

Abstract
Low levels of hydrogen peroxide (H2O2) are mitogenic to mammalian cells and stimulate the hyperphosphorylation of heterogeneous nuclear ribonucleoprotein C (hnRNP-C) by protein kinase CK1α. However, the mechanisms by which CK1α is regulated have been unclear. Here it is demonstrated that low levels of H2O2 stimulate the rapid dephosphorylation of CK1αLS, a nuclear splice form of CK1α. Furthermore, it is demonstrated that either treatment of endothelial cells with H2O2, or dephosphorylation of CK1αLS in vitro enhances the association of CK1αLS with hnRNP-C. In addition, dephosphorylation of CK1αLS in vitro enhances the kinase’s ability to phosphorylate hnRNP-C. While CK1α appears to be present in all metazoans, analysis of CK1α genomic sequences from several species reveals that the alternatively spliced nuclear localizing L-insert is unique to vertebrates, as is the case for hnRNP-C. These observations indicate that CK1αLS and hnRNP-C represent conserved components of a vertebrate-specific H2O2-responsive nuclear signaling pathway.
Keywords: casein kinase, protein kinase CK1, hydrogen peroxide, reactive oxygen species, RNA binding proteins, protein kinases, hnRNP-C, dephosphorylation, pre-mRNA processing
Introduction
While often viewed as a cytotoxic agent, low concentrations of hydrogen peroxide (H2O2) actually enhance the proliferation and survival of mammalian cells [1–4]. Application of low concentrations (<10µM) of H2O2 to cultured mammalian cells stimulates cell proliferation and enhances survival, while the application of catalase inhibits proliferation and stimulates cell death. Interestingly, these pro-growth functions of H2O2 have not been reported using invertebrate cells or yeast. In mammalian systems, H2O2 is generated by plasma membrane NADPH oxidases in response to receptor-ligand interactions by a wide variety of signaling agents including growth factors [5]. Many of the downstream effects of growth factors are in fact inhibited by treatment of the cells with catalase. In contrast to the effects of low concentrations of H2O2, the application of higher concentrations (>20µM) of H2O2 to cultured cells results in oxidative stress, and typically inhibits cell proliferation and stimulates cell death [4]. Although there have been many advances in understanding the pathways involved in the response of mammalian cells to oxidative stress, the biochemical pathways by which lower concentrations of H2O2 stimulate cell proliferation and enhance cell survival remain unclear [6].
Previously a functional proteomic analysis demonstrated that low concentrations of H2O2 stimulate the rapid phosphorylation of heterogeneous nuclear ribonucleoprotein-C (hnRNP-C)1 in human endothelial cells [7]. Although its function is not entirely understood, hnRNP-C is a nuclear pre-mRNA binding protein that appears to regulate pre-mRNA processing [8, 9] and to facilitate mammalian cell proliferation and differentiation [10]. H2O2–stimulated phosphorylation of hnRNP-C occurs on the C-terminal domain, and this phosphorylation regulates the mRNA binding ability of this protein [11, 12]. Importantly, the kinase responsible for the H2O2–stimulated phosphorylation of hnRNP-C was determined to be protein kinase CK1α (formerly casein kinase 1α), a kinase implicated in regulating cell cycle progression and pre-mRNA processing [11, 13, 14].
In mammals, the CK1 family of protein kinases is encoded by up to 7 distinct genes: CK1α, CK1β, CK1γ1, CK1γ2, CK1γ3, CK1δ, and CK1ε [15–17]. Although protein kinase CK1α was one of the first protein kinases discovered, its mechanism of regulation has remained obscure. Other members of the CK1 family, particularly protein kinases CK1δ, and CK1ε, can be activated by dephosphorylation of inhibitory autophosphorylation sites located primarily within a C-terminal extension [18–21]. However, protein kinase CK1α lacks this autoinhibitory C-terminal extension, and thus its mechanism of regulation has been less clear [15, 16]. Nonetheless, treatment of cells with various agents has been shown to specifically stimulate the phosphorylation of protein kinase CK1α substrates in the cells [11, 22], suggesting that the in vivo activity of the kinase is regulated. In some circumstances this regulation has appeared to involve the association of protein kinase CK1α into relatively stable complexes with its substrates [22–25].
In higher animals, protein kinase CK1α can be present in up to 4 distinct forms due to the presence of two alternatively spliced exons coding for the L-insert and the S-insert [26–35]. The L-insert is comprised of 28 amino acids and is located within the catalytic domain between protein kinase subdomains VIb and VII. The S-insert is comprised of 12 amino acids and is located at the C-terminus of the protein. The L-insert has been shown to mediate nuclear localization and contains a recognized nuclear localization sequence. Splice forms containing the L-insert (CK1αL and CK1αLS) are predominantly, if not entirely, nuclear in localization, while splice forms lacking the L-insert (CK1α and CK1αS) are predominantly, if not entirely, excluded from the nucleus during interphase [26, 28]. The function of the S-insert is not known. In humans, CK1α, CK1αS, and CK1αLS have also been referred to as CK1α1, CK1α2, and CK1α3, respectively [27, 30, 32].
Here we demonstrate that in primary human endothelial cells, the nuclear protein kinase CK1αLS is predominantly phosphorylated under resting conditions and that treatment with low levels of H2O2 leads to the rapid dephosphorylation of the kinase. Dephosphorylation of CK1αLS enhances its ability to both bind and phosphorylate hnRNP-C. Analysis of the genomic sequences of several species has revealed that, like hnRNP-C, the nuclear-localizing L-insert in CK1αLS is restricted to vertebrates, indicating that CK1αLS and hnRNP-C are components of a vertebrate-specific H2O2-responsive nuclear signaling pathway.
Materials and Methods
Endothelial Cell Culture
Pooled human umbilical vein endothelial cells (HUVECs) were obtained from VEC Technologies (Rensselaer, NY). Cells were cultured on 0.1% gelatin coated plates at 37°C with 5% CO2 in M199 medium (25mM HEPES, 2mM L-glutamine, 1.4 g/L NaHCO3, Cambrex, East Rutherford, NJ) containing 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA), 50 µg/mL bovine pituitary endothelial cell growth supplement (ECGS, Sigma, St. Louis, MO), 100 µg/mL heparin (Sigma), 100 units/mL penicillin, and 100 µg/mL streptomycin. The cells were provided new media the first day following passage, and then every 3rd day thereafter until all plates were 100% confluent. Cells were used at passage 3, 48 hrs after the last change of media, and 24–48 hrs after reaching confluency.
The confluent endothelial cells in M199 medium with FBS, heparin and ECGS were incubated in the absence or presence of 1–25 µM H2O2 for 5–20 min at 37 °C. After treatment, the cells were washed twice with ice cold 67 mM sodium phosphate, 150 mM NaCl, pH 7.4 (PBS), and harvested by scraping in Buffer A (10 mM Tris, 140 mM NaCl, 1 mM EDTA, pH 8.0). The cells were pelleted at 500 × g for 15 min. The cells were resuspended in 10 mM HEPES, 750 µM spermidine, 150 µM spermine, 20 mM NaF, 1 mM sodium orthovanadate, 2 mM EDTA, 5 mM DTT, complete protease inhibitor cocktail (CPI, 1 tablet/50 mL, Roche, Basel, Switzerland), pH 7.9 (Buffer B) containing 0.1% Nonidet P-40, incubated on ice for 10 min, and then centrifuged at 16,000 × g for 5 min. The supernatant was removed, and the nuclear pellet was retained.
2-Dimensional Electrophoresis
HUVEC nuclei were resuspended in isoelectric focusing (IEF) sample buffer [9 M urea, 65 mM DTT, 1% CHAPS, 0.1% Bio-Lyte 3/10 Ampholyte (Bio-Rad, Richmond, CA)], using ~200 µL / 107 cells. The suspension was centrifuged at 16,000 × g for 15 min, and the resulting supernatant was then applied to Bio-Spin 6 chromatography columns (Bio-Rad) equilibrated with IEF Sample Buffer, followed by centrifuging at 1000 × g for 4 min. The desalted nuclear extracts were supplemented with 1% pH 7–11 IPG Buffer (Amersham, Piscataway, NJ), and then subjected to IEF for 100,000 volt-hours using a PROTEAN IEF Cell (Bio-Rad) and 18cm pH 7–11 Immobiline DryStrip IPG strips (Amersham). Upon completion of the IEF, the IPG strips were incubated first with Equilibration Buffer (375 mM Tris, 6 M Urea, 2% SDS, 20% glycerol, pH 8.8) containing 130 mM DTT for 10 min, and then with Equilibration Buffer containing 135 mM iodoacetamide for 10 min. The strips were then placed on 10% polyacrylamide gels and electrophoresed at 30 mA per gel. In some experiments, the nuclear extracts were treated with alkaline phosphatase prior to 2-dimensional electrophoresis as described previously [7].
Co-Immunoprecipitation Studies
HUVEC nuclei from ~6×107 cells were lysed by sonication in 4 mL of Buffer B. The samples were incubated on ice for 1 hr to allow for particulate material to settle. The supernatant was removed, and to it was added 160 µL of agarose-conjugated anti-S-insert antibodies (Santa Cruz, Santa Cruz, CA). The samples were rotated for 1 hr at 4 °C, and then incubated on ice for 30 min to allow the resin to settle. The supernatant was set aside, and the resin was then washed 3 times, each with 1 mL of Buffer B, with precipitation of the resin by incubating on ice for 30 min. In some experiments, the resin was washed an additional 3 times with Buffer B, suspended in Laemmli sample buffer (Bio-Rad), and subjected to electrophoresis and immunoblotting for hnRNP-C and CK1α L-insert. In other experiments, the resin was then washed 3 times, each with 1 mL of 50 mM Tris pH 7.4. The resin was then divided into two equal samples and incubated for 1 hr at 37°C with either 20 mM NaF and 1 mM sodium orthovanadate, or with 10 mM MgCl2 and 0.1 Units of alkaline phosphatase (New England Biolabs, Ipswich, MA). The resin was then washed 3 times each with 1 mL of 50 mM Tris pH 7.4 followed by 3 washes, each with 1 mL of Buffer B. The resin was then added to the nuclear supernatant obtained above, and the samples rocked for 2 hr at 4 °C. The resin was then precipitated and washed 6 times each with 1 mL of Buffer B. The washed resin was suspended in Laemmli sample buffer and subjected to electrophoresis and immunoblotting for hnRNP-C and CK1α L-insert.
Immunoblotting
One and 2-dimensional gels were blotted onto polyvinylidene difluoride membranes. Membranes were blocked with 5% milk in 20 mM Tris, 500 mM NaCl, 0.1% Tween-20, pH 7.5 before application of primary and secondary antibodies. Blots were imaged using ECL-plus chemiluminescence detection kits (Amersham) with film and a Kodak X-omat film processor. Rabbit polyclonal antibodies specific for the L-insert of protein kinase CK1αLS were obtained from Cell Signaling Technology (Danvers, MA) and from Alpha Diagnostic (San Antonio, TX), and were used at dilutions of 1:1000 and 1:2000 respectively. Rabbit polyclonal antibodies specific for the S-insert of protein kinase CK1α were obtained from Abgent (San Diego, CA), and used at 1:300 dilution. Mouse monoclonal antibodies to hnRNP-C, clone 4F4, were generously provided by Dr. Gideon Dreyfuss, University of Pennsylvania, and used at 1:2000 dilution. Rabbit antibodies to hnRNP-C were obtained from Santa Cruz Biotechnology and used at a dilution of 1:200. Horseradish peroxidase-conjugated secondary antibodies, donkey anti-rabbit and goat anti-mouse, were obtained from Jackson Immunochemicals (West Grove, PA) and used at dilutions of 1:20,000 and 1:10,000 respectively.
32P Metabolic Labeling
Confluent HUVECs were exchanged into phosphate-deficient modified Eagle’s medium (Invitrogen) containing 15 mM HEPES (Sigma) and also supplemented with FBS, ECGS, Heparin and antibiotics as indicated above. The confluent cells in phosphate-deficient media were then incubated with 32P orthophosphate (1–2 mCi/ 106 cells, Perkin Elmer, Waltham, MA) for 48 hr. Cells were treated and harvested and nuclei were prepared as described above. The nuclei were lysed by sonication in Buffer B containing 1M NaCl. The homogenates were centrifuged at 16,000 × g for 30 min, and the supernantant was collected and diluted 1:2 with Buffer B. The samples were again centrifuged at 16,000 × g for 30 min. To each sample was added 100 µL of Macro-Prep High Q Support strong anion exchange resin (Bio-Rad). The samples were incubated for 30 min at 4 °C, and then centrifuged at 16,000 × g for 10 min. The supernatant was collected, and incubated with 40 µL of agarose-conjugated anti-S-insert antibodies (Santa Cruz) for 30 min at 4 °C. The samples were then centrifuged at 500 × g for the 5 min, and the pelleted resin was washed 10 × with Buffer B containing 0.5 M NaCl. Samples were subjected to electrophoresis, and 32P incorporation was assessed using a Perkin Elmer Cyclone Phosphor imaging device.
In vitro Kinase Assays
HUVEC nuclei were lysed by sonication in Buffer B containing 1M NaCl. The homogenates were centrifuged at 16,000 × g for 5 min, and the supernatant was collected and diluted 1:2 with Buffer B. The samples were again centrifuged at 16,000 × g for 5 min. The supernatant was collected, and incubated with 40 µL of agarose-conjugated anti-S-insert antibodies for 30 min at 4 °C. The samples were centrifuged at 500 × g for the 5 min, and the pelleted resin was washed 3× with Buffer B containing 0.5 M NaCl, and then 3× with 10mM Tris, 125 mM NaCl, pH 7.4. The kinase was treated with or without alkaline phosphatase as detailed above, and the resin was then washed 10× with 10 mM Tris 125 mM NaCl, pH 7.4. Intact hnRNP-C tetramers were immunoprecipitated from the above supernatants using 4F4 monoclonal antibodies and Protein A/G PLUS-agarose (Santa Cruz). Kinase reactions were performed at 37°C for 1 hour as previously detailed [11], except for addition of 1 mg/mL bovine serum albumin. The samples were then analyzed by phosphorescence imaging for 32P incorporation, and by immunblotting for CK1αLS L-insert and hnRNP-C, using rabbit antibodies. Kinase reactions were also performed in the absence of 32P, followed by assessment of hnRNP-C phosphorylation status by 2-dimensinonal immunoblotting as described previously [7,11].
Miscellaneous Methods
H2O2 concentrations of stock solutions were determined using an ε of 81M−1 at 230nm. UV-visible absorption spectra were recorded on a Cary 50 Bio UV-Visible spectrophotometer. For 1-dimensional electrophoresis, CK1α splice forms were separated on 20% acrylamide gels using a PROTEAN II XL electrophoresis cell (Bio-Rad), and coimmunoprecipitation samples were analyzed using a Criterion electrophoresis system (Bio-Rad) with 15% and 4–20% precast gels. Bands on film were quantitated with a GS-800 laser densitometer (Bio-Rad).
L-insert Sequence Analysis
Eukaryotic DNA, RNA and protein databases at the National Center for Biotechnology Information were Blast searched with the protein sequence for the human L-insert with the programs tblastn and blastp using the filter SEG and an Expect Value of 10. The databases were then subsequently searched with all nonidentical matches. Protein sequences were aligned using ClustalW. The genomic CK1α sequences for human (AADC01055869), chicken (AADN02074969), zebrafish (Danio rerio, NC_007125), transparent sea squirt (Ciona intestinalis, AABS01000001), purple sea urchin (Strongylocentrotus purpuratus, XP_786391), honey bee (Apis mellifera, XP_393612), red flour beetle (Tribolium castaneum, XP_973313), Drosophila melanogaster (NP_727631), Caenorhabditis briggsae (CAE72893), and Caenorhabditis elegans (NP_497818) were then inspected to identify the intron/exon boundaries in the region of the L-insert, in some cases by examination of the online gene tables, and in other cases by manual inspection of the shotgun genomic sequences.
Statistical Methods
For the data depicted in Figure 3, different treatment groups were compared by unpaired two-tailed t-test using GraphPad software. P values less then 0.05 were considered significant. For concentration dependency and time course analyses, the data were assessed by one-way Analysis of Variance (ANOVA), followed by post-hoc analysis using GraphPad’s post test calculator, which employs the Bonferroni correction to adjust for multiple comparisons.
Fig. 3.
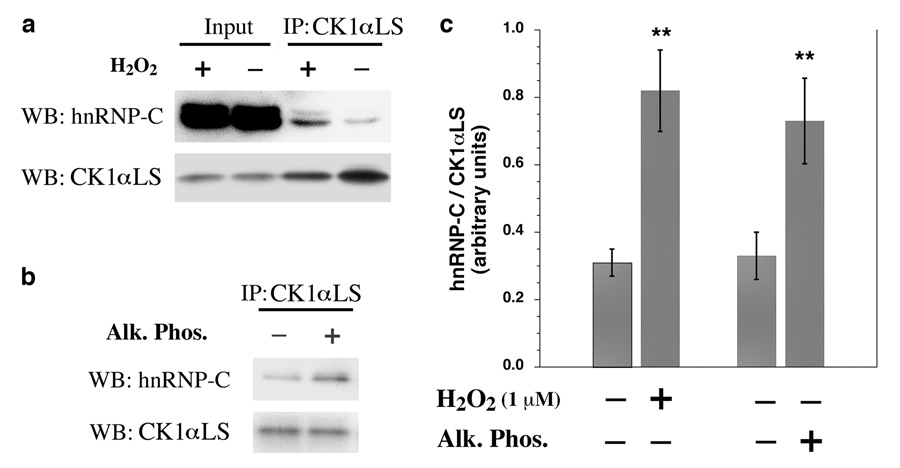
Dephosphorylation of CK1αLS Promotes Its Association with hnRNP-C. (a) HUVECs were treated with or without 1 µM H2O2 for 10 min. CK1αLS was immunoprecipitated from nuclear extracts using S-insert specific antibodies. The immunoprecipitates were subjected to immunoblotting for hnRNP-C and also for CK1αLS using L-insert specific antibodies. (b) CK1αLS was immunoprecipitated from the nuclear extracts of untreated HUVECs using S-insert specific antibodies and then incubated in vitro with or without alkaline phosphatase (Alk. Phos.). The immunopurified kinase was then added back to the nuclear extracts to associate with hnRNP-C. The complexes were immunoprecipitated and subjected to immunoblotting for hnRNP-C and also for CK1αLS using L-insert specific antibodies. (c) Quantitation of the experiments in (a) and (b) using three independent experiments. Error bars indicate the standard deviation; ** P<0.01 versus controls; ie no H2O2 for the comparison on the left and no Alk. Phos. for the comparison on the right.
Results
Protein Kinase CK1αLS is Dephosphorylated in Response to H2O2
The alternatively-spliced L-insert is known to direct nuclear localization of CK1α in higher animals [26, 28]. To determine which L-insert containing splice forms of CK1α are present in human endothelial cells, a crude nuclear / plasma-membrane fraction from human umbilical vein endothelial cells (HUVECs) was assessed by immunoblotting with antibodies specific for either the L-insert or the S-insert (Fig. 1a). A single band with an apparent mass of ~35 kDa was observed using antibodies specific to the L-insert. The S-insert specific antibodies revealed bands with apparent masses of 35 kDa and 32 kDa. Thus the 35 kDa band reactive to both the L-insert and S-insert specific antibodies is identified as CK1αLS. The lower band, reactive to the S-insert specific antibody is identified as CK1αS. In HUVECs, CK1αLS is present at much lower concentrations than CK1αS, consistent with previous observations with human brain tissue [32]. In addition, this analysis indicates that the predominant L-insert containing CK1αsplice form in HUVECs is CK1αLS, with no definitive CK1αL detected.
Fig. 1.
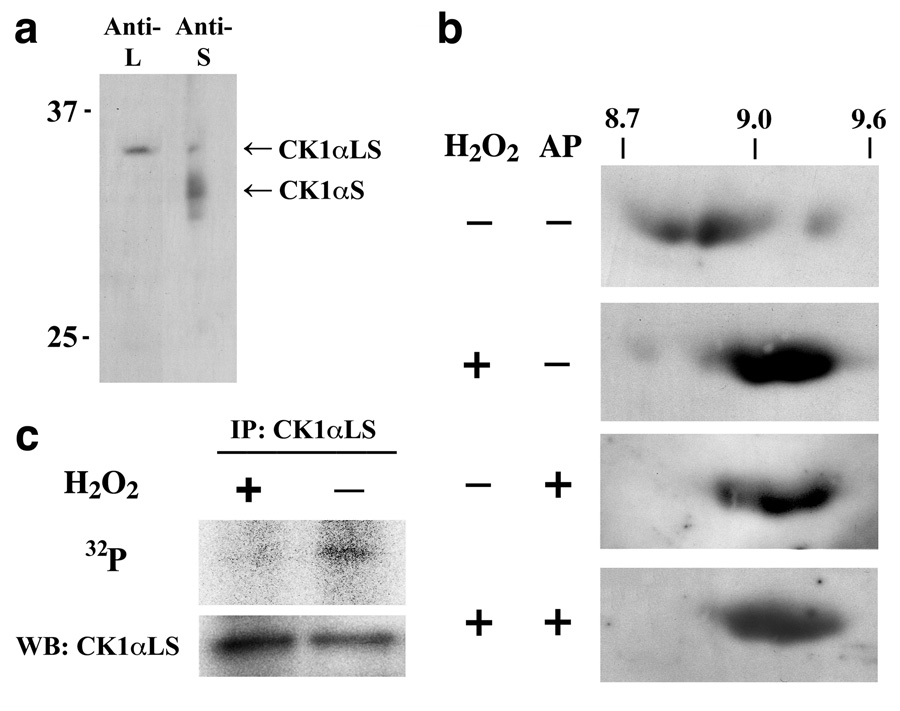
CK1αLS is Dephosphorylated in Response to H2O2. (a) 1-dimensional immunoblots of the crude HUVEC nuclear / plasma membrane fraction showing proteins containing the L-insert and the S-insert of CK1α. CK1αLS was identified as a ~35 kDa protein reactive to both L-insert and S-insert specific antibodies. CK1αS was identified as a ~32 kDa protein reactive only to the S-insert specific antibodies. (b) 2-dimensional immunoblots of HUVEC nuclear extracts probing for CK1αLS using L-insert specific antibodies. The scale at the top refers to the apparent isoelectric point. Under resting conditions, CK1αLS is present predominantly with a isoelectric point between 8.7 and 9.0. Treatment with 5 µM H2O2 for 15 min shifts the isoelectric point of CK1αLS to 9.0–9.4. The same change is seen upon incubation of the nuclear extracts with alkaline phosphatase (AP). (c) HUVECs metabolically labeled with 32P were treated with or without 5 µM H2O2 for 15 min. CK1αLS was immunoprecipitated from nuclear extracts and assessed for 32P incorporation by phosphorescence imaging and for total CK1αLS protein by immunoblotting using L-insert specific antibodies. All blots are representative of at least 3 independent experiments.
Analysis of HUVEC CK1αLS by 2-dimensional immunoblotting, revealed the protein to be present as multiple species, ranging in apparent isoelectric point from 8.7 to 9.5 (Fig. 1b). Treatment of the HUVECs with 5 µM H2O2 shifted the CK1αLS spots to the higher isoelectric points. An identical transition was observed if nuclear extracts from cells not exposed to H2O2, were treated in vitro with alkaline phosphatase. In fact, in vitro alkaline phosphatase treatment of the nuclear extracts resulted in no difference between the CK1αLS spot pattern of H2O2–treated and untreated cells, indicating that the low levels of H2O2 stimulate the dephosphorylation of CK1αLS. This observation was confirmed by metabolically labeling the cells with 32P. The 35 kDa CK1αLS immunoprecipitated from nuclear extracts had substantial 32P incorporation under resting conditions; however, treatment of the cells with 5 µM H2O2 for 15 min substantially decreased the amount of 32P incorporated into CK1αLS (Fig. 1c). Thus in resting HUVECs, CK1αLS is present in a predominantly phosphorylated state, with phosphates on potentially more than one site. Treatment of the cells with low levels of H2O2 stimulates the dephosphorylation of the kinase.
Dephosphorylation of CK1αLS Occurs Rapidly in Response to Low Levels of H2O2
To more fully characterize this H2O2–stimulated dephosphorylation of CK1αLS, the time course for the phenomenon was assessed (Fig. 2a). The dephosphorylation of CK1αLS was first noticeable at 5 min after treatment of the cells with H2O2, and was nearly complete by 10 min after treatment. In contrast, it was previously shown that the H2O2–stimulated CK1α-mediated phosphorylation of hnRNP-C is first noticeable at 10 min after treatment and complete by 20 min after treatment [7]. Thus following treatment of HUVECs with low concentrations of H2O2, dephosphorylation of CK1αLS occurs just prior to the phosphorylation of hnRNP-C. To determine the sensitivity of the phenomenon to H2O2, the cells were treated with various concentrations of H2O2 from 1 to 25 µM for 15 min (Fig. 2b). Substantial dephosphorylation of CK1αLS was observed with just 1 µM H2O2, and complete dephosphorylation of CK1αLS was seen with 5 µM H2O2. These extracellular concentrations of H2O2 correspond to steady state intracellular concentrations of 100 nM and 500 nM respectively [4], within the range known to be mitogenic for mammalian cells. Interestingly, increasing the amount of H2O2 to a mild stress level (25 µM) did not cause any enhanced CK1αLS dephosphorylation, and in fact was slightly less effective than the lower concentration.
Fig. 2.

CK1αLS Dephosphorylation Occurs Rapidly in Response to Low Levels of H2O2. Panels on the left are portions of 2-dimensional immunoblots of HUVEC nuclear extracts probing for CK1αLS using L-insert specific antibodies. The scales at the top refer to the isoelectric point. Dephosphorylation of CK1αLS was assessed by the shift to an isoelectric point > 8.9. The panels on the right depict quantitation of the changes from 3 independent experiments. Error bars indicate the standard deviation. (a) A time course analysis using 5 µM H2O2 indicates that the H2O2–induced dephosphorylation of CK1αLS is first apparent at 5 min after treatment and nearly complete by 10 min after treatment. ** P < 0.001 versus time 0 min. (b) Varying the concentration of applied H2O2 and assessing 15 min after treatment reveals substantial dephosphorylation of CK1αLS with just 1 µM H2O2, with the effect being maximal with 5 µM H2O2. ** P < 0.001 versus no H2O2 added.
Dephosphorylation of Protein Kinase CK1αLS Modulates Its Association with hnRNP-C
The functional significance of CK1αLS dephosphorylation was examined. Treating the cells with just 1 µM H2O2 for 10 min resulted in enhanced association of CK1αLS with hnRNP-C, as assessed by coimmunoprecipitation analysis of the endogenous proteins (Fig. 3a). This observation is in agreement with previous reports of regulated stable complex formation between CK1α and its target substrates [22–25]. To determine if dephosphorylation of CK1αLS is in part responsible for this enhanced association with hnRNP-C, CK1αLS was immunoprecipitated from nuclear extracts of untreated cells. The kinase was then dephosphorylated in vitro with alkaline phosphatase, and subsequently added back to the nuclear extracts to assess the effect of alkaline phosphatase treatment on the ability of CK1αLS to associate with hnRNP-C. It was observed that dephosphorylation of CK1αLS in vitro enhances its association with hnRNP-C (Fig. 3b), indicating that the dephosphorylation of the kinase that precedes hnRNP-C phosphorylation in vivo in fact facilitates the stable association of the kinase with its substrate.
Dephosphorylation of Protein Kinase CK1αLS Enhances Its Ability to Phosphorylate hnRNP-C
To determine if the enhanced interaction of dephosphorylated CK1αLS with hnRNP-C is associated with enhanced phosphorylation of hnRNP-C, intact hnRNP-C tetramers were immunoprecipitated from HUVEC nuclei. Subsequently, in vitro kinase reactions were performed using these intact hnRNP-C tetramers as the substrate, and using for the kinase immunopurified CK1αLS, which had been treated with or without alkaline phosphatase to remove all of the phosphates. Dephosphorylation of CK1αLS significantly enhanced its ability to phosphorylate hnRNP-C (Fig. 4). In addition, dephosphorylated CK1αLS showed enhanced ability to generate the hyperphosphorylated form of hnRNP-C, which on 2-dimensional gels has an isoelectric point of 4.9 [7,11–12].
Fig. 4.
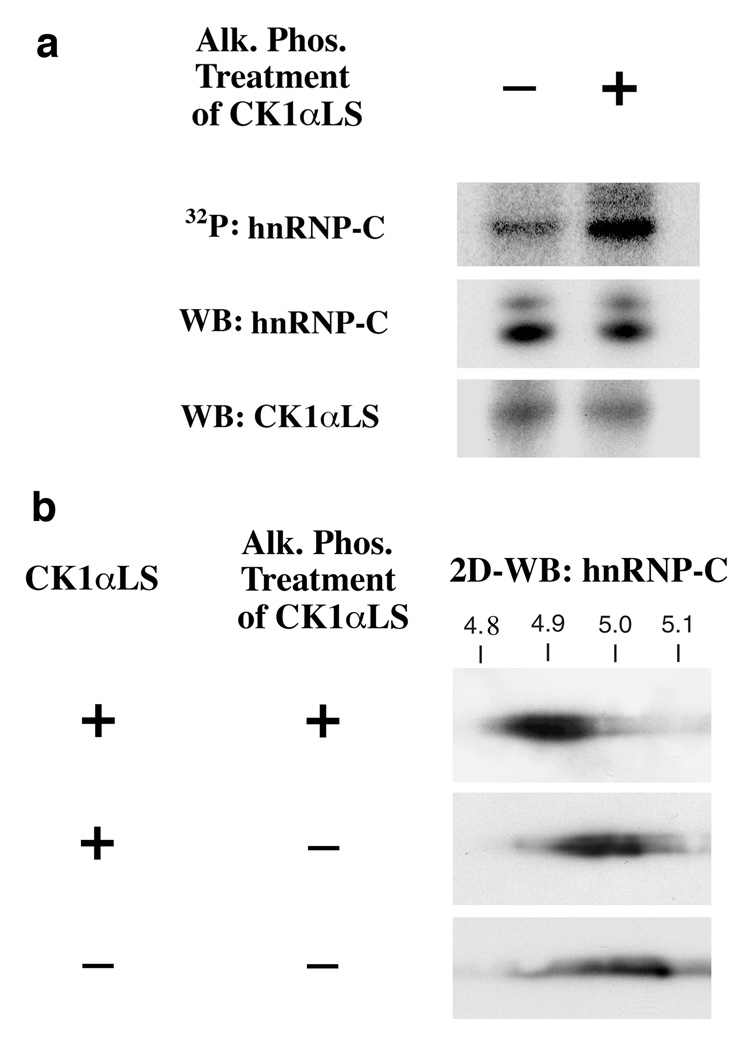
Dephosphorylation of CK1αLS Enhances the Phosphorylation of hnRNP-C by the Kinase. CK1αLS immunoprecipitated from HUVEC nuclei was treated with or without alkaline phosphatase (Alk. Phos.) to remove all phosphate groups. The kinase was then incubated with intact hnRNP-C tetramers immunoprecipitated from HUVEC nuclei, and the phosphorylation status of hnRNP-C was assessed by (a) 32P incorporation into hnRNP-C and also by (b) 2-dimensional immunoblotting for hnRNP-C. Dephosphorylation of CK1αLS enhanced the phosphorylation of hnRNP-C and specifically enhanced the formation of hyperphosphorylated forms of hnRNP-C, which have an isoelectric point of 4.9. All blots are representative of at least 3 independent experiments.
The L-Insert of Protein Kinase CK1αLS is Derived from a Vertebrate-Specific Alternatively Spliced Exon
Because hnRNP-C is highly abundant and predominantly nuclear restricted, it likely represents a predominant substrate for nuclear-localized L-insert containing CK1α splice forms. However, hnRNP-C appears to only be present in vertebrates, and orthologues have not been observed in invertebrates or yeast [11], while CK1α is likely present in all metazoans. However, the species distribution of the L-insert has not been reported. Blast searching of the public databases with the human L-insert sequence identified L-insert containing sequences from several vertebrate species, including mammals, amphibians, birds and fish. The L-insert is highly conserved among vertebrates, with 18 of the 28 residues being invariant (Fig. 5a). In addition L-insert like sequences are also present in yeast kinases, such as the vacuolar CK1, Yck3, in Saccharomyces cerevisiae [36]. The highly conserved KRKR nuclear localizing sequence is not conserved in the L-insert like segment of Yck3, and correspondingly, this kinase is predominantly associated with cytoplasmic vacuoles rather than the nucleus [37, 38]. Interestingly, no L-insert sequences were identified in invertebrate species when blasting protein, RNA or DNA databases.
Fig. 5.

CK1αLS is a Vertebrate-Specific Nuclear Kinase. (a) Depicted is an alignment of L-insert sequences identified in multiple vertebrate species as well as the L-insert like sequence from Saccharomyces cerevisiae Yck3. Green shading indicates completely invariant residues. Yellow shading indicates residues invariant throughout vertebrates but differing in yeast, and the red shading indicates the residues that differ from the human L-insert sequence. The accession numbers for the sequences are: Human, AADC01055869; Chimpanzee, AACZ020650861; Rhesus Monkey, AANU01185333; Cow, AAFC03126961; Elephant, AAGU01149089; Hedgehog, AAIY01297366; Dog, AAEX02012126; Opossum, AAFR03012999; Shrew, AALT01221290; Chicken, AADN02074969; Frog, EB476829; Mouse, AAHY01139286; Rat, AAHX01095469; Zebrafish, NC_007125; Goldfish, AB120746; Tiger Puffer Fish, CAAB01002152; Stickelback, DV014751; Medaka, BAAF02059097; and Yeast (Yck3), X87108. (b) An analysis of the intron/exon boundaries in the CK1α genes from several species in the region of the L-insert is shown. For all genes, the numbering is based on the human CK1α protein sequence. For numbering purposes, immediately adjacent to non-phase 0 introns, the amino acid was assigned to the exon containing 2 of the 3 nucleotides of the codon. Exons are depicted by thick black bars and are drawn to scale. The exon encoding the L-insert is shown in red. The introns are depicted by thin black lines and are not drawn to scale. Accession numbers are listed in the Experimental Procedures section.
To more thoroughly ascertain the species distribution of the CK1α L-insert, the CK1α genomic sequences from several species were analyzed with specific attention to the intron/exon boundaries. In the region of the L-insert, the intron/exon boundaries are completely conserved in the vertebrates including mammals, birds, and fish (Fig. 5b). However, there is substantial divergence of the intron/exon boundaries in many of the invertebrates. Interestingly, there is no intron/exon boundaries at all in the immediate location of the vertebrate L-insert in many of the invertebrate CK1α genes, including the Drosophila dmCK1α gene and the C. elegans kin-19 gene. In the honey bee, the sea urchin, and the transparent sea squirt, an intron is conserved at this location. However, in these species this intron shows no homology to the vertebrate L-insert. Also in these invertebrates this intron is relatively small, being composed of just 76 nucleotides in the honey bee, 449 nucleotides in the sea squirt, and 789 nucleotides in the sea urchin compared with 3,882 nucleotides in zebrafish and 7,080 nucleotides in humans. Thus the L-insert is encoded by an alternatively spliced exon that is unique to vertebrates.
Discussion
At extracellular concentrations of 1–5 µM, corresponding to intracellular concentrations of 100–500 nM, H2O2 exerts positive influences on cell growth and survival [1–4]. The biochemical mechanisms responsible for this effect are poorly understood. One of the very few pathways known to be activated in response to these low levels of H2O2 is the protein kinase CK1α-mediated phosphorylation of hnRNP-C [7, 11]. Although CK1α was one of the first protein kinases identified, the mechanisms regulating its activity have remained elusive. Other members of the mammalian protein kinase CK1 family, particularly protein kinases CK1δ and CK1ε, can be activated by dephosphorylation of inhibitory autophosphorylation sites located primarily within a C-terminal extension [18–21]. However, CK1α lacks the autophosphorylated C-terminal extension found in CK1δ, and CK1ε, and thus its mechanism of regulation has been less clear [15, 16].
Previous studies have suggested that the regulation of protein kinase CK1α activity in vivo involves the formation of specific protein-protein complexes principally with its target substrates. For example, retinoid X receptor (RXR) agonists stimulate the association of CK1α with RXRα, resulting in the phosphorylation of RXRα [22]. Also, through complex formation with axin and adenomatous polyposis coli (APC) tumor suppressor, CK1α binds and phosphorylates β-catenin, leading to β-catenin ubiquitination and degradation. Wnt signaling disrupts this β-catenin degrading complex resulting in the stabilization of β-catenin [24, 39, 40]. Likewise, CK1α binds to and phosphorylates the NFAT family of transcription factors, which along with the action of other kinases prevents NFAT translocation to the nucleus. Calcium-mobilizing stimuli disrupt this CK1α/NFAT complex facilitating, in conjunction with calcineurin activation, NFAT dephosphorylation and nuclear translocation [25, 41, 42]. Thus formation of relatively stable complexes containing CK1α and its various substrates may represent an important mechanism of regulation.
In higher animals, protein kinase CK1α is present in up to 4 distinct splice forms, CK1α, CK1αS, CK1αL, and CK1αLS, due to the alternative splicing of exons coding for the 28 amino acid L-insert and the 12 amino acid C-terminal S-insert. At the protein level, CK1α and CK1αS are typically present at substantially higher levels than CK1αL and CK1αLS [29, 31, 32, 35]. Also in interphase cells CK1α and CK1αS are predominantly if not entirely localized to the cytoplasm while CK1αL and CK1αLS are predominantly if not entirely localized to the nucleus, due to the presence of the nuclear localizing sequence within the L-insert [26, 28].
Defining the in vivo substrates specifically for the nuclear CK1αL and CK1αLS splice forms has been challenging. The cytoplasmic CK1α and/or CK1αS would appear to be the CK1α splice form(s) regulating the Wnt / β-catenin [24, 39, 40] and NFAT [25, 41, 42] pathways, as well as death receptor stimulated apoptotic signaling pathways [43, 44]. Since hnRNP-C is a largely nuclear localized pre-mRNA binding protein, it would necessitate that the H2O2-stimulated CK1α-mediated phosphorylation of the protein be accomplished by CK1αL or CK1αLS. Interestingly, in human endothelial cells, CK1αLS is the predominant L-insert containing CK1α splice variant, with no discernable CK1αL detected. This is in agreement with a previous study employing human brain [32]. Thus, in humans hnRNP-C is a substrate specifically for CK1αLS, and given that hnRNP-C is a highly abundant nuclear protein, it is likely that hnRNP-C is in fact a predominant substrate for CK1αLS.
Similar to other signaling pathways involving CK1α, the H2O2-stimulated CK1αLS-mediated phosphorylation of hnRNP-C involves the stable association of CK1αLS with its substrate, hnRNP-C (Fig. 3). Dephosphorylation of CK1αLS in vitro also enhances its association with hnRNP-C, indicating that the dephosphorylation of this kinase regulates its association into complexes with its substrate. Likewise dephosphorylation of CK1αLS in vitro, enhanced its ability to phosphorylate hnRNP-C (Fig. 4). This latter observation is in agreement with a previous report, concluding that dephosphorylation of the Drosophila CK1α orthologue dmCK1α, enhanced its ability to phosphorylate casein [45].
An interesting aspect of the pro-growth effects of H2O2 is that this phenomenon has only been observed with vertebrate cells [4]. Correspondingly, hnRNP-C appears to only be present in vertebrate cells [11]. In contrast, CK1α is known to be present in all animals, and L-insert like sequences are present within select CK1 isoforms in various yeast species [36]. Interestingly and somewhat unexpectedly, analysis of the genomic sequences of CK1α from several metazoans revealed the alternatively spliced exon encoding the nuclear-localizing L-insert to be present within all vertebrate genomic sequences examined, but not in any of the invertebrate genomic sequences. Thus CK1αLS is in effect a vertebrate-specific nuclear kinase. This observation would imply that in invertebrate cells, CK1α would be excluded from the nucleus during interphase. In fact in Drosophila, dmCK1α does appear to be excluded from the nucleus under normal conditions [45].
During mitosis of eukaryotic cells, there is a translocation of cytoplasmic CK1α/CK1αS to the centrosomes and mitotic spindles [46]. Correspondingly, antibodies to CK1α block cell cycle progression through M-phase in mouse oocytes [14]. In addition, in C. elegans the CK1α orthologue kin-19 also localizes to the centrosome during mitosis [47]. One important CK1α target at the mitotic spindle appears to be the Fas-associated death domain (FADD), such that CK1α-mediated phosphorylation on Ser-194 of FADD is important for regulating progression through M-phase [48]. Recently a phosphoproteome analysis of mitotic spindles from HeLa cells identified phosphorylation of Thr-321 in an S-insert containing CK1α, most likely CK1αS [49]. Precisely how this phosphorylation site relates to the phosphorylation site(s) regulating protein complex formation by CK1αLS is presently unclear.
Acknowledgements
This work was supported by NIH Grant HL074324.
Footnotes
Abbreviations used: 2D-PAGE,2-dimensional polyacrylamide gel electrophoresis; CPI, Complete protease inhibitor cocktail; DTT, dithiothreitol; ECGS, endothelial cell growth supplement; FADD, Fas-associated death domain; FBS, fetal bovine serum; hnRNP, heterogeneous nuclear ribonucleoprotein; HUVEC, human umbilical vein endothelial cell; IEF, isoelectric focusing; NFAT, nuclear factor of activated T cells; RXR, retinoid X receptor.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Burdon RH. Free Radical Biol. Med. 1995;18:775–794. doi: 10.1016/0891-5849(94)00198-s. [DOI] [PubMed] [Google Scholar]
- 2.Davis KJ. IUBMB Life. 1999;48:41–47. doi: 10.1080/713803463. [DOI] [PubMed] [Google Scholar]
- 3.Stone JR, Collins T. Endothelium. 2002;9:231–238. doi: 10.1080/10623320214733. [DOI] [PubMed] [Google Scholar]
- 4.Stone JR, Yang S. Antioxid. Redox Signal. 2006;8:243–270. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 5.Lambeth JD. Nat. Rev. Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 6.Stone JR. Arch. Biochem. Biophys. 2004;422:119–124. doi: 10.1016/j.abb.2003.12.029. [DOI] [PubMed] [Google Scholar]
- 7.Stone JR, Collins T. J. Biol. Chem. 2002;277:15621–15628. doi: 10.1074/jbc.M112153200. [DOI] [PubMed] [Google Scholar]
- 8.Dreyfuss G, Kim VN, Kataoka N. Nat. Rev. Mol. Cell Biol. 2002;3:195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- 9.Weighardt F, Biamonti G, Riva S. Bioessays. 1996;18:747–756. doi: 10.1002/bies.950180910. [DOI] [PubMed] [Google Scholar]
- 10.Williamson DJ, Banik-Maiti S, DeGregori J, Ruley HE. Mol. Cell. Biol. 2000;20:4094–4105. doi: 10.1128/mcb.20.11.4094-4105.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kattapuram T, Yang S, Maki JL, Stone JR. J. Biol. Chem. 2005;280:15340–15347. doi: 10.1074/jbc.M500214200. [DOI] [PubMed] [Google Scholar]
- 12.Stone JR, Maki JL, Collins T. Biochemistry. 2003;42:1301–1308. doi: 10.1021/bi0268091. [DOI] [PubMed] [Google Scholar]
- 13.Gross SD, Loijens JC, Anderson RA. J. Cell Sci. 1999;112:2647–2656. doi: 10.1242/jcs.112.16.2647. [DOI] [PubMed] [Google Scholar]
- 14.Gross SD, Simerly C, Schatten G, Anderson RA. J. Cell Sci. 1997;110:3083–3090. doi: 10.1242/jcs.110.24.3083. [DOI] [PubMed] [Google Scholar]
- 15.Gross SD, Anderson RA. Cell. Signal. 1998;10:699–711. doi: 10.1016/s0898-6568(98)00042-4. [DOI] [PubMed] [Google Scholar]
- 16.Knippschild U, Gocht A, Wolff S, Huber N, Lohler J, Stoter M. Cell. Signal. 2005;17:675–689. doi: 10.1016/j.cellsig.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 17.Vielhaber E, Virshup DM. IUBMB Life. 2001;51:73–78. doi: 10.1080/15216540117461. [DOI] [PubMed] [Google Scholar]
- 18.Cegielska A, Gietzen KF, Rivers A, Virshup DM. J. Biol. Chem. 1998;273:1357–1364. doi: 10.1074/jbc.273.3.1357. [DOI] [PubMed] [Google Scholar]
- 19.Gietzen KF, Virshup DM. J. Biol. Chem. 1999;274:32063–32070. doi: 10.1074/jbc.274.45.32063. [DOI] [PubMed] [Google Scholar]
- 20.Graves PR, Roach PJ. J. Biol. Chem. 1995;270:21689–21694. doi: 10.1074/jbc.270.37.21689. [DOI] [PubMed] [Google Scholar]
- 21.Rivers A, Gietzen KF, Vielhaber E, Virshup DM. J. Biol. Chem. 1998;273:15980–15984. doi: 10.1074/jbc.273.26.15980. [DOI] [PubMed] [Google Scholar]
- 22.Zhao Y, Qin S, Atangan LI, Molina Y, Okawa Y, Arpawong HT, Ghosn C, Xiao JH, Vuligonda V, Brown G, Chandraratna RA. J. Biol. Chem. 2004;279:30844–30849. doi: 10.1074/jbc.M404651200. [DOI] [PubMed] [Google Scholar]
- 23.Chen L, Li C, Pan Y, Chen J. Mol. Cell. Biol. 2005;25:6509–6520. doi: 10.1128/MCB.25.15.6509-6520.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Cell. 2002;108:837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 25.Okamura H, Garcia-Rodriguez C, Martinson H, Qin J, Virshup DM, Rao A. Mol. Cell. Biol. 2004;24:4184–4195. doi: 10.1128/MCB.24.10.4184-4195.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burzio V, Antonelli M, Allende CC, Allende JE. J. Cell. Biochem. 2002;86:805–814. doi: 10.1002/jcb.10263. [DOI] [PubMed] [Google Scholar]
- 27.Fish KJ, Cegielska A, Getman ME, Landes GM, Virshup DM. J. Biol. Chem. 1995;270:14875–14883. doi: 10.1074/jbc.270.25.14875. [DOI] [PubMed] [Google Scholar]
- 28.Fu Z, Chakraborti T, Morse S, Bennett GS, Shaw G. Exp. Cell Res. 2001;269:275–286. doi: 10.1006/excr.2001.5324. [DOI] [PubMed] [Google Scholar]
- 29.Fu Z, Green CL, Bennett GS. J. Neurochem. 1999;73:830–838. doi: 10.1046/j.1471-4159.1999.0730830.x. [DOI] [PubMed] [Google Scholar]
- 30.Green CL, Bennett GS. Gene. 1998;216:189–195. doi: 10.1016/s0378-1119(98)00291-1. [DOI] [PubMed] [Google Scholar]
- 31.Horiguchi R, Tokumoto M, Nagahama Y, Tokumoto T. Biochim. Biophys. Acta. 2005;1727:75–80. doi: 10.1016/j.bbaexp.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 32.Kuret J, Johnson GS, Cha D, Christenson ER, DeMaggio AJ, Hoekstra MF. J. Neurochem. 1997;69:2506–2515. doi: 10.1046/j.1471-4159.1997.69062506.x. [DOI] [PubMed] [Google Scholar]
- 33.Rowles J, Slaughter C, Moomaw C, Hsu J, Cobb MH. Proc. Natl. Acad. Sci. U.S.A. 1991;88:9548–9552. doi: 10.1073/pnas.88.21.9548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yong TJ, Gan YY, Toh BH, Sentry JW. Biochim. Biophys. Acta. 2000;1492:425–433. doi: 10.1016/s0167-4781(00)00146-9. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J, Gross SD, Schroeder MD, Anderson RA. Biochemistry. 1996;35:16319–16327. doi: 10.1021/bi9614444. [DOI] [PubMed] [Google Scholar]
- 36.Wang X, Hoekstra MF, DeMaggio AJ, Dhillon N, Vancura A, Kuret J, Johnston GC, Singer RA. Mol. Cell. Biol. 1996;16:5375–5385. doi: 10.1128/mcb.16.10.5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.LaGrassa TJ, Ungermann C. J. Cell Biol. 2005;168:401–414. doi: 10.1083/jcb.200407141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun B, Chen L, Cao W, Roth AF, Davis NG. Mol. Biol. Cell. 2004;15:1397–1406. doi: 10.1091/mbc.E03-09-0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dale T. Nat. Struct. Mol. Biol. 2006;13:9–11. doi: 10.1038/nsmb0106-9. [DOI] [PubMed] [Google Scholar]
- 40.Price MA. Genes Dev. 2006;20:399–410. doi: 10.1101/gad.1394306. [DOI] [PubMed] [Google Scholar]
- 41.Marin O, Burzio V, Boschetti M, Meggio F, Allende CC, Allende JE, Pinna LA. Biochemistry. 2002;41:618–627. doi: 10.1021/bi0112309. [DOI] [PubMed] [Google Scholar]
- 42.Zhu J, Shibasaki F, Price R, Guillemot JC, Yano T, Dotsch V, Wagner G, Ferrara P, McKeon F. Cell. 1998;93:851–861. doi: 10.1016/s0092-8674(00)81445-2. [DOI] [PubMed] [Google Scholar]
- 43.Desagher S, Osen-Sand A, Montessuit S, Magnenat E, Vilbois F, Hochmann A, Journot L, Antonsson B, Martinou JC. Mol. Cell. 2001;8:601–611. doi: 10.1016/s1097-2765(01)00335-5. [DOI] [PubMed] [Google Scholar]
- 44.Izeradjene K, Douglas L, Delaney AB, Houghton JA. Cancer Res. 2004;64:8036–8044. doi: 10.1158/0008-5472.CAN-04-0762. [DOI] [PubMed] [Google Scholar]
- 45.Santos JA, Logarinho E, Tapia C, Allende CC, Allende JE, Sunkel CE. J. Cell Sci. 1996;109:1847–1856. doi: 10.1242/jcs.109.7.1847. [DOI] [PubMed] [Google Scholar]
- 46.Brockman JL, Gross SD, Sussman MR, Anderson RA. Proc. Natl. Acad. Sci. U.S.A. 1992;89:9454–9458. doi: 10.1073/pnas.89.20.9454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walston T, Tuskey C, Edgar L, Hawkins N, Ellis G, Bowerman B, Wood W, Hardin J. Dev. Cell. 2004;7:831–841. doi: 10.1016/j.devcel.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 48.Alappat EC, Feig C, Boyerinas B, Volkland J, Samuels M, Murmann AE, Thorburn A, Kidd VJ, Slaughter CA, Osborn SL, Winoto A, Tang WJ, Peter ME. Mol. Cell. 2005;19:321–332. doi: 10.1016/j.molcel.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 49.Nousiainen M, Sillje HH, Sauer G, Nigg EA, Korner R. Proc. Natl. Acad. Sci. U.S.A. 2006;103:5391–5396. doi: 10.1073/pnas.0507066103. [DOI] [PMC free article] [PubMed] [Google Scholar]