where two short β-strands are bent because of the crystal contacts in this area. The magnesium binding site is shown in Fig. 4. The Mg2+ ion is coordinated by D64 and D116, whereas the third catalytic residue, E152, does not participate in metal binding. The binding mode of Mg2+ is very close to that observed in the ASV integrase (27). Two water molecules coordinate Mg2+ along with oxygens of D64 and D116 in the octahedral plane. The electron density that corresponds to the ion is elongated in the direction of octahedral axis, but the actual ligand peaks are poorly resolved. This might reflect high mobility of Mg2+ in this direction. It has been proposed, by analogy to the 3′-5′ exonuclease of E. coli polymerase I (28), that retroviral integrases bind two metal ion cofactors at each active site (8). It should be noted that in all integrase structures reported to date and solved in the presence of divalent metal ions, only one physiologically relevant metal ion can be located. Although the structure of ASV integrase has been solved with two bound zinc ions, these are unlikely to be the ions employed in vivo (29). If two metal ions are indeed required for catalysis, it is probable that the second ion binds only in the presence of the DNA substrate.
Figure 4.

Binding of magnesium ion by the core domain of HIV-1 integrase. (A) Difference omit map, contoured at 3.0 σ level. (B) Coordination of Mg2+ ion by D64 and D116.
In summary, the results reported here on seven crystallographically independent structures provide a consensus model for the active site of the HIV-1 core domain. In one of these structures the complete flexible loop can be traced and differs from a previously proposed model (19). The observation of a single bound Mg2+ ion at the active site has implications for possible approaches to inhibitor design. These results also demonstrate that cacodylate, in a reaction with cysteines, can distort the active site region, probably accounting for the previous inability to bind magnesium ions.
ABBREVIATIONS
- ASV
avian sarcoma virus
- rmsd
root mean square deviation
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, Biology Department, Brookhaven National Laboratory, Upton, NY 11973 (PDB ID codes 1bis, 1biu, 1biz).
References
- 1.Vink C, Plasterk R H A. Trends Genet. 1993;9:433–437. doi: 10.1016/0168-9525(93)90107-s. [DOI] [PubMed] [Google Scholar]
- 2.Katz R A, Skalka A M. Annu Rev Biochem. 1994;63:133–173. doi: 10.1146/annurev.bi.63.070194.001025. [DOI] [PubMed] [Google Scholar]
- 3.Andrake M D, Skalka A M. J Biol Chem. 1996;271:19633–19636. doi: 10.1074/jbc.271.33.19633. [DOI] [PubMed] [Google Scholar]
- 4.Mizuuchi K. Genes Cells. 1997;2:1–12. doi: 10.1046/j.1365-2443.1997.970297.x. [DOI] [PubMed] [Google Scholar]
- 5.Chow S A, Vincent K A, Ellison V, Brown P O. Science. 1992;255:723–726. doi: 10.1126/science.1738845. [DOI] [PubMed] [Google Scholar]
- 6.Bushman F D, Engelman A, Palmer I, Wingfield P, Craigie R. Proc Natl Acad Sci USA. 1993;90:3428–3432. doi: 10.1073/pnas.90.8.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engelman A, Craigie R. J Virol. 1992;66:6361–6369. doi: 10.1128/jvi.66.11.6361-6369.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kulkosky J, Jones K S, Katz R A, Mack J P G, Skalka A M. Mol Cell Biol. 1992;12:2331–2338. doi: 10.1128/mcb.12.5.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Gent D C, Oude Groeneger A A M, Plasterk R H A. Proc Natl Acad Sci USA. 1992;89:9598–9602. doi: 10.1073/pnas.89.20.9598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker T A, Luo L. Proc Natl Acad Sci USA. 1994;91:6654–6658. doi: 10.1073/pnas.91.14.6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jenkins T M, Hickman A B, Dyda F, Ghirlando R, Davies D R, Craigie R. Proc Natl Acad Sci USA. 1995;92:6057–6061. doi: 10.1073/pnas.92.13.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dyda F, Hickman A B, Jenkins T M, Engelman A, Craigie R, Davies D R. Science. 1994;266:1981–1986. doi: 10.1126/science.7801124. [DOI] [PubMed] [Google Scholar]
- 13.Davies J F, II, Hostomska Z, Hostomsky Z, Jordan S R, Matthews D A. Science. 1991;252:88–95. doi: 10.1126/science.1707186. [DOI] [PubMed] [Google Scholar]
- 14.Rice P, Mizuuchi K. Cell. 1995;82:209–220. doi: 10.1016/0092-8674(95)90308-9. [DOI] [PubMed] [Google Scholar]
- 15.Ariyoshi M, Vassylyev D G, Iwasaki H, Nakamura H, Shinagawa H, Morikawa K. Cell. 1994;78:1063–1072. doi: 10.1016/0092-8674(94)90280-1. [DOI] [PubMed] [Google Scholar]
- 16.Sherman P A, Fyfe J A. Proc Natl Acad Sci USA. 1990;87:5119–5123. doi: 10.1073/pnas.87.13.5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hickman A B, Palmer I, Engelman A, Craigie R, Wingfield P. J Biol Chem. 1994;269:29279–29287. [PubMed] [Google Scholar]
- 18.Bujacz G, Jaskolski M, Alexandratos J, Wlodawer A, Merkel G, Katz R A, Skalka A M. J Mol Biol. 1995;253:333–346. doi: 10.1006/jmbi.1995.0556. [DOI] [PubMed] [Google Scholar]
- 19.Bujacz G, Alexandratos J, Qing Z L, Clement-Mella C, Wlodawer A. FEBS Lett. 1996;398:175–178. doi: 10.1016/s0014-5793(96)01236-7. [DOI] [PubMed] [Google Scholar]
- 20.Engelman A, Hickman A B, Craigie R. J Virol. 1994;68:5911–5917. doi: 10.1128/jvi.68.9.5911-5917.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 22.Navaza J. Acta Cryst. 1994;50:157–163. [Google Scholar]
- 23.Kostrewa D, Winkler F K. Biochem. 1995;34:683–696. doi: 10.1021/bi00002a036. [DOI] [PubMed] [Google Scholar]
- 24.Orengo C A, Thornton J M. Structure. 1993;1:105–120. doi: 10.1016/0969-2126(93)90026-d. [DOI] [PubMed] [Google Scholar]
- 25.Scott N, Hatlelid K M, MacKenzie N E, Carter D E. Chem Res Toxicol. 1993;6:102–106. doi: 10.1021/tx00031a016. [DOI] [PubMed] [Google Scholar]
- 26.Noel J P, Hamm H E, Sigler P B. Nature (London) 1993;366:654–663. doi: 10.1038/366654a0. [DOI] [PubMed] [Google Scholar]
- 27.Bujacz G, Jaskolski M, Alexandratos J, Wlodawer A, Merkel G, Katz R A, Skalka A M. Structure. 1996;4:89–96. doi: 10.1016/s0969-2126(96)00012-3. [DOI] [PubMed] [Google Scholar]
- 28.Beese L S, Steitz T A. EMBO J. 1991;10:25–33. doi: 10.1002/j.1460-2075.1991.tb07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bujacz G, Alexandratos J, Wlodawer A, Merkel G, Andrake M, Katz R A, Skalka A M. J Biol Chem. 1997;272:18161–18168. doi: 10.1074/jbc.272.29.18161. [DOI] [PubMed] [Google Scholar]