

Abstract
We generated MRL/lpr mice deficient in the Activation Induced Deaminase (AID). Because AID is required for immunoglobulin hypermutation and class switch recombination, these mice lack hypermutated IgG antibodies. Unlike their AID wild-type littermates, AID-deficient MRL/lpr mice not only lacked autoreactive IgG antibodies, but also experienced a dramatic increase in the levels of autoreactive IgM. This phenotype in AID-deficient mice translated into a dramatic reduction in glomerulonephritis, minimal mononuclear cell infiltration in the kidney, and a dramatic increase in survival to levels comparable to previously reported for MRL/lpr mice completely lacking B cells and levels well below those of mice lacking secreted antibodies. Therefore, this study, wherein littermates with either high levels of autoreactive IgM or autorective IgG are directly examined, proves that autoreactive IgM antibodies alone are not sufficient to promote kidney disease in MRL/lpr mice. In addition, the substantial decrease in mortality combined with a dramatic increase in autoreactive IgM antibodies in AID-deficient MRL/lpr mice, suggest that autoreactive IgM antibodies might not only fail to promote nephritis, but may also provide a protective role in MRL/lpr mice. This novel mouse model containing high levels of autoreactive, unmutated IgM antibodies will help delineate the contribution of autoreactive IgM to autoimmunity.
Keywords: autoimmunity, B cells, Systemic Lupus Erythematosus, Autoantibodies
Introduction
MRL-Faslpr/lpr (MRL/lpr) mice develop a systemic autoimmune syndrome that shares many characteristics of human Systemic Lupus Erythematosus (SLE) [1–2]. Like the human disease, the MRL/lpr syndrome is characterized by polygenic inheritance, the presence of circulating autoantibodies, particularly to nuclear components, and lupus nephritis development through glomerular disease, mononuclear cell infiltration, and immune complex deposition [1–2]. MRL/lpr mice also develop splenomegaly and lymphadenopathy, with mononuclear cell infiltration in lungs, liver, and other tissues [3–4]. Unlike human SLE with low monozygotic twin concordance [5], all MRL/lpr mice eventually develop the syndrome [2]. Multiple loci contribute to autoimmunity in MRL/lpr mice, suggesting the involvement of various systems. Implicated are defects in B and T cell tolerance, complement activation, cytokine regulation, endothelial cell function, and apoptotic clearance [6–21].
There is compelling evidence for a role of B cells in the MRL/lpr syndrome, particularly affecting glomerulonephritis. B cell-deficient-MRL/lpr mice failed to develop glomerulonephritis [22–24]. Also important is a diverse lymphocyte repertoire, since MRL/lpr mice lacking terminal deoxynucleotidyl transferase, an enzyme that adds nucleotides to the V(D)J segments during recombination, have decreased glomerular disease [25–29]. However, how B cells contribute to lupus nephritis might be more complicated than previously appreciated. In addition to secreting autoantibodies, B cells might contribute to lupus nephritis as antigen-presenting cells to autoreactive T cells, and in promoting an inflammatory environment [23, 30–31]. MRL/lpr mice lacking secreted antibodies but with B cells bearing IgM receptors, still develop a milder form of kidney disease and experience higher mortality rates than mice completely lacking B cells [32]. A hallmark feature of MRL/lpr mice lacking B cells, is a dramatic increase in the proportion of naïve CD4+ T cells with a concomitant decrease in memory or activated T cells that was reconstituted to levels similar to conventional MRL/lpr mice in mice with B cells but without secreted antibodies [32]. These results suggest an additional, autoantibody-independent B cell role in the development of lupus nephritis in MRL/lpr mice, likely through the activation of autoreactive T cells.
An aspect of B cell biology that impacts autoimmunity is the memory B cell response. B cells jointly-activated by antigen and CD4+ T cells, seed germinal centers (GC)3 in secondary lymphoid tissues wherein their affinity to foreign antigen is enhanced by immunoglobulin (Ig) somatic hypermutation (SHM) and cellular selection [33–38]. Isotype-class switch recombination (CSR) also occurs in the GC environment, although not exclusively. In SHM, base-pair substitutions are introduced into the DNA encoding the variable (V) regions of Ig receptors. Follicular dendritic cells provide foreign antigen to B cells in the GC’s, selecting B cells with affinity-enhancing mutations to antigen in their receptors. Multiple rounds of division, mutation, and selection generate highly specific memory B cells. Interestingly, a majority of autoantibodies in patients with SLE and in MRL/lpr mice are hypermutated and isotype-switched [39–46]. In MRL/lpr mice, antibodies with mutations in the heavy chain Ig V correlate with anti-double-stranded DNA (dsDNA) specificity, particularly those introducing arginines into the CDRs [46, 47]. One could envision that since SHM is random in relation to affinity, occasionally new mutations increase affinity to self-antigens, or catastrophically, that self-antigen drives the affinity maturation reaction. Evidence of the latter scenario is found in diseases such as rheumatoid arthritis, myasthenia gravis, and Sjogren’s syndrome with ectopic GC formation resulting in high affinity autoantibodies against local self-antigens [48–50]. The recent discovery of AID, a molecule critical to SHM and CSR [51], can now provide a direct approach at examining the contribution of mutated, class-switched antibodies to the MRL/lpr mice syndrome. Since AID is required for SHM and CSR [51–55], AID-deficiency blocks formation of high affinity, isotype-switched antibodies in activated B cells, without impacting B or T cell development [51]. Herein, we generated MRL/lpr mice lacking AID and examined the impact of the lack of hypermutated, switched autoantibodies in the lupus syndrome of these mice.
Materials and Methods
Generation of AID-deficient MRL/lpr mice
AID-deficient C57BL/6 mice were provided by Tasuka Honjo and David Schatz. MRL/MpJ-Faslpr/J (MRL-lpr), C57BL/6J, and BALB/c strains were purchased from The Jackson Laboratory (Bar Harbor, Maine). AID−/− mice were backcrossed to MRL/lpr mice and at the 5th and 6th generation, AID−/+ MRL/lpr mice (with over 96% MRL/lpr background) were intercrossed to generate AID+/+-, AID+/−-, and AID−/− - MRL/lpr mice (N = 34, 33, 34, respectively). The mice were housed in specific pathogen-free facilities, maintained in microisolator cages on hardwood bedding and provided with autoclaved food and reversed osmosed, deionized water.
AID alleles were examined by PCR with primers: 811 (5′-CTG AGA TGG AAC CCT AAC CTC AGC C-3′) plus G4 (5′-CAC GAT TTT CTA CAA ATG TAT TCC AGC-3′) for wild-type allele and G3 (5′-GGG CCA GCT CAT TCC TCC ACT C-3′) plus G4 for mutant allele detection, as described [51]. Fas alleles were amplified by PCR following The Jackson Laboratory website protocol with primers: FASf1 (5′-GTA AAT AAT TGT GCT TCG TCA G -3′), FASr2 (5′-CAA ATC TAG GCA TTA ACA GTG -3′), and FASr1 (5′-TAG AAA GGT GCA CGG GTG TG -3′).
Lifespan analysis
In addition to the mice described above, 134 MRL/lpr mice from the F5 generation were used to examine survival: AID+/+MRl/lpr (N = 34) AID+/−MRL/lpr (N = 58), and AID−/−MRL/lpr, (N = 42). The non-backcrossed MRL/lpr mice (N = 39) were used as controls. Similar numbers of males and females were used in each group. The mice were closely monitored for at least 12 months and euthanized when moribund.
Histology
Formalin-fixed tissues were embedded in paraffin, sectioned at 5 μ, and stained with hematoxylin and Eosin. The severity of any abnormalities observed was graded as follows: 1= minimal, 2= mild, 3= moderate, and 4= marked. Additional sections of kidney were stained with Period Acid Schiff stain. Glomerular change severity was graded based upon an increase in the size of affected glomeruli due to increased cellularity and mesangial matrix. The severity of mononuclear cell infiltrate was graded based upon the total amount of infiltrate present.
The number of cells in each of 20 glomeruli per mouse was scored for the kidneys of each mouse. C57BL/6 and BALB/c mice of similar age were used as controls; the amount of mesangial matrix present in the glomeruli of controls, (approximately 10% of glomerulus), was considered the amount normally present. Lungs, lymph nodes, spleen, liver and bone marrow from each animal were examined for mononuclear cell infiltration.
Electron microscopy
Kidneys from 16–18 week-old mice collected in 3% paraformaldehyde were embedded in Spurr’s resin. Approximately 80nm sections from epoxy blocks were cut, mounted on 200-mesh copper grids, stained with methanolic uranyl acetate and Reynold’s lead citrate, and examined on a Zeiss 900 transmission electron microscope. A total of 40 photomicrographs from 2 representative mice from each genotype were evaluated.
Detection of urine protein level
Urine protein levels, collected monthly by expressing urine from the urethra directly, were tested with Multistix® 10 SG (Bayer, IN) and scored as: 0, negative; 1, trace; 2 (30 mg/dl); 3 (100mg/dl); 4 (300mg/dl); 5 (2000mg/dL or more).
Blood urea nitrogen and creatinine levels in the serum
Blood urea nitrogen (BUN) and creatinine were determined by urease with the glutamate dehydrogenase (GLDH) reaction and alkaline picrate (Jaffe Reaction), respectively. Both reagents were purchased from Olympus America Inc. (Melville, NY) and the determinations were run by Olympus AU400e Clinical Analyzer (Olympus America Inc., Melville, NY).
Immunofluorescence and Immunohistochemistry
To examine complement component 3 (C3) staining in glomeruli, kidneys from 16–18 week-old mice were frozen in Tissue Tek O.C.T. (Sakura, CA) and sectioned on Leica CM 3050 S cryostat (6 microns). Sections were fixed in acetone, washed in 1X automation buffer (Biomedia Corp, CA), and blocked with Dako Serum-Free Protein Block (Dakocytomation, CA). The slides were incubated in a 1:200 dilution of fluorescein-conjugated anti-mouse C3 antibodies (ICN Biomedicals, OH) for one hour, mounted with Vectashield Mounting Media (Vector Laboratories, CA) and viewed with a fluorescent microscope. For negative controls, FITC-conjugated goat serum (Caltag, CA) was used. IgG deposition analysis was similarly done except use of FITC-conjugated goat anti-mouse antibody (Sigma, St. Louis, MO) at a 1:100 dilution.
To examine GC morphology, biotin-labeled PNA (Vector Labs) was used following standard avidin-biotin-peroxidase protocols. Briefly, frozen spleens from 16–18 week-old F5 mice were sectioned on a cryostat (6 microns), affixed to slides, placed into Rapid Fix™ Solution (Shandon-Lipshaw) and into 1X Automation Buffer solution. The sections were placed in 3% H2O2, and rinsed in 1X Automation Buffer. Protein blocking was done with Avidin-Biotin Blocking Kit (Vector Labs, CA). Incubation with PNA was done at a 1:1000 dilution (1mM CaCl2, MgCl2, MnCl2) and labeling with Biogenex Streptavidin Label (Biogenex, CA). The stain was developed with diaminobenzidine (DAB) chromagen (Dakocytomation, CA), the slides counterstained with hematoxylin, dehydrated with graded ethanol, and visualized with a fluorescence microscope.
Flow cytometry
The following conjugated antibodies were used (BD PharMingen, California), following manufacturer’s instructions (1 ug/million cells): rat anti-mouse CD19 PE-Cy7, rat IgG2a, κ isotype control PE-Cy7, rat anti-mouse CD45R/B220-APC-Cy7, APC-Cy7-conjugated rat IgG2a, κ isotype control, FITC-conjugated rat anti-mouse CD21/CD35 monoclonal antibody, FITC-conjugated rat IgG2b, κ isotype control, biotin-conjugated rat anti-mouse CD23 (FcγRII) monoclonal antibody, biotin-conjugated IgG2a, κ isotype control, streptavidin-APC conjugate, rat anti-mouse CD4 PE, rat IgG2a, κ isotype control R-PE, rat anti-mouse CD8 APC, rat IgG2a, κ isotype control APC, anti-mouse CD44 PE-Cy5, rat IgG2b, κ isotype control PE-Cy5, anti-mouse CD62L FITC, rat IgG2a, κ isotype control FITC, anti-mouse CD40 R-PE, rat IgG2a, κ isotype control R-PE, anti-mouse I-Ak biotin, streptavidin-APC conjugate, mouse IgG2b, κ isotype control biotin, anti-mouse CD3 PE-Cy5, and rat IgG2b, κ isotype control PE-Cy5.
Splenic suspensions were made by squashing spleens between two frosted slides. Red blood cells were lysed by ACK lysing buffer (0.15 M NH4Cl, 10.0 mM KHCO3, 0.1mM Na2EDTA). After washing with DPBS, cells were resuspended in Staining Buffer (DPBS + 3% FBS + 0.09% sodium azide) at 1 × 106 cells/100ul, incubated with anti-FcγIII/IIR antibody (BD PharMingen), and stained with corresponding conjugated antibodies in the dark. Cells were resuspended in 1 ml staining buffer and were either analyzed on BD™ LSR II (BD Biosciences, CA) or fixed with 1 ml of 1% paraformaldehyde in PBS and stored at 4°C for next day analysis.
Detection of serum antibodies by ELISA
Beginning at 2 months of age, mice were bled monthly by retro-orbital puncture. Serum IgM, IgG and IgA were determined with commercial ELISA kits (Bethyl Labs, TX) following manufacturer’s instructions. Sera were diluted at 1:10,000, 1:50,000 and 1:10,000 for detection of total IgM, IgG, and IgA respectively. Mouse anti-dsDNA IgM and IgG antibodies were measured as previously reported with modification [47]. Briefly, Costar® 96 Well EIA/RIA Plates (Corning Inc, NY) were coated with streptavidin (Sigma, MO) in PBS at 100ul/well at 4°C overnight, and washed with wash buffer (phosphate buffered saline, pH7.4, 0.05% Tween 20). Salmon sperm DNA (Sigma) was phenol/chloroform-extracted, treated with S1 nuclease, and digested with Hae III. The DNA was biotinylated with Photoprobe Biotin Reagent (Vector Labs, CA) following manufacturer’s instructions. 100ul of biotin-dsDNA in PBS at 400ng/ml was added to wells and incubated at 4°C overnight. 200ul blocking buffer was added (PBS, pH7.4, with 1%BSA) at room temperature (RT) for 2 hours. Mouse sera were diluted with sample diluent (blocking buffer plus 0.05% Tween 20) at 1:2,000 (IgM) and 1:1,000 (IgG) and added to wells at 100ul/well. Pooled sera from four moribund MRL/lpr mice were serially diluted with sample diluent from 1:200~51,200 (IgM) and 1:400~102,400 (IgG) to be used as a standards. Background binding to streptavidin was determined. Goat anti-mouse IgG-HRP and goat anti-mouse IgM-HRP conjugators (Bethyl) were diluted at 1:10,000 and added at 100ul/well. Following incubation and washing, TMB enzyme substrate (KPL, MD) was added at 100ul/well and incubated for 30min at RT. The reaction was stopped by adding 100ml of 1M H2SO4. The absorbance at 450nm was measured in a microplate reader Multiskan Ascent (Thermo Electron Corp, Finland). The amount of anti-dsDNA IgM and IgG in wells was calculated according to the standard curve in which the pooled standard sera were defined as a value of 1.
ANA test
Antinuclear antibodies (ANA) were examined using a commercial indirect fluorescent antibody assay (The ANA HEp-2 Antigen Substrate Slide, BION Enterprises, IL) following manufacturer’s instructions. A drop of diluted mouse serum (1:200 in PBS) was applied to slide wells. After incubation for 30 min at RT, the slide was washed twice with PBS in Coplin jar. FITC conjugated-goat anti-mouse IgG (γ-chain specific) or IgM (μ-chain specific) antibodies (Sigma, at 1:200 in PBS) was added to wells. After two washes, slides were mounted with coverslip and examined under a fluorescence microscope.
Statistical analysis
Pairwise associations between the outcomes (lymphoid hyperplasia, glomerulo-nephritis, mononuclear cell infiltrate) were examined with the Kruskal-Wallis analysis of variance (K-W ANOVA) and Spearman’s correlation coefficient. When significant differences were detected, Mann-Whitney tests were used to compare them to the mild severity group. To assess which combination of measures best predicted outcome severity, stepwise linear regression analysis was used.
K-W ANOVA was used to test for differences among genotypes, followed by Mann-Whitney tests to identify the differing pair of genotypes. For urine data, paired measurements on days 12–14 and days 17–19 were compared using the Wilcoxon signed-ranks test.
Differences were considered statistically significant at the 0.05 level using the Bonferroni correction for multiple testing where appropriate.
Results
To examine the role of high affinity isotype-switched antibodies in the lupus-like syndrome of MRL/lpr mice, we backcrossed AID−/− mice to MRL/lpr mice and generated 5th (F5) and 6th (F6) backcrossed MRL/lpr mice that were wild-type, heterozygous, or deficient in AID. In analyses where both F5 and F6 mice were examined (kidney weights, urine protein, lymphocyte populations, and anti-dsDNA IgG and IgM), the trends were nearly identical between them.
Within each genotype, lesion severity was not significantly associated with antibody levels. However, among all genotypes combined, the following trends were consistently detected for glomerulonephritis scores: with increasing glomerulonephritis severity, anti-dsDNA IgG, urine protein, and mononuclear cell infiltrates increased. Among genotype comparisons, highly significant differences were detected for most measures and are discussed below.
AID deficiency in the MRL/lpr background alleviated glomerulonephritis and mononuclear cell infiltration in the kidneys
Multifocal mononuclear cell infiltration and glomerulonephritis were prominent findings in the kidney of MRL/lpr mice. The average severity of glomerulonephritis and mononuclear cell infiltrates among the F5 mice was significantly higher in the AID-wild type and AID-heterozygous MRL/lpr mice than in AID-deficient MRL/lpr mice littermates (Figure 1, K-W ANOVA, p <0.0001).
Figure 1.

AID deficiency-associated reduction in glomerulonephritis and mononuclear cell infiltrates in MRL/lpr mice. A. Average severity scores for amount of mesangial matrix, mononuclear cell infiltrates and overall glomerulonephritis in kidneys of AID+/+ MRl/lpr (N= 19), AID−/+ MRl/lpr (N = 18), and AID−/− MRl/lpr (N = 19) F5 mice. The increase in mesangial matrix and in glomerular change severity (due to increased cellularity and matrix) was graded as: 0= no increase (matrix occupied up to 10% of the glomerulus; 10% increased cellularity and mesangial matrix), 1= minimal (up to 25%), 2= mild (up to 50%), 3= moderate (up to 75%), and 4= marked (over 75%). B. The average number of glomerular cells in similar-size glomeruli is depicted. Non-autoimmune AID-deficient mice (N = 6), C57BL/6 mice (N = 4) and BALB/c mice (N = 3) were used as controls. Mesangial matrix, glomerulonephritis and mononuclear cell scores in control mice were set at 0 for comparison. C. Periodic Acid Schiff stain from representative AID wild-type and AID-deficient MRL/lpr mice are shown at 20X magnification. Arrows point to glomeruli. D. Hematoxylin and eosin stains of kidney cross-sections from representative AID wild-type and AID-deficient MRL/lpr mice. The arrows indicate areas of dense infiltrates.
Glomerulonephritis was characterized by varying increases in mesangial matrix due to a homogeneous eosinophilic material filling the mesangial spaces between glomerular capillary loops (Figure 1c). In glomeruli with severe glomerulonephritis, mesangial matrix increase was associated with increasing numbers of glomerular cells. Both the mesangial matrix average score and the number of glomerular cells were dramatically reduced with AID deficiency (Figure 1a, and 1b) wherein glomerular cell numbers in AID-deficient MRL/lpr mice were similar to observed in C57BL/6 and BALB/c mice (Figure 1b). The glomerular cell increase was due primarily to inflammatory cells, particularly mesangial macrophages.
Mononuclear cell infiltrates consisted of mixed mononuclear inflammatory cells, primarily lymphocytes and macrophages in the kidney interstitium. AID-deficient MRL/lpr mice mononuclear cell infiltrate scores were reduced compared to AID-wild type and heterozygous MRL/lpr littermates (Figure 1a, 1d; K-W ANOVA, p < 0.0001). These cells accumulated adjacent to the renal pelvis and in AID-deficient MRL/lpr mice, were seen only in that location (Figure 1d). As the amount of infiltrate increased, the cells formed large cuffs around arcuate arteries in the cortex, and in AID-wild type MRL/lpr mice, the cells were also scattered in the interstitium between clusters of tubules. Mononuclear cell infiltration in AID-deficient MRL/lpr mice, while reduced over that in AID-wild type littermates, was above background compared to AID/C57BL/6, C57BL/6 and BALB/c mice. The kidney weights of F5 and F6 AID−/− MRl/lpr mice were significantly reduced compared to AID-wild type littermates (data not shown).
Consistent with the reduced kidney pathology observed in the histology, urine protein levels in F5 AID-deficient MRL/lpr mice older than 10 weeks of age were significantly lower than AID- wild type MRL/lpr mice (Figure 2a, K-W ANOVA, p = 0.0002) and undistinguishable from either non-autoimmune AID-deficient mice in the Bl6 background or conventional C57Bl/6 mice. Urine protein scores from AID wild-type MRL/lpr mice of the F6 backcrossed generation were also significantly higher than AID-deficient MRL/lpr siblings at 17–19 weeks (N = 24, mean = 2.8±1.32 in wild type compared to 1.39 ±0.41 for AID-deficient, p < 0.01, Wilcoxon signed-ranks test). Similarly, F7 and F9 combined data from mice with more than 99.22% MRL/lpr background that were between 20–30 weeks old, displayed the same trend (N = 17, mean = 3.11±1.19 for AID-competent mice versus 1.5 ± 0.40 for AID-deficient MRL/lpr mice; p < 0.01, Wilcoxon signed-ranks test). Furthermore, this difference in correlates of kidney pathology persisted over time as significantly higher levels of blood urea nitrogen and creatinine in the serum of AID wild type and heterozygous MRL/lpr mice were detected when compared to AID-deficient MRL/lpr siblings of the F5 generation at 52 weeks of age (Figure 2b; p < 0.05 for creatinine analysis and p < 0.01 for blood urea nitrogen analysis, Wilcoxon ranks test).
Figure 2.

A. Accumulation of protein in the urine of F5 AID -deficient and wild type MRL/lpr mice. Sample sizes were: 22 AID +/+MRL/lpr, 19 AID −/−MRL/lpr. In addition, 5 non-autoimmune AID −/−, and 6 C57BL/6 mice were used. Error bars represent standard deviations. The differences between genotypes at various time points remained identical when gender was considered. B. The levels of blood urea nitrogen and creatinine in the serum are reduced in 52-week old AID-deficient MRL/lpr mice. Each circle depicts data from an individual mouse.
AID-deficient MRL/lpr mice kidneys revealed lower C3 levels in their glomeruli than AID-wild type MRL/lpr littermates (Figure 3a; K-W ANOVA, p < 0.02) suggesting that the abrogation of glomerulonephritis is associated with a reduction in immune complex deposition. Also, IgG deposition in the glomeruli of AID-wild type MRl/lpr mice was detected at 16–18 weeks of age, which, as expected, was absent in glomeruli from AID-deficient MRL/lpr mice (Figure 3b). IgM deposition was detected in AID-wild type, heterozygous, and deficient MRL/lpr mice but no differences among the groups were seen (data not shown). To determine whether any evidence of early stages of glomeruli damage could be observed in AID-deficient MRL/lpr mice, electron micrographs of glomeruli from representative mice were taken. While glomeruli from AID-wild type MRL/lpr mice had severe lesions consisting of fusion of the glomerular podocytes’s foot processes and infiltration by intravascular macrophages, the glomeruli from AID-deficient MRL/lpr mice were intact and undistinguishable from that of non-autoimmune C57BL/6 mice (Figure 3c).
Figure 3.

Reduced immune complex deposition in AID-deficient MRL/lpr mice accompanied by normal morphology of podocyte’s foot processes and reduced infiltration of macrophages. A. AID+/+MRL/lpr (N = 17), and AID−/−MRL/lpr (N = 18), mice were examined for C3 levels in the glomeruli. For scoring: (+) represents weak staining with limited localization in the glomerulus; (++) represents moderate stain intensity where localization in the glomerulus was more diffuse; (+++) represents strong stain intensity with diffused and homogeneous stain localization in the glomerulus. The panels above the pie charts are representative of the various types of staining observed and the respective scores. B. IgG deposition in the glomeruli of AID-wild type-MRL/lpr mice but not in AID-deficient MRL/lpr mice. The results are representative of at least 3 mice per group of the F5 generation at 16–18 weeks of age. C. Electron microscopy views reveal normal appearance of glomeruli from AID-deficient MRL/lpr, and C57BL/6 mice, while AID wild-type MRL/lpr mice displayed marked fusion of the foot podocyte’s processes and infiltration of macrophages. Notched arrows point to the podocyte’s foot processes (fingerlike structure in normal glomeruli but notice fused pattern in AID wild-type MRL/lpr mice), while striped arrows point to the nuclei of intravascular infiltrating macrophages seen in AID wild-type MRL/lpr mice. Two representative mice from each group were used and 40 micrographs were evaluated.
Glomerulonephritis scores were similar between males and females (K-W ANOVA, p > 0.15) but females tended to have more severe mononuclear cell infiltrate scores (K-W ANOVA, p = 0.007). Gender differences in mononuclear cell infiltrates cannot account for the differences among genotypes since similar gender ratios were used, and when analyzed separately for gender, the differences between the various MRL/lpr littermates remained intact.
Pathological manifestations in other tissues were similar among all MRL/lpr mice and different from normal mice
In the liver, a small degree of mononuclear cell infiltrates was observed in some mice of the MRL/lpr background regardless of AID status. Also, nearly all MRL/lpr mice had lymphoid hyperplasia in the spleen, and myeloid hyperplasia of the bone marrow, although the latter was reduced with AID deficiency. There was lymphoid hyperplasia in the lung that was characterized primarily by an increase in the number of lymphocytes normally present around vessels and airways and it generally affected primarily one or two lung lobes rather than diffusely affecting all lobes. However, there was no difference in lung hyperplasia in AID-deficient MRL/lpr mice when compared to AID-wild type or heterozygous siblings. Also, no significant difference was detected among all MRL/lpr mice in spleen or lymph node weights (data not shown).
Improved survival with AID deficiency in MRL/lpr mice
To examine the impact of AID deficiency on lifespan, a group of F5 mice were allowed to live until multiple signs of impending death were evident as determined by at least two veterinarians (i.e. decreased activity, lowered body temperature, respiratory distress, weigh loss, etc). After 50 weeks, over 75% of the AID-wild type MRL/lpr mice; 65% of AID-heterozygous MRL/lpr mice, and 75% of the non-backcrossed MRL/lpr controls had perished while only 22% of the AID-deficient MRL/lpr had died, indicating a dramatic increase in lifespan with AID deficiency in MRL/lpr mice (Figure 4; Wilcoxon test; p < 0.0001).
Figure 4.

Increased lifespan in AID-deficient MRL/lpr mice. AID+/+MRL/lpr (N = 34), AID+/−MRL/lpr (N = 58), AID−/−MRL/lpr (N = 42) and non-backcrossed MRl/lpr mice (N = 39) were set aside for lifespan study. Mice were euthanized when they reached moribund stage as determined by two veterinarians. At 52 weeks (not shown), the results were nearly identical to survival at 50 weeks.
B and T cell subsets in AID-deficient MRL/lpr mice
The total numbers of CD19+B220+ B cells, and, among CD19+ B cells, the percentage of naïve and activated B cells (based on expression of CD40, I-Ak, Peanut Agglutinin (PNA), or CD44) from spleen and lymph nodes were similar among F5 and F6 MRL/lpr mice regardless of AID status (data not shown). As reported previously [7] marginal zone B cells (based on CD21/CD23 expression) in all MRL/lpr mice were increased over those of BALB/c and C57Bl/6 mice (~ 26% in MRL/lpr vs. ~10% in C57BL/6 mice) with a concomitant reduction in follicular zone B cells. The germinal centers of AID-deficient MRL/lpr mice were similar in morphology and number to AID wild type MRL/lpr littermates, as revealed by PNA staining of GC B cells (data not shown).
Within T cells, the fractions of CD4+ or CD8+ T cells were similar among MRL/lpr mice regardless of AID status. The intriguing CD4−, CD8−, B220+ T cell population characteristic of MRL/lpr mice [56], was similar among all MRL/lpr mice, (about 50% of CD3+ cells in the spleen and lymph nodes). B cell-deficient MRL/lpr mice had been previously reported to have a large increase in the percentage of naïve CD4+ T cells with a concomitant decrease in activated or memory T cells [23], suggesting B cell-mediated activation of autoreactive T cells. Since in mice with B cells but lacking secreted antibodies, the alteration in the proportions of naïve, activated and memory T cells were restored to those seen in MRL/lpr mice, (naïve T cell population was reduced by over 90%), this effect on splenic T cells was directly attributed to an antibody-independent role by B cells [32]. F5 and F6 AID-deficient MRL/lpr mice consistently displayed a slight increase in the splenic naïve CD4+T cell population that was significant in the F6 mice but this increase was only 2-fold (3% in AID +/+ versus 6%, in AID −/−, K-W ANOVA test, p < 0.05). No consistent pattern emerged in the mean values for a concomitant decrease in the memory or activated CD4+ T cell population in neither F5 (10.4% activated, 69.4% memory in AID+/+ versus 10.8% activated and 68.6% memory in AID−/−) nor F6 mice (15% activated, 77.1% memory in AID+/+ versus 17.8% activated and 71.8% memory in AID−/−). Further analysis of the lymph nodes from these mice revealed no difference in the levels of naïve, activated or memory CD4+ T cells in AID-deficient versus AID wild-type, MRL/lpr mice.
Autoreactive IgG and IgM levels in AID−/−, AID −/+, and AID +/+ MRL/lpr mice
Serum anti-dsDNA IgG antibodies are characteristic of MRL/lpr mice and of humans with SLE. These autoantibodies contribute to glomerulonephritis via their deposition in glomeruli as part of immune complexes [39–41]. We first examined total serum Ig antibodies and as expected, AID-deficient MRL/lpr mice had increased levels of IgM but lacked total IgG and IgA, as they lack the ability to undergo CSR (Figure 5a). Anti-dsDNA IgG antibodies levels in the sera of F5 and F6 AID heterozygous- and AID wild type- MRL/lpr mice increased over time while, as expected, AID-deficient MRL/lpr mice had undetectable levels of anti-dsDNA IgG (Figure 5b). Intriguingly, AID heterozygous MRL/lpr mice had significantly lower levels of anti-dsDNA IgG than their AID wild-type MRL/lpr littermates (Figure 5b, K-W ANOVA, p = 0.0001). In fact, there was a small, statistically insignificant, but consistent trend for AID heterozygous to display lower severity scores in the histology measures of kidney pathology (Figure 1), which disappeared with age (data not shown). These combined data suggest an AID dosage effect in MRL/lpr mice that is being examined by ongoing experiments.
Figure 5.

High levels of autoreactive IgG were observed in AID wild-type MRL/lpr mice, while the sera from AID-deficient MRL/lpr mice contained high levels of autoreactive IgM antibodies. A. AID-deficient MRL/lpr mice have increased levels of IgM in the serum and as expected lack any IgG or IgA antibodies. Each circle depicts data from an individual mouse of the F5 generation (16–18 weeks of age). B. Anti-dsDNA IgG scores for AID +/+MRL/lpr (N=34); AID−/+MRL/lpr (N=35); and AID −/−MRL/lpr (N=27). C. Anti-dsDNA IgM scores in same mice. D. Anti-dsDNA IgM in non-autoimmune AID −/− and C57BL/6 mice. Each individual symbol in figures 5a–d is a single mouse score and the line across each lane depicts the average for that genotype. 5b–d depict data combined for F5 and F6 mice.
We also looked at levels of anti-dsDNA IgM. Strikingly, AID-deficient MRL/lpr mice displayed a 5-fold increase in the levels of IgM autoantibodies (Figure 5c, K-W ANOVA, p = 0.0001). To address if this is strictly due to the AID defect, we examined anti-dsDNA IgM levels in AID-deficient C57BL/6 mice. There was a modest increase in the levels of anti-dsDNA IgM (less than 2-fold) in the AID deficient mice compared to C57BL/6 mice (Figure 5d) but that was 60-fold lower than in AID-deficient MRL/lpr mice, suggesting that it is the combination of AID deficiency in the MRL/lpr background contributing to exaggerated anti-dsDNA IgM levels.
To confirm serum autoantibody results, ANA tests using HEp-2 cells were done. Sera from AID wild-type MRL/lpr mice showed bright staining for IgG in the nucleus of cells, while sera from AID deficient MRL/lpr mice were negative (Figure 6). Confirming high levels of autoreactive IgM antibodies, the ANA IgM stain was bright for serum from all AID−/−MRL/lpr mice, displaying two distinct patterns: strictly nuclear, and nuclear with punctate cytoplamic. There was weak IgM staining for AID wild-type MRL/lpr mice in most samples, with a few showing moderate staining (shown in Figure 6).
Figure 6.
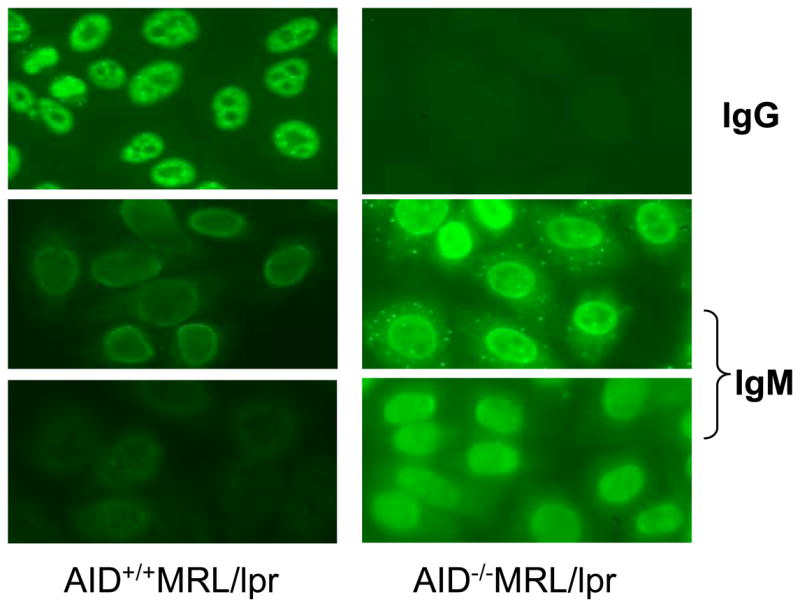
Autoantibodies in sera from AID wild-type and deficient MRL/lpr mice stained nuclear and/or cytoplasmic components of cells. When FITC-conjugated anti-IgG antibodies were used to detect binding, all AID- wild type MRL/lpr mice were positive, while none of the AID deficient mice were. However, all AID-deficient MRL/lpr mice showed bright staining when anti-IgM antibodies were used. Two panels are shown for each group for the IgM analysis to reflect different patterns observed. Approximately half of AID-deficient MRL/lpr mice also stained a cytoplasmic protein/structure with a punctate distribution. A few AID wild-type MRL/lpr mice displayed moderate staining but most displayed weak staining with IgM. The exposure time for all slides incubated with anti-IgM antibody were 4.2 seconds and 5 seconds for anti-IgG antibody, except for anti-IgG in AID-deficient MRL/lpr mice, which were exposed for 25 seconds, since the short exposure was completely black. Sera from C57BL/6 mice were negative in all analyses (not shown).
Discussion
AID deficiency in MRL/lpr mice resulted in a dramatic decrease in glomerulonephritis and mononuclear cell infiltrates in the kidneys, with a concomitant increase in survival. Since these mice cannot undergo SHM or CSR, these results directly implicate high affinity switched antibodies in kidney damage and mortality in MRL/lpr mice. Affinity maturation and SHM in the GC reaction have been implicated in the lupus-like syndrome of MRL/lpr mice since many MRL/lpr mice-derived autoantibodies originate from oligoclonal populations of B cells with mutations selected for self-antigen binding [39, 41, 42, 44, 46, 57–59]. These results confirm the observation that antibodies derived from B cells activated to undergo CSR and SHM, play a critical role in the development of kidney disease in MRL/lpr mice.
AID-deficient MRL/lpr mice displayed a 5-fold increase in anti-dsDNA IgM levels in the serum and their antibodies stained nuclear and cytoplasmic components when incubated with HEp-2 cells. The increased levels of autoreactive IgM antibodies required both AID deficiency and the MRL/lpr background since conventional AID knockout mice had only a 2-fold increase over conventional C57Bl/6 mice in autoreactive IgM, and both had over 60-fold lower levels than AID-deficient MRL/lpr mice. It may be that autoreactive surface IgM-bearing B cells that escape central tolerance are normally mutated away from polyreactivity as they switch to IgG-secreting cells in GC’s. If true and since these mice are not capable of undergoing SHM, these autoreactive B cells would be predicted to accumulate in AID-deficient MRL/lpr mice. Preliminary data from hybridomas generated from these mice showed a 7-fold increase in the number of autoreactive clones when compared to hybridomas derived from AID wild-type littermates, suggesting that the increase in autoreactive IgM antibodies in the serum originates from an increase in autoreactive IgM-secreting B cells (Jiang et al., unpublished data).
The importance of IgG autoantibodies in kidney pathology is suggested by reduced glomerular injury in mice deficient for the activating receptors FcRγ and FcγRIII, but increased severity in mice deficient in the inhibitory receptor FcRγII [60–63]. The role of IgM in lupus nephritis is more controversial. Several studies have identified IgM autoantibodies that significantly contribute to immune complex deposition and are nephritic [64–66], while others have found either a negative correlation between secreted autoreactive IgM and kidney pathology or that monoclonal autoreactive IgM-autoantibodies might even mediate alleviation of nephritis [67–70]. This study provides a direct demonstration wherein MRL/lpr mice secreting high levels of autoreactive IgG can be compared directly to littermates secreting high levels of autoreactive IgM antibodies (without any IgG). AID-deficient MRL/lpr mice, in spite of having high levels of autoreactive IgM, experienced a near complete abrogation of glomerulonephritis, proving that autoreactive IgM antibodies derived from a full B cell repertoire are not sufficient to induce kidney disease in MRL/lpr mice. One possibility is that the deposition of IgM-containing immune complexes might be altered and their trapping in glomeruli might be inhibited due to the larger size of the IgM pentamer compared to IgG. However, others and we have observed IgM deposition in the glomeruli of kidneys of MRL/lpr mice [65]. More likely explanations for the non-pathogenic effect of autoreactive IgM reported here include the recruitment of different clearance pathways activated by IgM versus IgG, and the impact of these pathways on inflammation (see below).
If IgG vs IgM antibodies was the entire story, one would expect that the mortality rates of AID-deficient MRL/lpr mice would be similar to MRL/lpr mice with B cells but lacking secreted antibodies (mIgM) [32]. However, the mortality rates of AID-deficient MRL/lpr mice were much lower than previously reported mortality rates for mIgM mice and similar to MRL/lpr completely lacking B cells (JHD), even though mortality for AID wild-type MRL/lpr, and non-backcrossed MRL/lpr mice in this and the previous study were nearly identical (Table 1). This led us to speculate that the lack of high affinity autoreactive B cells (and not just their antibodies) in AID-deficient MRL/lpr mice might also contribute to reduced disease and mortality in these mice. Shlomchik and colleagues [23, 32], demonstrated that an indication of a direct role of B cells in the lupus syndrome of MRL/lpr mice is the activation of splenic CD4+ T cells. For example, MRL/lpr mice lacking B cells displayed a near 10-fold increase in the proportion of naïve T cells with a concomitant decrease in memory T cells, which was restored in the non-secreting mice [32]. There was only a modest increase (2-fold) in the proportion of naïve T cells in AID-deficient MRL/lpr mice, and no consistent pattern emerged suggesting a decrease in the proportion of activated or memory T cells. Also, although B cells from the AID-deficient MRL/lpr mice cannot hypermutate, these mice have a full naïve B cell repertoire, while the mIgM mice have a limited repertoire. These results suggest, at best, a modest contribution by B cells to the difference in mortality between AID-deficient and wild-type MRL/lpr littermates. Instead, since non-hypermutated germline-encoded natural IgM has been implicated in apoptotic-cell clearance at injury sites, minimizing inflammation [71–74], it may be that AID-deficient MRL/lpr mice have high levels of natural IgM antibodies, and these antibodies play a protective role as suggested by some studies using monoclonal autoreactive IgM antibodies [69]. Indeed, the dramatic reduction in lymphocyte infiltration in the kidneys of AID-deficient MRL/lpr mice is consistent with decreased inflammation. Interestingly, Nemazee and colleagues recently reported that lpr mice lacking secreted IgM but with high levels of secreted IgG and other isotypes (μMT/lpr mice), the converse situation than for AID-deficient MRL/lpr mice, experienced accelerated autoimmunity and autoantibody production compared to lpr mice, possibly suggesting a protective role for IgM [75]. Ongoing studies examining the effects of passive transfer of autoreactive polyclonal and monoclonal IgM antibodies from AID-deficient MRL/lpr mice into AID wild-type MRL/lpr mice and autoreactive IgG into AID-deficient MRL/lpr mice, will enable us to directly examine these hypotheses and perhaps help delineate the characteristics of protective and/or pathogenic IgM antibodies.
Table 1.
Mortality rates of MRL/lpr mice with various B cell defects
| Mouse Model | B-lymphocyte phenotype | Survival at 52 weeks | Reference |
|---|---|---|---|
| JhD | Lack B cells | ~85% | Chan et al., 1999 |
| mIgM | Cannot secrete antibodies, limited B cell repertoire (derived from IgM transgene), somatic hypermutation presumably normal | ~50% | Chan et al., 1999 |
| MRL/lpr | Controls | ~ 25%, (50% at 32 weeks) | Chan et al., 1999 |
| AID+/+MRL/lpr | Littermate controls | 25%, (50% at 32 weeks) | this study |
| AID−/−MRL/lpr | Only secrete IgM, full naïve B cell repertoire but no somatic hypermutation or class switch recombination | 78% | this study |
| MRL/lpr | Controls | 25%, (50% at 32 weeks) | this study |
Acknowledgments
We are indebted to Silvia Bolland, Perry Blackshear, Laurent Verkoczy, and Steve Kleeberger for critical readings of the manuscript and suggestions. Special thanks to Warren Lieuallen for electron microscopy work, Ralph Wilson for help with hematology analysis, and Carl Bortner and Maria Sifre for assistance with flow cytometry. We are grateful to Sue Pierce, Tony Xiao and Jonathan Weiss for suggestions.
Footnotes
This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
List of abbreviations: AID, activation-induced deaminase; GC, germinal center, SHM, somatic hypermutation, CSR, class switch recombination
References
- 1.Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol. 1985;37:269–390. doi: 10.1016/s0065-2776(08)60342-9. [DOI] [PubMed] [Google Scholar]
- 2.Andrews BS, Eisenberg RA, Theofilopoulos AN, Izui S, Wilson CB, McConahey PJ, Murphy ED, Roths JB, Dixon FJ. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J Exp Med. 1978;148:1198–1215. doi: 10.1084/jem.148.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morse HC, 3rd, Davidson WF, Yetter RA, Murphy ED, Roths JB, Coffman RL. Abnormalities induced by the mutant gene Ipr: expansion of a unique lymphocyte subset. J Immunol. 1982;129:2612–2615. [PubMed] [Google Scholar]
- 4.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 5.Deapen D, Escalante A, Weinrib L, Horwitz D, Bachman B, Roy-Burman P, Walker A, Mack TM. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum. 1992;35:311–318. doi: 10.1002/art.1780350310. [DOI] [PubMed] [Google Scholar]
- 6.Culton DA, O’Conner BP, Conway KL, Diz R, Rutan J, Vilen BJ, Clarke SH. Early preplasma cells define a tolerance checkpoint for autoreactive B cells. J Immunol. 2006;176:790–802. doi: 10.4049/jimmunol.176.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y, Li H, Ni D, Weigert M. Anti-DNA B cells in MRL/lpr mice show altered differentiation and editing pattern. J Exp Med. 2002;196:1543–1552. doi: 10.1084/jem.20021560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mandik-Nayak L, Seo SJ, Sokol C, Potts KM, Bui A, Erikson J. MRL-lpr/lpr mice exhibit a defect in maintaining developmental arrest and follicular exclusion of anti-double-stranded DNA B cells. J Exp Med. 1999;189:1799–1814. doi: 10.1084/jem.189.11.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pisetsky DS, Caster SA, Roths JB, Murphy ED. Ipr gene control of the anti-DNA antibody response. J Immunol. 1982;128:2322–2325. [PubMed] [Google Scholar]
- 10.Peng SL, Craft J. T cells in murine lupus: propagation and regulation of disease. Mol Biol Rep. 1996;23:247–251. doi: 10.1007/BF00351176. [DOI] [PubMed] [Google Scholar]
- 11.Bao L, Osawe I, Haas M, Quigg RJ. Signaling through up-regulated C3a receptor is key to the development of experimental lupus nephritis. J Immunol. 2005;175:1947–1955. doi: 10.4049/jimmunol.175.3.1947. [DOI] [PubMed] [Google Scholar]
- 12.Trouw LA, Seelen MA, Duijs JM, Benediktsson H, Van Kooten C, Daha MR. Glomerular deposition of C1q and anti-C1q antibodies in mice following injection of antimouse C1q antibodies. Clin Exp Immunol. 2003;132:32–39. doi: 10.1046/j.1365-2249.2003.02108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Potter PK, Cortes-Hernandez J, Quartier P, Botto M, Walport MJ. Lupus-prone mice have an abnormal response to thioglycolate and an impaired clearance of apoptotic cells. J Immunol. 2003;170:3223–3232. doi: 10.4049/jimmunol.170.6.3223. [DOI] [PubMed] [Google Scholar]
- 14.Bao L, Haas M, Boackle SA, Kraus DM, Cunningham PN, Park P, Alexander JJ, Anderson RK, Culhane K, Holers VM, Quigg RJ. Transgenic expression of a soluble complement inhibitor protects against renal disease and promotes survival in MRL/lpr mice. J Immunol. 2002;168:3601–3607. doi: 10.4049/jimmunol.168.7.3601. [DOI] [PubMed] [Google Scholar]
- 15.Pickering MC, Botto M, Taylor PR, Lachmann PJ, Walport MJ. Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol. 2000;76:227–324. doi: 10.1016/s0065-2776(01)76021-x. [DOI] [PubMed] [Google Scholar]
- 16.Watanabe H, Garnier G, Circolo A, Wetsel RA, Ruiz P, Holers VM, Boackle SA, Colten HR, Gilkeson GS. Modulation of renal disease in MRL/lpr mice genetically deficient in the alternative complement pathway factor B. J Immunol. 2000;164:786–794. doi: 10.4049/jimmunol.164.2.786. [DOI] [PubMed] [Google Scholar]
- 17.Ghebrehiwet B, Peerschke EI. Role of C1q and C1q receptors in the pathogenesis of systemic lupus erythematosus. Curr Dir Autoimmun. 2004;7:87–97. doi: 10.1159/000075688. [DOI] [PubMed] [Google Scholar]
- 18.Botto M, Dell’Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, Loos M, Pandolfi PP, Walport MJ. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19:56–59. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- 19.Pascual V, Banchereau J, Palucka AK. The central role of dendritic cells and interferon-alpha in SLE. Curr Opin Rheumatol. 2003;15:548–556. doi: 10.1097/00002281-200309000-00005. [DOI] [PubMed] [Google Scholar]
- 20.Theofilopoulos AN, Lawson BR. Tumour necrosis factor and other cytokines in murine lupus. Ann Rheum Dis. 1999;58(Suppl 1):I49–55. doi: 10.1136/ard.58.2008.i49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hickey MJ. Alterations in leucocyte trafficking in lupus-prone mice: an examination of the MRL/faslpr mouse. Immunol Cell Biol. 2003;81:390–396. doi: 10.1046/j.1440-1711.2003.01186.x. [DOI] [PubMed] [Google Scholar]
- 22.Chan OT, Madaio MP, Shlomchik MJ. B cells are required for lupus nephritis in the polygenic, Fas-intact MRL model of systemic autoimmunity. J Immunol. 1999;163:3592–3596. [PubMed] [Google Scholar]
- 23.Chan O, Shlomchik MJ. A new role for B cells in systemic autoimmunity: B cells promote spontaneous T cell activation in MRL-lpr/lpr mice. J Immunol. 1998;160:51–59. [PubMed] [Google Scholar]
- 24.Shlomchik MJ, Madaio MP, Ni D, Trounstein M, Huszar D. The role of B cells in lpr/lpr-induced autoimmunity. J Exp Med. 1994;180:1295–1306. doi: 10.1084/jem.180.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watson LC, Moffatt-Blue CS, McDonald RZ, Kompfner E, Ait-Azzouzene D, Nemazee D, Theofilopoulos AN, Kono DH, Feeney AJ. Paucity of V-D-D-J rearrangements and VH replacement events in lupus prone and nonautoimmune TdT−/− and TdT+/+ mice. J Immunol. 2006;177:1120–1128. doi: 10.4049/jimmunol.177.2.1120. [DOI] [PubMed] [Google Scholar]
- 26.Robey IF, Peterson M, Horwitz MS, Kono DH, Stratmann T, Theofilopoulos AN, Sarvetnick N, Teyton L, Feeney AJ. Terminal deoxynucleotidyltransferase deficiency decreases autoimmune disease in diabetes-prone nonobese diabetic mice and lupus-prone MRL-Fas(lpr) mice. J Immunol. 2004;172:4624–4629. doi: 10.4049/jimmunol.172.7.4624. [DOI] [PubMed] [Google Scholar]
- 27.Molano ID, Redmond S, Sekine H, Zhang XK, Reilly C, Hutchison F, Ruiz P, Gilkeson GS. Effect of genetic deficiency of terminal deoxynucleotidyl transferase on autoantibody production and renal disease in MRL/lpr mice. Clin Immunol. 2003;107:186–197. doi: 10.1016/s1521-6616(03)00035-4. [DOI] [PubMed] [Google Scholar]
- 28.Feeney AJ, Lawson BR, Kono DH, Theofilopoulos AN. Terminal deoxynucleotidyl transferase deficiency decreases autoimmune disease in MRL-Fas(lpr) mice. J Immunol. 2001;167:3486–3493. doi: 10.4049/jimmunol.167.6.3486. [DOI] [PubMed] [Google Scholar]
- 29.Molano ID, Wloch MK, Alexander AA, Watanabe H, Gilkeson GS. Effect of a genetic deficiency of terminal deoxynucleotidyl transferase on autoantibody production by C57BL6 Fas(lpr) mice. Clin Immunol. 2000;94:24–32. doi: 10.1006/clim.1999.4797. [DOI] [PubMed] [Google Scholar]
- 30.Mamula MJ, Lin RH, Janeway CA, Jr, Hardin JA. Breaking T cell tolerance with foreign and self co-immunogens. A study of autoimmune B and T cell epitopes of cytochrome c. J Immunol. 1992;149:789–795. [PubMed] [Google Scholar]
- 31.Lin RH, Mamula MJ, Hardin JA, Janeway CA., Jr Induction of autoreactive B cells allows priming of autoreactive T cells. J Exp Med. 1991;173:1433–1439. doi: 10.1084/jem.173.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan OT, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. 1999;189:1639–1648. doi: 10.1084/jem.189.10.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eisen HN, Siskind GW. Variations In Affinities Of Antibodies During The Immune Response. Biochemistry. 1964;3:996–1008. doi: 10.1021/bi00895a027. [DOI] [PubMed] [Google Scholar]
- 34.Klinman NR. The mechanism of antigenic stimulation of primary and secondary clonal precursor cells. J Exp Med. 1972;136:241–260. doi: 10.1084/jem.136.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weigert MG, I, Cesari M, Yonkovich SJ, Cohn M. Variability in the lambda light chain sequences of mouse antibody. Nature. 1970;228:1045–1047. doi: 10.1038/2281045a0. [DOI] [PubMed] [Google Scholar]
- 36.Crews S, Griffin J, Huang H, Calame K, Hood L. A single VH gene segment encodes the immune response to phosphorylcholine: somatic mutation is correlated with the class of the antibody. Cell. 1981;25:59–66. doi: 10.1016/0092-8674(81)90231-2. [DOI] [PubMed] [Google Scholar]
- 37.Clarke SH, Huppi K, Ruezinsky D, Staudt L, Gerhard W, Weigert M. Inter- and intraclonal diversity in the antibody response to influenza hemagglutinin. J Exp Med. 1985;161:687–704. doi: 10.1084/jem.161.4.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berek C, Berger A, Apel M. Maturation of the immune response in germinal centers. Cell. 1991;67:1121–1129. doi: 10.1016/0092-8674(91)90289-b. [DOI] [PubMed] [Google Scholar]
- 39.Shlomchik M, Mascelli M, Shan H, Radic MZ, Pisetsky D, Marshak-Rothstein A, Weigert M. Anti-DNA antibodies from autoimmune mice arise by clonal expansion and somatic mutation. J Exp Med. 1990;171:265–292. doi: 10.1084/jem.171.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Radic MZ, Mascelli MA, Erikson J, Shan H, Shlomchik M, Weigert M. Structural patterns in anti-DNA antibodies from MRL/lpr mice. Cold Spring Harb Symp Quant Biol. 1989;54(Pt 2):933–946. doi: 10.1101/sqb.1989.054.01.108. [DOI] [PubMed] [Google Scholar]
- 41.Shlomchik MJ, Aucoin AH, Pisetsky DS, Weigert MG. Structure and function of anti-DNA autoantibodies derived from a single autoimmune mouse. Proc Natl Acad Sci U S A. 1987;84:9150–9154. doi: 10.1073/pnas.84.24.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shlomchik MJ, Marshak-Rothstein A, Wolfowicz CB, Rothstein TL, Weigert MG. The role of clonal selection and somatic mutation in autoimmunity. Nature. 1987;328:805–811. doi: 10.1038/328805a0. [DOI] [PubMed] [Google Scholar]
- 43.Wellmann U, Letz M, Herrmann M, Angermuller S, Kalden JR, Winkler TH. The evolution of human anti-double-stranded DNA autoantibodies. Proc Natl Acad Sci U S A. 2005;102:9258–9263. doi: 10.1073/pnas.0500132102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Es JH, Gmelig Meyling FH, van de Akker WR, Aanstoot H, Derksen RH, Logtenberg T. Somatic mutations in the variable regions of a human IgG anti-double-stranded DNA autoantibody suggest a role for antigen in the induction of systemic lupus erythematosus. J Exp Med. 1991;173:461–470. doi: 10.1084/jem.173.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winkler TH, Fehr H, Kalden JR. Analysis of immunoglobulin variable region genes from human IgG anti-DNA hybridomas. Eur J Immunol. 1992;22:1719–1728. doi: 10.1002/eji.1830220709. [DOI] [PubMed] [Google Scholar]
- 46.Radic MZ, Mackle J, Erikson J, Mol C, Anderson WF, Weigert M. Residues that mediate DNA binding of autoimmune antibodies. J Immunol. 1993;150:4966–4977. [PubMed] [Google Scholar]
- 47.Radic MZ, Mascelli MA, Erikson J, Shan H, Weigert M. Ig H and L chain contributions to autoimmune specificities. J Immunol. 1991;146:176–182. [PubMed] [Google Scholar]
- 48.Sims GP, Shiono H, Willcox N, Stott DI. Somatic hypermutation and selection of B cells in thymic germinal centers responding to acetylcholine receptor in myasthenia gravis. J Immunol. 2001;167:1935–1944. doi: 10.4049/jimmunol.167.4.1935. [DOI] [PubMed] [Google Scholar]
- 49.Stott DI, Hiepe F, Hummel M, Steinhauser G, Berek C. Antigen-driven clonal proliferation of B cells within the target tissue of an autoimmune disease. The salivary glands of patients with Sjogren’s syndrome. J Clin Invest. 1998;102:938–946. doi: 10.1172/JCI3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim HJ, Berek C. B cells in rheumatoid arthritis. Arthritis Res. 2000;2:126–131. doi: 10.1186/ar77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 52.Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, Tezcan I, Ersoy F, Kayserili H, Ugazio AG, Brousse N, Muramatsu M, Notarangelo LD, Kinoshita K, Honjo T, Fischer A, Durandy A. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–575. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 53.Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol. 2002;12:1748–1755. doi: 10.1016/s0960-9822(02)01215-0. [DOI] [PubMed] [Google Scholar]
- 54.Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002;418:99–103. doi: 10.1038/nature00862. [DOI] [PubMed] [Google Scholar]
- 55.Odegard VH, Schatz DG. Targeting of somatic hypermutation. Nat Rev Immunol. 2006;6:573–583. doi: 10.1038/nri1896. [DOI] [PubMed] [Google Scholar]
- 56.Santoro TJ, Portanova JP, Kotzin BL. The contribution of L3T4+ T cells to lymphoproliferation and autoantibody production in MRL-lpr/lpr mice. J Exp Med. 1988;167:1713–1718. doi: 10.1084/jem.167.5.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.William J, Euler C, Christensen S, Shlomchik MJ. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science. 2002;297:2066–2070. doi: 10.1126/science.1073924. [DOI] [PubMed] [Google Scholar]
- 58.Brard F, Shannon M, Prak EL, Litwin S, Weigert M. Somatic mutation and light chain rearrangement generate autoimmunity in anti-single-stranded DNA transgenic MRL/lpr mice. J Exp Med. 1999;190:691–704. doi: 10.1084/jem.190.5.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Putterman C, Limpanasithikul W, Edelman M, Diamond B. The double edged sword of the immune response: mutational analysis of a murine anti-pneumococcal, anti-DNA antibody. J Clin Invest. 1996;97:2251–2259. doi: 10.1172/JCI118666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sylvestre DL, Ravetch JV. Fc receptors initiate the Arthus reaction: redefining the inflammatory cascade. Science. 1994;265:1095–1098. doi: 10.1126/science.8066448. [DOI] [PubMed] [Google Scholar]
- 61.Hazenbos WL, Gessner JE, Hofhuis FM, Kuipers H, Meyer D, Heijnen IA, Schmidt RE, Sandor M, Capel PJ, Daeron M, van de Winkel JG, Verbeek JS. Impaired IgG-dependent anaphylaxis and Arthus reaction in Fc gamma RIII (CD16) deficient mice. Immunity. 1996;5:181–188. doi: 10.1016/s1074-7613(00)80494-x. [DOI] [PubMed] [Google Scholar]
- 62.Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV. Augmented humoral and anaphylactic responses in Fc gamma RII-deficient mice. Nature. 1996;379:346–349. doi: 10.1038/379346a0. [DOI] [PubMed] [Google Scholar]
- 63.Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19:275–290. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 64.Katz MS, Foster MH, Madaio MP. Independently derived murine glomerular immune deposit-forming anti-DNA antibodies are encoded by near-identical VH gene sequences. J Clin Invest. 1993;91:402–408. doi: 10.1172/JCI116214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ito MR, Terasaki S, Kondo E, Shiwaku H, Fukuoka Y, Nose M. Experimental lupus nephritis in severe combined immunodeficient (SCID) mice: remodelling of the glomerular lesions by bystander IgM antibodies. Clin Exp Immunol. 2000;119:340–345. doi: 10.1046/j.1365-2249.2000.01133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Youd ME, Luus L, Corley RB. IgM monomers accelerate disease manifestations in autoimmune-prone Fas-deficient mice. J Autoimmun. 2004;23:333–343. doi: 10.1016/j.jaut.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 67.Forger F, Matthias T, Oppermann M, Becker H, Helmke K. Clinical significance of anti-dsDNA antibody isotypes: IgG/IgM ratio of anti-dsDNA antibodies as a prognostic marker for lupus nephritis. Lupus. 2004;13:36–44. doi: 10.1191/0961203304lu485oa. [DOI] [PubMed] [Google Scholar]
- 68.Witte T, Hartung K, Sachse C, Matthias T, Fricke M, Deicher H, Kalden JR, Lakomek HJ, Peter HH, Schmidt RE. IgM anti-dsDNA antibodies in systemic lupus erythematosus: negative association with nephritis. SLE Study Group. Rheumatol Int. 1998;18:85–91. doi: 10.1007/s002960050063. [DOI] [PubMed] [Google Scholar]
- 69.Werwitzke S, Trick D, Kamino K, Matthias T, Kniesch K, Schlegelberger B, Schmidt RE, Witte T. Inhibition of lupus disease by anti-double-stranded DNA antibodies of the IgM isotype in the (NZB × NZW)F1 mouse. Arthritis Rheum. 2005;52:3629–3638. doi: 10.1002/art.21379. [DOI] [PubMed] [Google Scholar]
- 70.Boes M, Schmidt T, Linkemann K, Beaudette BC, Marshak-Rothstein A, Chen J. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc Natl Acad Sci U S A. 2000;97:1184–1189. doi: 10.1073/pnas.97.3.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gaipl US, Kuhn A, Sheriff A, Munoz LE, Franz S, Voll RE, Kalden JR, Herrmann M. Clearance of apoptotic cells in human SLE. Curr Dir Autoimmun. 2006;9:173–187. doi: 10.1159/000090781. [DOI] [PubMed] [Google Scholar]
- 72.Ehrenstein MR, Cook HT, Neuberger MS. Deficiency in serum immunoglobulin (Ig)M predisposes to development of IgG autoantibodies. J Exp Med. 2000;191:1253–1258. doi: 10.1084/jem.191.7.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ogden CA, Kowalewski R, Peng Y, Montenegro V, Elkon KB. IGM is required for efficient complement mediated phagocytosis of apoptotic cells in vivo. Autoimmunity. 2005;38:259–264. doi: 10.1080/08916930500124452. [DOI] [PubMed] [Google Scholar]
- 74.Binder CJ, Silverman GJ. Natural antibodies and the autoimmunity of atherosclerosis. Springer Semin Immunopathol. 2005;26:385–404. doi: 10.1007/s00281-004-0185-z. [DOI] [PubMed] [Google Scholar]
- 75.Melamed D, Miri E, Leider N, Nemazee D. Unexpected autoantibody production in membrane Ig-mu-deficient/lpr mice. J Immunol. 2000;165:4353–4358. doi: 10.4049/jimmunol.165.8.4353. [DOI] [PubMed] [Google Scholar]