

Abstract
Mitochondrial DNA is replicated and repaired by DNA polymerase γ (pol γ), encoded by the POLG gene. The Y955C substitution in POLG leads to autosomal dominant progressive external ophthalmoplegia (PEO) with other severe phenotypes. PEO patients with this mutation can further develop parkinsonism or premature ovarian failure. Mouse and yeast models with this mutation show enhanced amounts of oxidative lesions and increased mtDNA damage. In DNA pol γ, Tyr955 plays a critical role in catalysis and high fidelity DNA synthesis. 7,8-dihydro-8-oxo-2′-deoxyguanosine (8-oxo-dG) is one of the most common oxidative lesions in DNA and can promote transversion mutations. Mitochondria are thought to be a major source of endogenous reactive oxygen species that can react with dG to form 8-oxo-dG as one of the more common products. DNA polymerases can mitigate mutagenesis by 8-oxo-dG through allosteric interactions from amino acid side chains, which limit the anti-conformation of the 8-oxo-dG template base during translesion DNA synthesis. Here, we show that the Y955C pol γ displays relaxed discrimination when either incorporating 8-oxo-dGTP or translesion synthesis opposite 8-oxo-dG. Molecular modeling and biochemical analysis suggest that this residue, Tyr955, in conjunction with Phe961 helps attenuate the anti-conformation in human pol γ for error free bypass of 8-oxo-dG and substitution to Cys allows the mutagenic syn conformation. Collectively, these results offer a biochemical link between the observed oxidative stress in model systems and parkinsonism in patients, suggesting that patients harboring the Y955C POLG mutation may undergo enhanced oxidative stress and DNA mutagenesis.
Introduction
Mitochondrial DNA is replicated and repaired by DNA polymerase γ (pol γ), encoded by the nuclear POLG gene (1,2). Mutations in the POLG are known to cause deletions and depletion of mtDNA and will manifest as a wide array of diseases, including progressive external ophthalmoplegia (PEO), ataxia-neuropathy symptoms and Alpers syndrome (3–5). The Y955C mutation in POLG is commonly associated in patients with a fully penetrant autosomal dominant form of PEO and patients with this mutation may develop proximal myopathy, dysphagia, progressive neurological deterioration with ataxia and terminal dementia (6,7). In several individuals with the Y955C allele, there is significant cosegregation of parkinsonism with adPEO (8). In addition, many women with PEO due to the Y955C mutation develop early menopause and suffer from high gonadotropin and low estrogen concentrations, indicative of premature ovarian failure (8,9).
We previously reported that Y955C mutant pol γ retains less than 1% polymerase activity, catalyzes error-prone DNA synthesis and replication stalling (10,11), an observation that is consistent with the accumulation of mtDNA point mutations and deletions found in PEO patients. Heart specific expression of the human Y955C cDNA in a mouse transgenic model causes cardiomyopathy with decreased mtDNA and mitochondrial ultrastructural defects (12). Interestingly, the mtDNA from these TG hearts displayed a 2–3-fold increase in 7,8-dihydro-8-oxo-2′-deoxyguanosine (8-oxo-dG) indicative of either increased oxidative stress or relaxed incorporation of this oxidized nucleotide into DNA (12). The analogous mutation in the yeast pol γ gene, Y757C in MIP1, causes loss of mtDNA, high petite frequency and enhanced mtDNA damage consistent with an increase in oxidative-generated mtDNA lesions (13). This high-petite frequency can be rescued by treatment with the ROS scavenger, dihydrolipoic acid (14). These results indicate that oxidative damage to mtDNA may be a part of the pathological mechanism associated with the Y955C mutation in POLG. This is especially relevant since parkinsonism has been shown to be strongly associated with this mutation (8).
Mitochondria are sites of endogenous production of reactive oxygen species and the subsequent formation of 8-oxo-dG as a byproduct of oxidative phosphorylation in mitochondrial DNA (mtDNA) has been reported (15,16). 8-oxo-dG is a common oxidative DNA lesion, found both in nuclear and mitochondrial DNA, where it accumulates during aging. The 8-oxo-dG base can form two conformations in DNA, the anti conformation which favors Watson-Crick base pairing with dCMP and the syn-conformation which allows Hoogsteen base pairing with adenine (Fig. 1). The Hoogsteen 8-oxo-dG:A base mispair will cause the G:C to T:A transversion mutation if extended and not repaired. Consistent with this hypothesis, a study with the Xenopus laevis pol γ has demonstrated translesion synthesis opposite 8-oxo-dG where the correct dCMP is incorporated 73% of the time while dAMP is incorporated the remaining 27% (17).
Figure 1.
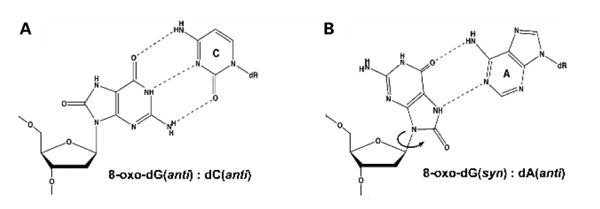
Rotamer conformations of 8-oxo-dG template base. (A) Watson-Crick hydrogen bonding between the 8-oxo-dG base and the dCTP base when 8-oxo-dG assumes the anti conformation. (B) Hoogsteen hydrogen bonding between the 8-oxo-dG base and the dATP base when 8-oxo-dG assumes the syn conformation.
The nucleotide incorporation specificity opposite 8-oxo-dG varies depending on which polymerase copies the lesion and how that polymerase attenuates the anti conformation. In Bacillus stearothermophilus DNA polymerase, the syn conformation of the 8-oxo-dG template base prefers to hydrogen bond with adenine and appears to the polymerase as a correct base pair, evading the normal proofreading mechanism (18). Several recent structural studies with DNA polymerase β and T7 DNA polymerase have implicated a few key amino acid residues involved in attenuating mutagenesis by 8-oxo-dG during translesion DNA synthesis. In DNA pol β Lys280 appears to reposition itself due to the carbonyl at the C8 position (19). This residue and Asp276 form van der Waals contact with the templating and incoming bases, respectively. In the high fidelity T7 DNA polymerase, 8-oxo-dG in the template makes contact with Lys536 at the C8 carbonyl position. This interaction is thought to keep the 8-oxo-dG rotamer in the anti position, thus limiting Hoogsteen base pairing with dATP (20). When 8-oxo-dG was modeled in the mutagenic syn conformation in T7 DNA pol, the C8 carbonyl group exhibited steric clashes with Tyr530 and Lys536 (20). Thus, the combined efforts of Lys536 and Tyr530 help to sterically attenuate against the syn conformation. These structural observations in T7 DNA polymerase are in agreement with biochemical evidence that indicates T7 DNA pol inserts dATP much less favorably than dCTP (20–23).
Bacillus stearothermophilus DNA pol, T7 DNA pol and DNA pol γ belong to the family A class of DNA polymerases. In human DNA pol γ, Tyr955 is structurally homologous to Tyr530 of T7 DNA polymerase. The highly conserved Tyr955 plays a crucial role in forming the hydrophobic pocket that accommodates and stabilizes the incoming dNTP. Tyr955 with Glu895 and Tyr951 help provide faithful nucleotide selectivity, the permissive incorporation of dideoxyribonucleotides, and the discrimination against ribonucleotides that characterize pol γ (24).
Here we analyzed DNA synthesis efficiency and fidelity of wild type and Y955C human DNA pol γ opposite template 8-oxo-dG and during incorporation of 8-oxo-dGTP. Following the kinetics studies, we developed a model structure of both wild type and Y955C forms of human pol γ with the mutagenic 8oxoG lesion in the active site and incorporation of different dNTPs. Human pol γ displayed limited ability to insert opposite 8-oxo-dG in the template as well as limited incorporation of 8-oxo-dGMP. This suggests that, although 8-oxo-dG levels are widely believed to be higher in mitochondrial DNA, the human pol γ largely prevents these adducts from becoming mutations by limiting incorporation opposite this lesion. However, the Y955C substitution allows more room in the active site for the formation of the dATP:8-oxo-dG Hoogsteen base pairing.
Results
Insertion of 8-oxo-dGTP into DNA by pol γ
We first tested the ability of the wild type and Y955C mutant pol γ to incorporate 8-oxo-dGTP into DNA by performing primer extension reactions and analyzed the products by gel electrophoresis. 8-oxo-dG can hydrogen-bond with dC through normal Watson-Crick pairing but can also form a Hoogsteen base pair with the dA base when the 8-oxo-dG base is rotated into the syn conformation. The incorporation of 8-oxo-dGTP into DNA opposite template dC or template dA with oligonucleotide primer templates was analyzed. We used the exonuclease deficient DNA pol γ in this assay to avoid degradation of the primer by the proofreading function and to simplify interpretation of results. Additionally, it was recently determined that the human pol γ exonuclease does not recognize 8-oxo-dG paired with either dC or dA (25).
Rates were determined as the fraction of primer extended by one nucleotide per unit time and Michaelis-Menten kinetic constants were determined by plotting the rate as a function of 8-oxo-dGTP or dGTP concentration (26). Steady-state assumptions allow the overall efficiency of each enzyme on a homopolymeric template to be estimated as kcat/Km(dTTP) (also referred to as the specificity factor), a parameter comparable to the pre-steady-state indicator of enzymatic efficiency kpol/Kd(dNTP) (27,28).
Table 1 compares the results of these steady-state kinetic parameters. Comparing the insertion of 8-oxo-dGTP with dGTP opposite dC in the template revealed that the wild-type pol γ discriminates against 8-oxo-dGTP insertion by 10 000-fold (kcat/Km = 70 min−1μM−1 for dGTP insertion compared to 0.007 min−1μM−1 for 8-oxo-dGTP insertion). Hanes et al. (25) recently reported a similar analysis of 8-oxo-dGTP insertion with the human mitochondrial DNA polymerase using pre-steady-state kinetics. They also determined that the discrimination by pol γ on 8-oxo-dGTP insertion was 10 000-fold. As stated above, this equivalent value determined by two different kinetic methods underscores the validity in using steady-state kinetics in evaluating the efficiency of insertion by pol γ. Compared to dGTP insertion opposite dC, the specificity of insertion of dGTP opposite dA was 14 000-fold less. The discrimination of insertion of 8-oxo-dGTP opposite dA was determined to be 50 000, or 5-times less likely than insertion opposite the correct pairing dC. Thus, when 8-oxo-dGTP is inserted into DNA 80% of the time this is correctly paired with dC and 20% of the time, it is incorrectly inserted opposite dA.
Table 1.
Incorporation of dGTP/8-oxo-dGTP by wild-type exo- and Y955C pol γ opposite dA or dC in the template
| Exo- enzyme | A in the template | C in the template | ||||||
|---|---|---|---|---|---|---|---|---|
| Km (μM) | kcat (min −1) | kcat/Km (min μM)−1 | DFa | Km (μM) | kcat (min −1) | kcat/Km (min μM)−1 | DFa | |
| Wild-type | ||||||||
| dGTP | 70 (±11.98) | 0.36 (±0.007) | 0.005 | 1.4×l04 | 0.04 (±0.01) | 2.8 (±0.77) | 70 | 1 |
| 8-oxo-dGTP | 84 (±14.09) | 0.12 (±0.007) | 0.0014 | 4.8×l04 | 43 (±11) | 0.29 (±0.04) | 0.007 | 1.0×104 |
| Y955C | ||||||||
| dGTP | 264 (±68.3) | 0.042 (±0.005) | 0.0002 | 90 | 9.5 (±1.24) | 0.014 (±0.004) | 0.018 | 1 |
| 8-oxo-dGTP | 323 (±60.1) | 0.07 (±0.014) | 0.0002 | 90 | 185 (±32.4) | 0.08 (±0.012) | 0.0004 | 45 |
DF, discrimination factor (kcat/Km)dGTP with the C template/(kcat/Km)dGTP or 8-oxo-dGTP with either template (Discrimination factor for the incorporation of 8-oxo-dGTP or incorrect nucleotide compared to incorporation of normal dGTP across dC for each enzyme set). All reactions were done in the presence of the accessory subunit.
The Y955C mutant pol γ has previously been determined to have poor rates of catalysis and error prone DNA synthesis due to an increase in Km(dNTP) (10,11). In the gel-based incorporation assay, the Y955C pol γ also demonstrated a reduced ability to incorporate dCTP in which the kcat/Km for dCTP was reduced nearly 4000-fold (kcat/Km= 0.018 (min μM)−1). The kcat/Km of 8-oxo-dGTP insertion by the Y955C pol γ was further reduced by another 45-fold. Compared to WT, the ability of Y955C pol γ to discriminate against 8-oxo-dGTP relative to dGTP was reduced by over 200-fold [discrimination factor (DF) for WT was 10 000 while the DF for Y955C was only 45]. This lack of discrimination was also evident for inserting either dGTP or 8-oxo-dGTP opposite dA in the template. (DF was 90 for both). Thus, the poor specificity and loss of discrimination displayed by the Y955C pol γ emphasizes the critical role of this side chain not only in normal replication fidelity but also in discrimination against 8-oxo-dGTP insertion.
Insertion opposite template 8-oxo-dG
To assess the preference of dNTP inserted by human DNA pol γ opposite 8-oxo-dG in template DNA, primer extension reactions were carried out with oligonucleotides in which 8-oxo-dG resided in the template. Reactions were carried out either in the presence of all four dNTPs or a single dNTP (dATP or dTTP or dGTP or dCTP) and products of the reaction visualized by PhosphorImage analysis after separation by polyacrylamide gel electrophoresis. The reactions were performed either with the pol γ catalytic subunit alone (Fig. 2A), or with the pol γ holoenzyme (catalytic subunit plus the accessory subunit—Fig. 2B). The total dNTP concentration was 200 μM.
Figure 2.
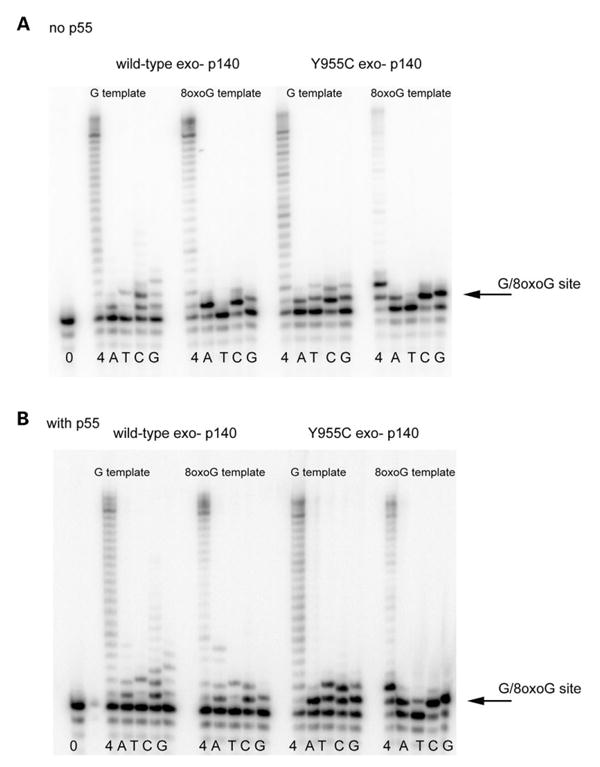
Translesion synthesis past 8-oxo-dG by wild type and Y955C. (A) Incorporation of depicted nucleotides opposite a normal dG or a 8-oxo-dG lesion in the template by the wild type or Y955C mutant pol γ in the absence of the p55 accessory subunit. (B) Incorporation of depicted nucleotides opposite a normal dG or a 8-oxo-dG lesion in the template by the wild type or Y955C mutant pol γ in the presence of the p55 accessory subunit. In both panels, reactions were performed in the presence of all four dNTPs (4) or by the single dNTPs (dATP or dTTP or dGTP or dCTP). The total dNTP concentration was 200 μM. Products of the reactions were separated through a 15% polyacrylamide sequencing gel and analyzed as described in the ‘Materials and Methods’. The arrows depict the site of either the dG or 8-oxo-dG lesion.
Without the accessory subunit (which is known to increase enzyme processivity but decrease fidelity slightly), and in the presence of all four dNTPs, DNA pol γ was capable of efficient primer extension beyond the 8-oxo-dG site in the template. Reactions performed in the presence of only one dNTP revealed the following efficiency of dNTPs incorporation opposite 8-oxo-dG dATP ≈ dCTP ≫ dGTP. No dTTP incorporation was observed (Fig. 2A). These results were qualitatively consistent with incorporation opposite 8-oxo-dG by the Xenopus pol γ (17).
To test the consequence of the Y955C substitution on incorporation opposite dG or 8-oxo-dG in the template, the identical reaction was carried out with the Y955C mutant pol γ, except that 10-fold more enzyme was utilized in the reactions due to the lower activity of this enzyme. The Y955C mutant pol γ was unable to efficiently produce full-length product of primer extension on the 8-oxo-dG containing template and a strong replication block was observed one nucleotide beyond the 8-oxo-dG site. When only one dNTP was present, the efficiency of incorporation was as follows: dCTP > dGTP ≫ dATP. Again, no dTTP incorporation was observed (Fig. 2A). Thus, in the absence of the accessory subunit, the Y955C substitution appeared to switch the specificity of incorporation by reducing the relative dAMP incorporation while at the same time increasing dGMP incorporation opposite the 8-oxo-dG lesion.
The presence of the accessory subunit (p55) resulted in the ability to incorporate dTTP opposite 8-oxo-dG, by both wild-type pol γ and its Y955C form (Fig. 2B).
Also, in the presence of p55, the Y955C was able to extend the primer more efficiently, although the strong replication block one base pair beyond 8-oxo-dG site in the template was still observed.
Specificity of nucleotide insertion opposite 8-oxo-dG in template DNA
The experiment described in Figure 2 results from the use of saturating dNTP concentrations in order to detect incorporation events. To evaluate the in vitro selectivity of nucleotide insertion by DNA pol γ opposite 8-oxo-dG, we determined the steady-state kinetic parameters of incorporation of a single nucleotide onto a 3′-terminally matched primer template for wild type and the Y955C mutant pol γ. In the presence of limiting but increasing amounts of either next correct or non-cognate dNTP, the amount of incorporation opposite either dG or 8-oxo-dG was determined (Table 2). With the normal dG template, the WT enzyme had high discrimination (between 3300 and 30 000-fold) against the incorrectly paired incoming nucleotides similar to what has been observed for human pol γ (29). When the WT enzyme was assayed for incorporation opposite 8-oxo-dG in the template, the only significant insertion detected was dCMP. Relative to incorporation of dCMP opposite a normal dG in the template, incorporation of dCMP across an 8-oxo-dG occurs only 4.7% of the time [kcat/Km of 4.3 (min μM)−1 for 8-oxo-dG template versus 305 for dG template]. This can be inferred to mean that incorporation opposite the 8-oxo-dG lesion is blocked 95% of the time. Incorporation of dAMP, dGMP and dTMP was also detected opposite 8-oxo-dG but this incorporation appeared to roughly follow the discrimination opposite normal dG. However, the insertion of both dAMP and dGMP was increased nearly 10-fold opposite 8-oxo-dG when compared with the insertion of dCMP (compare the DF for dAMP and dGMP with 8-oxo-dG and dG in the template). Thus, the wild type was less accurate when incorporating opposite 8-oxo-dG when compared with the normal nucleotide even though the absolute specificity of incorporation was generally less opposite 8-oxo-dG by nearly 10-fold.
Table 2.
Insertion/misinsertion of dNTPs opposite 8-oxo-dG by wild-type exo- and the Y955C pol γ
| Enzyme | dG in the template | 8-oxo-dG in the template | ||||||
|---|---|---|---|---|---|---|---|---|
| Km (μM) | kcat (min −1) | kcat/Km (min μM)−1 | DFa | Km (μM) | kcat (min −1) | kcat/Km (min μM)−1 | DFa | |
| Wild-type | ||||||||
| A | 41.8 (±9.2) | 0.07 (±0.01) | 0.018 | 1.7×l04 | 54.0 (±5.3) | 0.31 (±0.06) | 0.0057 | 2.2×103 |
| T | 18.7 (±3.7) | 0.18 (±0.02) | 0.096 | 3.3×103 | 76.0 (±12.4) | 0.069 (±0.01) | 0.0009 | 1.6×104 |
| C | 0.02 (±0.004) | 6.1 (±1.7) | 305 | 1 | 0.3 (±0.02) | 4.3 (±0.2) | 14.3 | 1 |
| G | 53.1 (±10.2) | 0.53 (±0.06) | 0.01 | 3.0×104 | 62 (±5.4) | 0.43 (±0.08) | 0.0069 | 2.1×103 |
| Y955C | ||||||||
| A | 232 (±21.5) | 0.51 (±0.07) | 0.0022 | 33 | 280 (±15.0) | 0.34 (±0.02) | 0.0012 | 24 |
| T | 303 (±17.3) | 0.14 (±0.02) | 0.0005 | 150 | 1530 (±171) | 0.08 (±0.01) | 0.00005 | 590 |
| C | 6.2 (±0.61) | 0.46 (±0.05) | 0.074 | 1 | 18 (±2.7) | 0.52 (±0.07) | 0.029 | 1 |
| G | 257 (±35.3) | 0.19 (±0.03) | 0.0007 | 105 | 350 (±41.5) | 0.20 (±0.03) | 0.00057 | 50 |
DF, discrimination factor (kcat/Km)correct dNTP/(kcat/Km)incorrect dNTP; values compared within each of the following categories: (i) exo- wt p140 utilizing G template, (ii) exo- wt p140 utilizing 8-oxo-dG template, (iii) exo-Y955C utilizing G template and (iv) exo-Y955C utilizing 8-oxo-dG template. All reactions were done in the presence of the accessory subunit.
We next compared incorporation by Y955C pol γ of normal nucleotides opposite the cognate dG or 8-oxo-dG in the template. As expected for the Y955C pol γ (10), the enzyme misincorporates nucleotides more readily than wild type on the normal dG template. Compared to the WT enzyme, the Y955C enzyme misincorporated dAMP, dTMP and dGMP 500-, 220- and 285-fold more readily (Table 2). Although the activity and incorporation of the Y955C pol γ is low, the Y955C does incorporate dCMP opposite the 8-oxo-G template 39% of the time (compare 0.029 with 0.074) relative to normal dG in the template (Table 2). This is a 8-fold relative increase of dCMP insertion compared with the WT enzyme. The incorporation of the mismatched nucleotides opposite 8-oxo-dG was also increased relative to the WT enzyme. The misincorporation of dAMP opposite 8-oxo-dG was highest relative to the correctly matched dCMP incorporation with a DF of only 24. This misincorporation by the Y955C enzyme represents a 100-fold increase of dAMP misincorporation compared to the WT enzyme (compare the DF of 2200 for WT and 24 for Y955C). The misincorporation of dGMP was also high opposite 8-oxo-dG with Y955C and nearly 42-fold greater than the WT enzyme (compare the DF of 50 by Y955C with 2100 by WT). Unlike the WT situation, the misincorporation was not significantly increased opposite the 8-oxo-dG template. This may represent the already poor fidelity displayed by the Y955C and the inability to discriminate between correct or incorrect nucleotide opposite either a normal or damaged base. Thus, the Y955C enzyme appeared to display enhanced incorporation of nucleotides opposite the 8-oxo-dG lesion even though the Y955C misincorporates readily opposite either the normal or 8-oxo-dG lesion.
Molecular modeling of 8-oxo-dG template in pol γ
To understand at the molecular level, the consequence of the Y955C substitution on the enhanced incorporation opposite 8-oxo-dG and the ability to readily misincorporate opposite this lesion, we probed the three-dimensional structural model of pol γ. Based on solved T7 crystal structures reported by Brieba et al. (20), structural models of wild type pol γ and Y955C pol γ were developed, demonstrating incorporation of dCTP or dATP opposite 8-oxo-dG in the template. For comparison to the situation in DNA pol γ, Figure 3A displays the T7 DNA polymerase with 8-oxo-dG in the template with the discriminating residues (Tyr530 and Lys536) labeled. Breiba et al. (20) previously demonstrated that in the wild type T7 pol structure the rotation of the 8-oxo-dG base from anti to syn conformation is blocked by an allosteric gate composed of Lys536 facing the C8 carbonyl on one side and Tyr530 on the other side of the 8-oxo-dG template base (Fig. 3A). Substitution of Lys536 to Ala relieves this allosteric interaction on one side allowing formation of the mutagenic syn conformation (Fig. 3B) (23). This situation leads to the incorporation of dATP via Hoogsteen basepairing with the 8-oxo-dG base (23).
Figure 3.
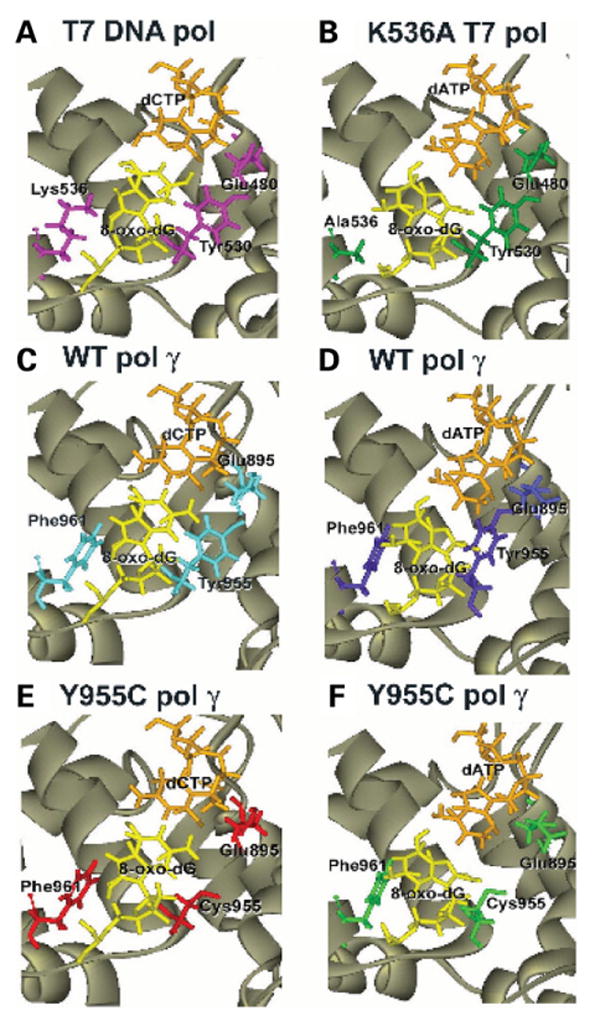
Modeled structure of the wild type, Y955C pol γ or the T7 DNA pol structure with 8-oxo-dG in the template. (A) T7 DNA pol with incoming dCTP. (B) T7 DNA pol with incoming dATP. (C) Wild-type pol γ with incoming dCTP. (D) Wild-type pol γ with incoming dATP. (E) Y955C mutant pol γ with incoming dCTP. (F) Y955C mutant pol γ with incoming dATP. The 8-oxo-dG is labeled in each panel and is shown in the anti-conformation in A, C and E, and shown in the syn-conformation in B, D, and F. The structure of the wild-type T7 pol with 8-oxo-dG in panel A was derived from PDB: 1TKO (20) with Lys536 and Tyr530 labeled. The structure of the K536A derivative of T7 pol in B was derived from PDB: 1ZYQ (23) with Ala536 labeled.
The T7 pol residues Tyr530 and Lys536 correspond to Tyr955 and Phe961, respectively, in human pol γ as shown in Figure 3A and C. Tyr955 is located 2.5 angstroms from the DNA base with which it stacks. With a normal template base, the –OH group of the tyrosine is positioned within hydrogen bonding interaction with the template base (11). The Tyr955 –OH also hydrogen bonds with Glu895 to coordinate the glutamate side chain in discrimination against ribonucleotides (24). The side chains of Tyr955, Glu895 and Tyr951 form a hydrophobic pocket that accommodates and stabilizes the incoming dNTP. The observed hydrogen-bonding interactions between these residues help to explain the high fidelity of nucleotide incorporation by pol γ (29). Modeling of 8-oxo-dG into the WT pol γ with incoming dCTP was easily accommodated into the active site with no apparent steric clashes or Van der Waals overlaps (Fig. 3C). Note that the rotamer of the Lys536 in T7 (Fig. 3A) and pol γ Phe961 (Fig. 3C) are oriented with the same respect to the 8-oxo-dG. The Tyr955 side chain was 2.7 angstroms from 8-oxo-dG as measured in this 8-oxo-dG:dCTP model structure.
We next modeled dATP into the WT pol γ active site with 8-oxo-dG as the template base. These two bases interact via Hoogsteen base-pairing when 8-oxo-dG assumes the syn rotamer. In the wild-type model, replacement of the incoming dCTP with dATP, with 8-oxo-dG as the template base in the syn conformation shows close steric clashes and Van der Waals overlaps with the Tyr955 side chain (Fig. 3D). This steric interaction appears to prevent the dATP insertion by attenuating the syn conformation of the 8-oxo-dG base.
Mutation of this Tyr955 to Cys disrupts the stacking interactions between the guanine template base and the aromatic ring of the tyrosine and also alters the hydrophobic environment offered by the phenyl ring (11). This aromatic ring, either phenylalanine or tyrosine, is critical for selectivity of incorporating antiviral nucleoside analogs (24). Mutation to cysteine dramatically decreases fidelity of nucleotide incorporation (10) and exhibits high Km for nucleotides, low catalytic activity and low processivity (11). This change is associated with the most severe form of autosomal dominant PEO (7,11).
In the Y955C mutant pol γ model structure (Fig. 3E), the 8-oxo-dG template base with incoming dCTP appears similar to the wild-type model with 8-oxo-dG (compare with Fig. 3C). However, the substitution of cysteine not only weakens some of these interactions as seen with a normal template base but the smaller residue now allows for more room for the rotation of the 8-oxo-dG base. This extra room in the mutant Y955C active site now allows for dATP to base pair with the syn-conformation of the 8-oxo-dG base (Fig. 3F).
Superimposition of these models allows for the direct comparison between the T7 structure and the pol γ models (Fig. 4). Superimposition of the models with incoming dCTP shows no obvious structural hindrance to the dCTP:8-oxo-dG hydrogen bonding (Fig. 4A). However, examination of the WT pol γ model with incoming dATP shows close Van der Waals overlaps and steric hindrance between the Tyr955 and Phe961 and the syn conformation of the 8-oxo-dG template base (blue in Fig. 4B). But when either Tyr955 is substituted for Cys in human pol γ or Lys536 is substituted for Ala in T7 pol, then enough space exists for the syn conformation of the 8-oxo-dG template base (green and yellow in Fig. 4B). This orientation of the 8-oxo-dG can now bind with the incoming dATP by Hoogsteen base-pairing. Thus, the allosteric gate offered in the wild-type structure that prevents rotation of the 8-oxo-dG base by the combined efforts of Phe961 and Tyr955 side chains is lost in the Y955C situation.
Figure 4.
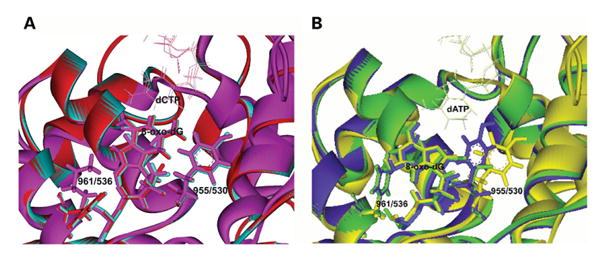
Superimposition of the T7 DNA polymerase structure and the human DNA pol γ model with 8-oxo-dG in the template and an incoming dCTP or dATP. (A) Superimposition of WT T7, WT pol γ and Y955C pol γ with incoming dCTP showing 8-oxo-dG in the anti-conformation. (B) Superimposition of K536A T7, WT pol γ and Y955C pol γ with incoming dATP showing 8-oxo-dG in the syn-conformation. The color scheme follows that shown in Fig. 3 and are as follows; WT pol γ with incoming dCTP (turquoise), Y955C pol γ with incoming dCTP (red) and WT T7 DNA pol with incoming dCTP (pink), WT pol γ with incoming dATP (blue), Y955C pol γ with incoming dATP (green) and K536A T7 pol (yellow). The amino acid residues are labeled by their amino acid number with the pol γ amino acid position preceding T7 pol. The structure of the wild type T7 pol with 8-oxo-dG was derived from PDB: 1TKO (20) with Lys536 and Tyr530 labeled. The structure of the K536A derivative of T7 pol was derived from PDB: 1ZYQ (23) with Ala536 labeled.
Discussion
Oxidative respiration in the mitochondria is thought to be the main source of endogenous oxidative stress and the generation of reactive oxygen species, which ultimately generates 8-oxo-dG as one of the main products. Because pol γ is the only known DNA polymerase in the mitochondria, the burden of dealing with this oxidized base by attenuating the incorporation of 8-oxo-dGMP and preventing incorrect synthesis past the 8-oxo-dG lesion once it is in DNA falls on pol γ. The mitochondria reduce the chance for oxidative mutagenesis through concerted action of the base excision repair machinery. The MTH1 gene product is imported in the mitochondria and nucleus and functions to sanitize 8-oxo-dGTP pools (30), thus reducing the chance for pol γ to encounter this modified nucleotide. If 8-oxo-dGTP is incorporated into mtDNA or oxidation of the base occurs in DNA, the OGG1 and MUTYH gene products function in the mitochondria to excise 8-oxo-dG opposite cytosine or opposite adenine, respectively (31). However, if the 8-oxo-dG in DNA escapes this repair pathway pol γ must deal with the lesion. We demonstrate here that incorporation opposite a 8-oxo-dG template base by the wild-type human DNA pol γ holoenzyme was blocked 95% of the time and that the remaining 5% nucleotide incorporation occurred in an error-free manner. Nonetheless, insertion of the other three nucleotides was detected and followed the general order: dCTP > > > dATP > dGTP≫ dTTP. This is in agreement with earlier work with the Xenopus pol γ and more recent work with the human enzyme (17,25). DNA pol γ belongs to the family A class of DNA polymerases which includes T7 DNA polymerase. The relative error-free incorporation opposite 8-oxo-dG followed a similar trend of bypass and error-free replication past 8-oxo-dG by T7 DNA polymerase (20–22).
Previously, the error rate of the chick embryo pol γ with equal molar 8-oxo-dGTP and dNTPs (each at 100 μM) was determined to be 3.7 × 10−3 with the M13 gap filling assay followed by transfection into E. coli (32). We found that pol γ incorporated 8-oxo-dGTP only 1 in every 10 000 dGMP incorporation events opposite dC. This level of discrimination is based on equal molar concentrations of 8-oxo-dGTP and dGTP which suggests that DNA mutagenesis should be even lower at the expected low physiological concentrations of 8-oxo-dGTP relative to dGTP levels. However, recent pre-steady-state kinetic analysis suggests that the level of incorporation of 8-oxo-dGTP opposite dA may be even higher opposite dC (25). If the 8-oxo-dGTP does get incorporated opposite a template dA, then it is likely to be fixed as a mutations because this mispair appears to evade proofreading by the pol γ exonuclease (25,33).
Examination of the origins of mtDNA mutations suggests that most of the fixed mutations may occur from spontaneous DNA replication errors and less from DNA damage (34). The inefficient incorporation efficiencies seen in our study by the wild-type pol γ support this hypothesis. This suggests that the endogenous base excision glycosylases, OGG1 and MUTYH, as well as the MTH1 activity to remove 8-oxo-dGTP from dNTP precursor pools, are sufficient to handle the normal load of oxidized lesions. However, studies in yeast and mammalian cell systems do indicate that a defect in one or more steps in these repair pathways results in enhanced mutation rate (35–38). Thus, unlike the nucleus which has redundant base excision repair pathways, mitochondria do not and any genetic mutation that attenuates this process would be predicted to accelerate mtDNA mutations as well as aging. This is reinforced by animal models such as the MTH1-null mice which exhibit a greater accumulation of 8-oxo-dG in mtDNA when compared with nuclear DNA (39) or OGG1 null mice that show accumulation of mtDNA mutations (40,41). A naturally occurring mutation in the POLG gene could also show a similar effect of enhanced susceptibility to oxidative lesions. The Y955C disease mutation appears to fit this scenario.
Structural implications of the Tyr to Cys substitution
Substitution of Tyr955 with cysteine severely diminished the overall enzyme performance while at the same time leading to lower selectivity when performing synthesis opposite 8-oxo-dG. The order of efficiency of dNTP incorporation was similar to that of the wild-type enzyme, dCTP ≫ dATP > dGTP > dTTP, but incorporation events were higher compared to the normal nucleotide. Substitution of tyrosine to the smaller cysteine increased misinsertions opposite 8-oxo-dG lesion, when compared with incorporation opposite dG in the template. This is consistent with what we described previously for this disease variant (10).
In DNA pol β, the interaction of Lys280 and Tyr271 with the 8-oxo-dG base seem to fix the rotamer in the anti conformation to enable error-free bypass of the lesion (19). The unfavorable interactions of Tyr271 and the incoming dATP form a steric gate that prevents the mutagenic syn conformation of the 8-oxo-dG residue and help explain why pol β prefers to insert dCTP opposite 8-oxo-dG (42). Recently, Brieba et al. (20,23) determined in T7 DNA polymerase that Lys536 is important in attenuating the mutagenic potential of 8-oxo-dG in the template. The Lys536 side chain is normally in a position to prevent the syn conformation of the 8-oxo-dG base, which confers the mutagenic dA:8-oxo-dG Hoogsteen base pair. Replacement of Lys536 by Ala removes the steric block and the 8-oxo-dG base can rotate into the syn conformation. This substitution caused T7 DNA pol to make dA:8-oxo-dG mutations more readily than the wild-type enzyme (23).
The analogous position of T7 DNA pol Lys536 in the human pol γ is Phe961. Like the Lys536 residue in T7 DNA pol, the Phe961 in the human pol γ model appears to be in close contact to the 8-oxo-dG template. As in T7 pol, this larger Phe residue most likely attenuates against the syn conformation which prevents the dAMP:8-oxo-dG Hoogsteen base pairing. On the opposite side of the 8-oxo-dG base, the Tyr955 also helps attenuate against the syn rotamer when dA attempts to bind via Hoogsteen hydrogen bonding. In the wild-type structure, this is seen as a steric clash between the 8-oxo-dG base and the Tyr955 side chain. These close Van der Waals interactions account for the 2200-fold discrimination against dATP insertion opposite 8-oxo-dG in the wild-type enzyme. The molecular model of the pol γ demonstrates that substitution of Tyr to Cys permits greater flexibility and space in the active site which can easily accommodate the syn rotamer and the dATP:8-oxo-dG Hoogsteen base pair. This is revealed biochemically by a 100-fold loss of discrimination from 2200 to 24. The relaxation of nucleotide incorporation by the Y955C was not limited to incorporation opposite 8-oxo-dG in the template. The Y955C pol γ also displayed over a 200-fold increase in incorporating 8-oxo-dGTP relative to dGTP opposite dC in the template. Thus, the Y955C pol γ displays enhanced 8-oxo-dGTP insertion as well as increased incorporation opposite 8-oxo-dG relative to the normal nucleotides.
Biological consequence of the Y955C and 8-oxo-dG
Although the Y955C enzyme predominantly causes replication stalling (11,43), our results suggest that patients harboring the Y955C mutation have an added complication and are more likely to suffer from 8-oxo-dG mutagenesis as compared to an individual with wild-type pol γ. Our data offer a molecular and kinetic explanation for part of this oxidative mutagenesis. It is unclear whether the inability of the Y955C pol γ to discriminate against oxidative lesions has a role in the parkinsonism observed in these patients. Of the >100 disease mutations in POLG, adPEO with parkinsonism has been associated with the G737R, Y831C, R853W, Y955C and A1105T mutations (8,44,45). In one study, four out of seven families with POLG mutations causing PEO with parkinsonism contained the Y955C mutation (8). Y831C was also found in non-parkinsonism control groups suggestive of other factors needed in conjunction with the Y831C mutation to cause parkinsonism (46). The link between Parkinson disease and mitochondrial dysfunction in the substantia nigra has been previously suggested (47) and mtDNA deletions were recently shown to accumulate to a higher degree in the dopaminergic neurons of Parkinson patients when compared with control subjects (48,49). Furthermore, inhibition of the mitochondrial complex I by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or rotenone is well established (50). Inhibition of complex I by MPTP or rotenone promotes electron leakage increasing the formation of reactive oxygen species and leads to Parkinson disease. In addition, the substantia nigra neurons have been shown to have a high oxidative capacity and oxidative stress (51). Several studies have found significant increases of 8-oxo-dG in mtDNA from the substantia nigra and dopaminergic neurons of Parkinson patients (52–56). These observations suggest that mitochondrial dysfunction and oxidative stress is a strong component in Parkinson and parkinsonism. The benefits of Coenzyme Q10 as an electron acceptor and potent antioxidant have shown promise as a therapy in early Parkinson disease (57) and help to strengthen the role of oxidative stress in these diseases. Since mtDNA deletions are characteristic of generalized PEO, it is tantalizing to speculate that the additional inability of the DNA pol γ to discriminate against oxidative lesions may partially account for the parkinsonism symptoms.
In yeast, the equivalent substitution of Tyr to Cys at this position (Y757C) in the MIP1 gene not only causes depletion of mtDNA but also displayed elevated mtDNA damage (13). Furthermore, it has been shown recently that many of the phenotypical results associated with the Y757C mutations in the yeast MIP1 gene can be partially rescued by antioxidants (14). Interestingly, in the mouse transgenic model that overexpresses the Y955C human POLG cDNA we found elevated levels of 8-oxo-dG in heart mtDNA (12). Collectively, these reports suggest that patients harboring this mutation may have elevated levels of oxidative stress and DNA damage due to 8-oxo-dG incorporation and/or translesion synthesis. The parkinsonism symptoms associated specifically with the Y955C mutation may be partly due to the inability of this mutant polymerase to discriminate against oxidative lesions. Finally, anti-oxidative therapy may be beneficial for these patients in alleviating some of the symptoms.
Materials and Methods
Enzymes
The exonuclease deficient form of wild type human DNA pol γ, as well as Y955C mutant form of the enzyme, were purified to homogeneity from baculoviral-infected insect cells as described (10,58). The accessory subunit (p55) was purified to homogeneity from E. coli, and the heterotrimeric forms of the polymerase were reconstituted as previously described (59).
Oligonucleotides
Following oligonucleotides were purchased from The MIDLAND Certified Reagent Company: primers (25-mers) 5′-AATTTCTGCAGGTCGACTACATAGG-3′ and 5′-AATTTCTGCAGGTCGACTACATACC-3′ and templates (45-mers). 3′-TTAAAGACGTCCAGCTGATGTATCCGATTGGGCCAT GGCTCGACC-5′ (‘G’ template), 3′-TTAAAGACGTCCAG CTGATGTATCC8-oxo-dGATTGGGCCATGGCTCGACC-5′ (‘8-oxo-dG’ template), 5′-CCAGCTCGGTACCGGGTTGACC TATGTAGTCGACCTGCAGAAATT-3′(‘C’ template) and 5′-CCAGCTCGGTACCGGGTTACGGTATGTAGTCGACCT GCAGAAATT-3′ (‘A’ template).
DNA polymerase reactions
DNA pol γ reactions (10 μl final volume) contained 25 mM Hepes-OH (pH 7.5), 5 mM MgCl2, various concentrations of the four dNTPs, 2 mM 2-mercaptoethanol, 50 μg/ml bovine serum albumin, 0.1 mM EDTA, 1 pmol 5′-32P-labeled 25-mer annealed to 1.1 pmol of 45-mer template and various concentrations of dNTP similar to what has been previously described (11,60). Reactions were started by addition of DNA pol γ (30 fmol of the wild-type enzyme and 300 fmol of its Y955C form) in complex with the accessory subunit (1:2 molar ratio). Samples were incubated for 10 min at 37°C. For kinetic studies of each of the four dNTPs incorporation opposite 8-oxo-dG in the template, as well as incorporation of 8-oxo-dGTP opposite A or C by wild-type and Y955C form of DNA pol γ, incubations of 5 min for the wild-type enzyme and 10 min for its Y955C form were performed. Reactions were stopped by addition of 10 μl of 95% aqueous formamide containing 25 mM EDTA and gel sequencing dye solution, followed by heating in a boiling water bath.
Michaelis-Menten kinetic constants for the incorporation of 8-oxo-dGTP or dGTP were determined by non-linear plot of rate versus dNTP concentration as described (26). The apparent kcat values represent the aggregate contributions of the competing association (kon), catalytic (kpol), and dissociation (koff) rate constants. The relative contributions of kon and koff are small for unrestricted DNA synthesis, so kpol values are closely approximated by apparent kcat values. Because kcat/Km(dNTP) ≈ kpol/Kd(dNTP), Km(dNTP) values are a good indicator of nucleotide affinity during unrestricted DNA synthesis, even though Kd(dNTP) values are not strictly accessible through steady-state measurements. In contrast, during distributive or otherwise restricted DNA synthesis, dissociation of the enzyme from the primer-template becomes rate limiting, and koff becomes the principle component of the measured kcat value. Now kcat ≠ kpol, and Km(dNTP) values are no longer good indicators of nucleotide affinity. In fact, since Km(dNTP) ≈ kcat(Kd(dNTP)/kpol), one can easily see how Km(dNTP) could increase due to a decrease in kpol with no change in Kd(dNTP). Thus, restriction of DNA synthesis limits the utility of kcat and Km(dNTP) values as independent parameters. However, with saturating primer-template concentrations the ratio of kcat/Km(dNTP) does not change upon restriction of DNA synthesis (61), and kcat/Km(dNTP) remains a valid parameter for comparing mutant derivatives of pol γ. Similarly, since steady-state parameters do not account for the expected differences in Kd(dNTP) for normal nucleotides and 8-oxo-dGMP, comparisons of differential insertion by a given enzyme are best described as, DF = (kcat/Km)8oxoGTP /(kcat/Km)dNTP. Individual kinetic parameters are presented with the relevant kcat/Km(dNTP) ratios and DF values.
Incorporation analysis
Reaction products were separated on 15% polyacrylamide/7 M urea gels. Following electrophoresis, the gels were exposed to a PhosphorImager screen and analyzed on a Storm 860 PhosphorImager (Molecular Dynamics). Radioactive bands were quantified with NIH Image software (version 1.63). Linear least-squares curve fitting of double-reciprocal plots yielding kcat and Km values were calculated using PRISM software for a nonlinear regression. Relative values of kcat/Km were used to calculate misincorporation efficiency and 8-oxo-dGTP incorporation opposite A or C in the template.
Molecular modeling
The wild-type pol γ and Y955C mutant pol γ model structures, with native dG:dCTP nucleotide insertion, were developed based on coordinates from the solved T7 polymerase structure coordinates with native dG:dCTP insertion (PDB entry: 1T8E) (20), using the Schrodinger Prime version 1.2 Homology Modeling Software Suite. A structural alignment of family A polymerase crystal structures was performed using the DALI structural alignment program (62). The sequences of Thermus aquaticus DNA polymerase I (Klentaq), Bacillus stearothermophilus DNA polymerase I, E. coli DNA polymerase I (Klenow) and T7 DNA polymerase (PDB structures: 2KTQ, 3DBP, 1D8Y, and 1T7P, 1T8E respectively) were utilized in the structural alignment and the pol γ sequence was aligned to the sequences of T7 and to the other family A polymerase structures using the T-Coffee sequence alignment algorithm (63).
Models of the wild type and Y955C mutant pol γ structure with mutagenic 8-oxo-7,8-dihydro-2′–deoxyguanosine (8-oxo-dG) lesions: 8-oxo-dG:dCTP, insertion, 8-oxo-dG:dC post insertion and 8-oxo-dG:dATP post insertion, were also developed based on the coordinates for the reported structures for T7 polymerase (PDB entries 1TK0, 1TKD and 1TK8, respectively) (20). Energy minimization and model structure refinement were performed using the Schrodinger Prime protein-optimized OPLS2000 all-atom force field with a Surface Generalized Born continuum solvation model. Protein structure validation checks were performed using the program WHATIF (64,65).
Models of the wild type and Y955C mutant pol γ structure with mutagenic 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dG):dATP insertion were developed based on the crystal structure of the K536A T7 mutant (PDB entry: 1ZYQ) (23). This pol γ model with dATP insertion opposite the lesion was developed using the same sequence alignment as the previous models. Modeler 9.0 Protein Homology Modeling algorithm (66) as implemented in the Accelrys Discovery Studio 1.7 modeling software package was used to develop the wild type and Y955C mutant pol γ 8-oxo-dG:dATP model. The Protein Health implementation within the Accelrys DS 1.7 software package of the Profiles-3D method (67) was used to verify the quality of the model along with the DOPE energy method (68). In all the pol γ model structures, there are three major loop insertions relative to the T7 DNA polymerase structures, between residues 916–925, 1005–1023 and 1053–1094. These loop insertions do not involve residues involved in catalysis and do not structurally impact the configuration of the polymerase active site.
Acknowledgments
We thank Drs Zachary Pursell and Thomas Kunkel for critically reading this manuscript.
Footnotes
FUNDING: Division of Intramural Research, National Institute of Environmental Health Sciences, NIH, DHHS.
Conflict of Interest statement. None declared.
References
- 1.Kaguni LS. DNA polymerase gamma, the mitochondrial replicase. Annu Rev Biochem. 2004;73:293–320. doi: 10.1146/annurev.biochem.72.121801.161455. [DOI] [PubMed] [Google Scholar]
- 2.Graziewicz MA, Longley MJ, Copeland WC. DNA polymerase gamma in Mitochondrial DNA Replication and Repair. Chem Rev. 2006;106:383–405. doi: 10.1021/cr040463d. [DOI] [PubMed] [Google Scholar]
- 3.Longley MJ, Graziewicz MA, Bienstock RJ, Copeland WC. Consequences of mutations in human DNA polymeraseγ. Gene. 2005;354:125–131. doi: 10.1016/j.gene.2005.03.029. [DOI] [PubMed] [Google Scholar]
- 4.Hudson G, Chinnery PF. Mitochondrial DNA polymerase-gamma and human disease. Hum Mol Genet. 2006;15 Spec no 2:R244–R252. doi: 10.1093/hmg/ddl233. [DOI] [PubMed] [Google Scholar]
- 5.DiMauro S, Davidzon G, Hirano M. A polymorphic polymerase. Brain. 2006;129:1637–1639. doi: 10.1093/brain/awl169. [DOI] [PubMed] [Google Scholar]
- 6.Van Goethem G, Dermaut B, Lofgren A, Martin JJ, Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet. 2001;28:211–212. doi: 10.1038/90034. [DOI] [PubMed] [Google Scholar]
- 7.Lamantea E, Tiranti V, Bordoni A, Toscano A, Bono F, Servidei S, Papadimitriou A, Spelbrink H, Silvestri L, Casari G, et al. Mutations of mitochondrial DNA polymerase gamma are a frequent cause of autosomal dominant or recessive Progressive External Ophthalmoplegia. Ann Neurol. 2002;52:211–219. doi: 10.1002/ana.10278. [DOI] [PubMed] [Google Scholar]
- 8.Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, Chalmers RM, Oldfors PA, Rautakorpi I, Peltonen PL, Majamaa PK, et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet. 2004;364:875–882. doi: 10.1016/S0140-6736(04)16983-3. [DOI] [PubMed] [Google Scholar]
- 9.Pagnamenta AT, Taanman JW, Wilson CJ, Anderson NE, Marotta R, Duncan AJ, Bitner-Glindzicz M, Taylor RW, Laskowski A, Thorburn DR, et al. Dominant inheritance of premature ovarian failure associated with mutant mitochondrial DNA polymerase gamma. Hum Reprod. 2006;21:2467–2473. doi: 10.1093/humrep/del076. [DOI] [PubMed] [Google Scholar]
- 10.Ponamarev MV, Longley MJ, Nguyen D, Kunkel TA, Copeland WC. Active site mutation in DNA polymerase gamma associated with progressive external ophthalmoplegia causes error-prone DNA synthesis. J Biol Chem. 2002;277:15225–15228. doi: 10.1074/jbc.C200100200. [DOI] [PubMed] [Google Scholar]
- 11.Graziewicz MA, Longley MJ, Bienstock RJ, Zeviani M, Copeland WC. Structure-function defects of human mitochondrial DNA polymerase in autosomal dominant progressive external ophthalmoplegia. Nat Struct Mol Biol. 2004;11:770–776. doi: 10.1038/nsmb805. [DOI] [PubMed] [Google Scholar]
- 12.Lewis W, Day BJ, Kohler JJ, Hosseini SH, Chan SSL, Green E, Haase CP, Keebaugh E, Long R, Ludaway T, et al. MtDNA depletion, oxidative stress, cardiomyopathy, and death from transgenic cardiac targeted human mutant polymerase gamma. J Clin Invest. 2007;87:326–335. doi: 10.1038/labinvest.3700523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stuart GR, Santos JH, Strand MK, Van Houten B, Copeland WC. Mitochondrial DNA defects in S. cerevisiae with mutations in DNA polymerase gamma associated with Progressive External Ophthalmolplegia. Hum Mol Genet. 2006;15:363–374. doi: 10.1093/hmg/ddi454. [DOI] [PubMed] [Google Scholar]
- 14.Baruffini E, Lodi T, Dallabona C, Puglisi A, Zeviani M, Ferrero I. Genetic and chemical rescue of the Saccharomyces cerevisiae phenotype induced by mitochondrial DNA polymerase mutations associated with progressive external ophthalmoplegia in humans. Hum Mol Genet. 2006;15:2846–2855. doi: 10.1093/hmg/ddl219. [DOI] [PubMed] [Google Scholar]
- 15.Beckman KB, Saljoughi S, Mashiyama ST, Ames BN. A simpler, more robust method for the analysis of 8-oxoguanine in DNA. Free Radic Biol Med. 2000;29:357–367. doi: 10.1016/s0891-5849(00)00316-6. [DOI] [PubMed] [Google Scholar]
- 16.Beckman KB, Ames BN. Endogenous oxidative damage of mtDNA. Mutat Res. 1999;424:51–58. doi: 10.1016/s0027-5107(99)00007-x. [DOI] [PubMed] [Google Scholar]
- 17.Pinz KG, Shibutani S, Bogenhagen DF. Action of mitochondrial DNA polymerase gamma at sites of base loss or oxidative damage. J Biol Chem. 1995;270:9202–9206. doi: 10.1074/jbc.270.16.9202. [DOI] [PubMed] [Google Scholar]
- 18.Hsu GW, Ober M, Carell T, Beese LS. Error-prone replication of oxidatively damaged DNA by a high-fidelity DNA polymerase. Nature. 2004;431:217–221. doi: 10.1038/nature02908. [DOI] [PubMed] [Google Scholar]
- 19.Krahn JM, Beard WA, Miller H, Grollman AP, Wilson SH. Structure of DNA polymerase Beta with the mutagenic DNA lesion 8-oxodeoxyguanine reveals structural insights into its coding potential. Structure (Camb) 2003;11:121–127. doi: 10.1016/s0969-2126(02)00930-9. [DOI] [PubMed] [Google Scholar]
- 20.Brieba LG, Eichman BF, Kokoska RJ, Doublie S, Kunkel TA, Ellenberger T. Structural basis for the dual coding potential of 8-oxoguanosine by a high-fidelity DNA polymerase. EMBO J. 2004;23:3452–3461. doi: 10.1038/sj.emboj.7600354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furge LL, Guengerich FP. Analysis of nucleotide insertion and extension at 8-oxo-7,8-dihydroguanine by replicative T7 polymerase exo- and human immunodeficiency virus-1 reverse transcriptase using steady-state and pre-steady-state kinetics. Biochemistry. 1997;36:6475–6487. doi: 10.1021/bi9627267. [DOI] [PubMed] [Google Scholar]
- 22.Furge LL, Guengerich FP. Pre-steady-state kinetics of nucleotide insertion following 8-oxo-7,8-dihydroguanine base pair mismatches by bacteriophage T7 DNA polymerase exo. Biochemistry. 1998;37:3567–3574. doi: 10.1021/bi9722094. [DOI] [PubMed] [Google Scholar]
- 23.Brieba LG, Kokoska RJ, Bebenek K, Kunkel TA, Ellenberger T. A lysine residue in the fingers subdomain of T7 DNA polymerase modulates the miscoding potential of 8-oxo-7,8-dihydroguanosine. Structure (Camb) 2005;13:1653–1659. doi: 10.1016/j.str.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 24.Lim SE, Ponamarev MV, Longley MJ, Copeland WC. Structural determinants in human DNA polymerase gamma account for mitochondrial toxicity from nucleoside analogs. J Mol Biol. 2003;329:45–57. doi: 10.1016/s0022-2836(03)00405-4. [DOI] [PubMed] [Google Scholar]
- 25.Hanes JW, Thal DM, Johnson KA. Incorporation and replication of 8-Oxo-deoxyguanosine by the human mitochondrial DNA polymerase. J Biol Chem. 2006;281:36241–36248. doi: 10.1074/jbc.M607965200. [DOI] [PubMed] [Google Scholar]
- 26.Boosalis MS, Petruska J, Goodman MF. DNA polymerase insertion fidelity. Gel assay for site-specific kinetics. J Biol Chem. 1987;262:14689–14696. [PubMed] [Google Scholar]
- 27.Lim SE, Copeland WC. Differential incorporation and removal of antiviral deoxynucleotides by human DNA polymerase gamma. J Biol Chem. 2001;276:23616–23623. doi: 10.1074/jbc.M101114200. [DOI] [PubMed] [Google Scholar]
- 28.Johnson AA, Ray AS, Hanes J, Suo Z, Colacino JM, Anderson KS, Johnson KA. Toxicity of antiviral nucleoside analogs and the human mitochondrial DNA polymerase. J Biol Chem. 2001;276:40847–40857. doi: 10.1074/jbc.M106743200. [DOI] [PubMed] [Google Scholar]
- 29.Longley MJ, Nguyen D, Kunkel TA, Copeland WC. The fidelity of human DNA polymerase gamma with and without exonucleolytic proofreading and the p55 accessory subunit. J Biol Chem. 2001;276:38555–38562. doi: 10.1074/jbc.M105230200. [DOI] [PubMed] [Google Scholar]
- 30.Kang D, Nishida J, Iyama A, Nakabeppu Y, Furuichi M, Fujiwara T, Sekiguchi M, Takeshige K. Intracellular localization of 8-oxo-dGTPase in human cells, with special reference to the role of the enzyme in mitochondria. J Biol Chem. 1995;270:14659–14665. doi: 10.1074/jbc.270.24.14659. [DOI] [PubMed] [Google Scholar]
- 31.Nakabeppu Y, Sakumi K, Sakamoto K, Tsuchimoto D, Tsuzuki T, Nakatsu Y. Mutagenesis and carcinogenesis caused by the oxidation of nucleic acids. Biol Chem. 2006;387:373–379. doi: 10.1515/BC.2006.050. [DOI] [PubMed] [Google Scholar]
- 32.Pavlov YI, Minnick DT, Izuta S, Kunkel TA. DNA replication fidelity with 8-oxodeoxyguanosine triphosphate. Biochemistry. 1994;33:4695–4701. doi: 10.1021/bi00181a029. [DOI] [PubMed] [Google Scholar]
- 33.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 34.Zheng W, Khrapko K, Coller H, Thilly WG, Copeland WC. Origins of human mitochondrial point mutations as DNA polymerase gamma-mediated errors. Mutat Res. 2006;599:11–20. doi: 10.1016/j.mrfmmm.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 35.Bohr VA. Repair of oxidative DNA damage in nuclear and mitochondrial DNA, and some changes with aging in mammalian cells. Free Radic Biol Med. 2002;32:804–812. doi: 10.1016/s0891-5849(02)00787-6. [DOI] [PubMed] [Google Scholar]
- 36.O'Rourke TW, Doudican NA, Mackereth MD, Doetsch PW, Shadel GS. Mitochondrial dysfunction due to oxidative mitochondrial DNA damage is reduced through cooperative actions of diverse proteins. Mol Cell Biol. 2002;22:4086–4093. doi: 10.1128/MCB.22.12.4086-4093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doudican NA, Song B, Shadel GS, Doetsch PW. Oxidative DNA damage causes mitochondrial genomic instability in Saccharomyces cerevisiae. Mol Cell Biol. 2005;25:5196–5204. doi: 10.1128/MCB.25.12.5196-5204.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Croteau DL, Stierum RH, Bohr VA. Mitochondrial DNA repair pathways. Mutat Res. 1999;434:137–148. doi: 10.1016/s0921-8777(99)00025-7. [DOI] [PubMed] [Google Scholar]
- 39.Yamaguchi H, Kajitani K, Dan Y, Furuichi M, Ohno M, Sakumi K, Kang D, Nakabeppu Y. MTH1, an oxidized purine nucleoside triphosphatase, protects the dopamine neurons from oxidative damage in nucleic acids caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Cell Death Differ. 2006;13:551–563. doi: 10.1038/sj.cdd.4401788. [DOI] [PubMed] [Google Scholar]
- 40.de Souza-Pinto NC, Eide L, Hogue BA, Thybo T, Stevnsner T, Seeberg E, Klungland A, Bohr VA. Repair of 8-oxodeoxyguanosine lesions in mitochondrial DNA depends on the oxoguanine dna glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial DNA of OGG1-defective mice. Cancer Res. 2001;61:5378–5381. [PubMed] [Google Scholar]
- 41.Karahalil B, de Souza-Pinto NC, Parsons JL, Elder RH, Bohr VA. Compromised incision of oxidized pyrimidines in liver mitochondria of mice deficient in NTH1 and OGG1 glycosylases. J Biol Chem. 2003;278:33701–33707. doi: 10.1074/jbc.M301617200. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Reddy S, Beard WA, Wilson SH, Schlick T. Differing conformational pathways before and after chemistry for insertion of dATP vs. dCTP opposite 8-oxoG in DNA polymerase β. Biophys J. 2007;92:3063–3070. doi: 10.1529/biophysj.106.092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wanrooij S, Luoma P, van Goethem G, van Broeckhoven C, Suomalainen A, Spelbrink JN. Twinkle and POLG defects enhance age-dependent accumulation of mutations in the control region of mtDNA. Nucleic Acids Res. 2004;32:3053–3064. doi: 10.1093/nar/gkh634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mancuso M, Filosto M, Oh SJ, DiMauro S. A novel polymerase gamma mutation in a family with ophthalmoplegia, neuropathy, and Parkinsonism. Arch Neurol. 2004;61:1777–1779. doi: 10.1001/archneur.61.11.1777. [DOI] [PubMed] [Google Scholar]
- 45.Davidzon G, Greene P, Mancuso M, Klos KJ, Ahlskog JE, Hirano M, Dimauro S. Early-onset familial parkinsonism due to POLG mutations. Ann Neurol. 2006;59:859–862. doi: 10.1002/ana.20831. [DOI] [PubMed] [Google Scholar]
- 46.Tiangyou W, Hudson G, Ghezzi D, Ferrari G, Zeviani M, Burn DJ, Chinnery PF. POLG1 in idiopathic Parkinson disease. Neurology. 2006;67:1698–1700. doi: 10.1212/01.wnl.0000238963.07425.d5. [DOI] [PubMed] [Google Scholar]
- 47.DiMauro S. Mitochondrial involvement in Parkinson's disease: the controversy continues. Neurology. 1993;43:2170–2172. doi: 10.1212/wnl.43.11.2170. [DOI] [PubMed] [Google Scholar]
- 48.Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 49.Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38:518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- 50.Vyas I, Heikkila RE, Nicklas WJ. Studies on the neurotoxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: inhibition of NAD-linked substrate oxidation by its metabolite, 1-methyl-4-phenylpyridinium. J Neurochem. 1986;46:1501–1507. doi: 10.1111/j.1471-4159.1986.tb01768.x. [DOI] [PubMed] [Google Scholar]
- 51.Greene JG, Dingledine R, Greenamyer JT. Gene expression profiling of rat midbrain dopamine neurons: implications for selective vulnerability in parkinsonism. Neurobiol Dis. 2005;18:19–31. doi: 10.1016/j.nbd.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 52.Nakabeppu Y, Tsuchimoto D, Yamaguchi H, Sakumi K. Oxidative damage in nucleic acids and Parkinson's disease. J Neurosci Res. 2007;85:919–934. doi: 10.1002/jnr.21191. [DOI] [PubMed] [Google Scholar]
- 53.Fukae J, Mizuno Y, Hattori N. Mitochondrial dysfunction in Parkinson's disease. Mitochondrion. 2007;7:58–62. doi: 10.1016/j.mito.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 54.Shimura-Miura H, Hattori N, Kang D, Miyako K, Nakabeppu Y, Mizuno Y. Increased 8-oxo-dGTPase in the mitochondria of substantia nigral neurons in Parkinson's disease. Ann Neurol. 1999;46:920–924. [PubMed] [Google Scholar]
- 55.Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, Jenner P, Halliwell B. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem. 1997;69:1196–1203. doi: 10.1046/j.1471-4159.1997.69031196.x. [DOI] [PubMed] [Google Scholar]
- 56.Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, Montine TJ. Parkinson's disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol. 1999;154:1423–1429. doi: 10.1016/S0002-9440(10)65396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shults CW, Oakes D, Kieburtz K, Beal MF, Haas R, Plumb S, Juncos JL, Nutt J, Shoulson I, Carter J, et al. Effects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol. 2002;59:1541–1550. doi: 10.1001/archneur.59.10.1541. [DOI] [PubMed] [Google Scholar]
- 58.Longley MJ, Ropp PA, Lim SE, Copeland WC. Characterization of the native and recombinant catalytic subunit of human DNA polymerase gamma: identification of residues critical for exonuclease activity and dideoxynucleotide sensitivity. Biochemistry. 1998;37:10529–10539. doi: 10.1021/bi980772w. [DOI] [PubMed] [Google Scholar]
- 59.Lim SE, Longley MJ, Copeland WC. The mitochondrial p55 accessory subunit of human DNA polymerase gamma enhances DNA binding, promotes processive DNA synthesis, and confers N-ethylmaleimide resistance. J Biol Chem. 1999;274:38197–38203. doi: 10.1074/jbc.274.53.38197. [DOI] [PubMed] [Google Scholar]
- 60.Graziewicz MA, Sayer JM, Jerina DM, Copeland WC. Nucleotide incorporation by human DNA polymerase gamma opposite benzo[a]pyrene and benzo[c]phenanthrene diol epoxide adducts of deoxyguanosine and deoxyadenosine. Nucleic Acids Res. 2004;32:397–405. doi: 10.1093/nar/gkh213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reardon JE, Miller WH. Human immunodeficiency virus reverse transcriptase. Substrate and inhibitor kinetics with thymidine 5′-triphosphate and 3′-azido-3′-deoxythymidine 5′-triphosphate. J Biol Chem. 1990;265:20302–20307. [PubMed] [Google Scholar]
- 62.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 63.Notredame C, Higgins DG, Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 64.Hooft RW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 65.Rodriguez R, Chinea G, Lopez N, Pons T, Vriend G. Homology modeling, model and software evaluation: three related resources. Bioinformatics. 1998;14:523–528. doi: 10.1093/bioinformatics/14.6.523. [DOI] [PubMed] [Google Scholar]
- 66.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 67.Luthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 68.Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]